

Abstract
N -glycosylation in the endoplasmic reticulum (ER) consists of the transfer of a pre-assembled glycan conserved among species (Glc3Man9GlcNAc2) from a lipid donor to a consensus sequence within a nascent protein that is entering the ER. The protein-linked glycans are then processed by glycosidases and glycosyltransferases in the ER producing specific structures that serve as signalling molecules for the fate of the folding glycoprotein: to stay in the ER during the folding process, to be retrotranslocated to the cytosol for proteasomal degradation if irreversibly misfolded, or to pursue transit through the secretory pathway as a mature glycoprotein. In the ER, each glycan signalling structure is recognized by a specific lectin. A domain similar to that of the mannose 6-phosphate receptors (MPRs) has been identified in several proteins of the secretory pathway. These include the beta subunit of glucosidase II (GII), a key enzyme in the early processing of the transferred glycan that removes middle and innermost glucoses and is involved in quality control of glycoprotein folding in the ER (QC), the lectins OS-9 and XTP3-B, proteins involved in the delivery of ER misfolded proteins to degradation (ERAD), the gamma subunit of the Golgi GlcNAc-1-phosphotransferase, an enzyme involved in generating the mannose 6-phosphate (M6P) signal for sorting acidic hydrolases to lysosomes, and finally the MPRs that deliver those hydrolytic enzymes to the lysosome. Each of the MRH-containing proteins recognizes a different signalling N-glycan structure. Three-dimensional structures of some of the MRH domains have been solved, providing the basis to understand recognition mechanisms.
Keywords: Endoplasmic reticulum, Glucosidase II, glycoprotein, MRH domain, glycosylation, N-glycan, Quality control, Secretory pathway
1) N-glycosylation
One third of the proteins synthesized by eukaryotic cells will be part of the secretory pathway. Proteins enter this pathway through the endoplasmic reticulum (ER), either co- or post-translationally. Once in the ER, proteins acquire their native tertiary structure, oligomerize and undergo post-translational modifications such as signal peptide removal, disulfide bridge formation, proline isomerization and N-glycosylation. Almost 70 % of the proteins of the secretory pathway are N-glycosylated [1]. N-linked glycosylation is a protein modification specific to asparagine residues whose basic features have been known for many years [2]. However, in recent years the surprising discovery was made that N-glycosylation occurs not only in Eukaryotic cells but also in Bacteria and Archaea [3]. During N-glycosylation in eukaryotic cells a glycan (Figure 1A) is transferred by the translocon-associated oligosaccharyltransferase complex (OST) to the Asn residue in the consensus sequence Asn-X-Ser/Thr, in which X cannot be Pro [4]. This consensus sequence is N-glycosylated co-translationally as the nascent polypeptide emerges into the ER lumen through the translocon [5], although in some cases N-glycosylation may occur post-translationally [6]. N-glycans are then modified in the ER and the Golgi complex by removal and addition of different sugar residues to produce a high variety of structures.
Figure 1. Processing of N-glycans in the ER.

A. Oligosaccharide Glc3Man9GlcNAc2 (G3M9) is transferred to Asn residues on nascent polypeptides by oligosaccharyltransferase. Lettering a–n indicates the order of addition of the monosaccharides during in vivo synthesis of the Dolichol-PP-Glc3Man9GlcNAc2 precursor. Arm A, B and C indicate the oligosaccharide branch. During biosynthesis residues a–g are added on the cytosolic face of the ER membrane from nucleotide-sugar precursors, while residues h–n are added from Dol-P-Glc or Dol-P-Man precursors after the oligosaccharide has flipped across the membrane. B. After glycan transfer to proteins, Glucosidase I (GI) removes glucose n, Glucosidase II (GII) removes glucose m and l, and UDP-Glc:glycoprotein glucosyltransferase (UGGT) adds glucose l. ER mannosidases may remove mannoseI and k. Monoglucosylated N-glycans are able to interact with ER lectins calnexin (CNX) and/or calreticulin (CRT). ER mannosidase I (ERManI) (MNSI in yeasts) acts as a timer of permanence of glycoproteins in the ER and removes residue I generating M8B. Subsequently, ERManI or other ER mannosidases (probably EDEM in mammals or Htm1in yeasts) may remove residue k, generating M7. Demannosylated glycans constitute the ERAD signal. Dotted lines are used to indicate that GII activity is reduced toward demmanosylated species.
N-glycosylation is one of the most relevant post-translational protein modifications. First, N-glycans have key roles in molecular recognition events, and due to their diverse composition they increase the cell’s proteome. Second, N-glycans may change the biophysical behavior of glycoproteins by increasing protein solubility, reducing aggregation, and influencing the folding process by modulating the local conformational preferences of the glycosylated sequence [7]. Third, immediately after their transfer to proteins, N-glycans are modified by several ER-resident glycosylhydrolases and glucosyltransferases and the resulting glycans serve as signals to indicate the folding status and the fate of a glycoprotein within the cell. Specific glycan structures are recognized by lectins that participate in cycles of quality control during glycoprotein folding in the ER (QC), in the retro-translocation to the cytosol of terminally misfolded proteins to be degraded by the ubiquitin-proteasome system (ERAD, for ER-associated degradation), or in the sorting of mature proteins to the different compartments in the secretory pathway [8, 9].
a) Biosynthesis of Dol-PP-oligosaccharides
The N-glycosylation pathway initiates with the transfer of a pre-built glycan that in most eukaryotic cells has the composition Glc3Man9GlcNAc2 (G3M9) [10] from a dolichol-P-P derivative to the consensus sequence Asn-X-Ser/Thr of nascent proteins. The biosynthesis of this precursor starts in the cytosolic side of the ER membrane, where nucleotide-activated sugars (UDP-GlcNAc, GDP-Man) serve as substrates for different glycosyltransferases coded by alg (for asparagine-linked glycosylation) genes that act in an ordered manner to produce a Man5GlcNAc2-Dol-PP precursor. This precursor is then translocated across the ER membrane where four mannoses and three glucoses are added from Dol-P-Man or Dol-P-Glc substrates in the luminal side of the ER membrane. The order of addition of each monosaccharide is depicted in Figure 1A [11]. Although the G3M9 structure of the glycan that is transferred during N-glycosylation is conserved in animal, plant and fungal species, protozoans transfer unglucosylated glycans to proteins during N-glycosylation (Man9GlcNAc2 (M9) in Trypanosoma, Man7GlcNAc2 (M7) in Critidia and Man6GlcNAc2 (M5) in Leishmania are some examples) [12, 13].
b) Transference of glycans to proteins by the OST complex
OST transfers the glycan G3M9 en bloc to proteins as soon as the glycosylation consensus sequence emerges from the luminal side of the ER at a distance of 12–13 amino acids from the ER membrane [5]. The presence of an emerging Asn-X-Ser/Thr sequence at the appropriate distance is not sufficient for N-glycosylation. There is evidence suggesting that this covalent modification is produced in sequons that are less prone to acquire a secondary structure quickly or are located in a flexible region of the polypeptide, (i.e., loop and turn regions) [11, 14]. In addition, N-glycosylation at noncanonical Asn-X-Cys sequences has been reported in mammals, yeast and plants [15–17]. In budding yeasts and mammals, OST is a multisubunit protein bearing one catalytic subunit (STT3) and seven-eight regulatory subunits that is associated in many eukaryotic systems not only with the translocon, but also with the ribosome [18]. In these organisms, OST transfers G3M9 about 10- to 25-fold faster than M9. Mutant cells unable to elongate Dolichol-PP-M9 (alg mutants) exhibit underglycosylation of sequons. This, in turn, produces deleterious effects on the proper folding of many glycoproteins [11, 19]. Several variants of a human disease called Congenital Disorder of Glycosylation type I are due to the underglycosylation of proteins [20]. As mentioned above, nonglucosylated oligosaccharides are the natural glycan transferred in some protozoan species [13]. Their OST (as well as in Bacteria and Archaea), however, consists of only the catalytic subunit STT3 and has broader spectra of specificity for the glycan to be transferred [21].
2) Processing in the ER of the protein-linked oligosaccharides
The half-life of the G3M9 form of the protein-bound oligosaccharide has is very short, as its outermost glucose (residue n in Figure 1) is immediately removed by glucosidase I (GI) after the transfer of the oligosaccharide to the polypeptide nascent chain, thus forming Glc2Man9GlcNAc2 (G2M9) (Figure 1B). GI is a type II membrane α1,2 exoglucosidase associated with the translocon complex in close proximity to OST that belongs to the glycosyl hydrolase family 63 [22]. The remaining two glucoses of the protein-linked G2M9 are removed by glucosidase II (GII), an ER soluble α(1,3) glucosidase belonging to the glycosyl hydrolase family 31. GII possesses a dual activity, successively trimming middle (Glcα1,3Glc bond, GII first cleavage) and innermost (Glcα1,3Man bond, GII second cleavage) glucoses (residues l and m in Figure 1), thus generating Glc1Man9GlcNAc2 (G1M9) and M9 [1, 23]. In addition, the G1M9 glycan structure may be produced in the ER by the activity of a soluble UDP-Glc:glycoprotein glucosyltransferase (UGGT) that adds a glucose (residue l, Figure 1B) only if the protein moiety of the glycoprotein has not yet acquired its native tertiary structure [24–26]. Finally, one or more mannoses may be removed from the N-glycan by ER-resident mannosidases, ER α-mannosidase I and EDEM (Mns1 and Htm1 in Saccharomyces cerevisiae), which progressively remove terminal α1,2-bonded mannose residues of the B arm and the C arm (residues i and k in Figure 1B) generating demannosylated species bearing 8, 7, and in some cases 6 mannose units (M8 isomer B, lacking terminal mannose i of arm B; M7 and M6) [27–29].
What signal(s) is triggered by each glycan structure produced in the ER by the activity of GI, GII, UGGT and ER-mannosidases that impacts protein maturation and sorting? As it will be discussed in the following section, specific N-glycan structures are recognized by QC, ERAD or secretory pathway sorting machineries.
3) Quality control of glycoprotein folding in the endoplasmic reticulum
Protein folding is a complex process and there are several mechanisms in eukaryotic cells to ensure that proteins acquire their native conformation, and protein N-glycosylation is one of them. Each glycan structure generated by a specific glycosidase or glucosyltransferase in the ER provides information that determines the fate of the glycoprotein. The glycan G3M9 is recognized and transferred to proteins by OST during the N-glycosylation reaction. This glycan is deglucosylated immediately after its addition to proteins: the glycan G1M9 formed by the successive action of GI and GII is recognized by lectins/chaperones of the ER, calnexin (CNX), a membrane protein, and calreticulin (CRT) its soluble homologue (Figure 2). This interaction facilitates protein folding by preventing aggregation of folding intermediates and by favoring interaction with ERp57 (a protein disulfide isomerase which forms a complex with CNX) that results in proper disulfide bridge formation. This interaction also prevents an early exit from the ER of intermediates and misfolded glycoproteins [1]. Once deglucosylated by GII’s second cleavage reaction, M9-bearing glycoproteins can no longer interact with CNX or CRT, and depending on its folding status the glycoproteins may then be re-glucosylated by UGGT or continue their transit through the secretory pathway. If not yet completely folded, molten globule structures and hydrophobic surfaces are recognized by UGGT on M9-bearing glycoproteins and the same G1M9 structure is formed again [26, 30]. Cycles of deglucosylation by GII, interaction with CNX or CRT, and re-glucosylation by UGGT (CNX/CRT cycles) continue until the protein is folded and continues its way through the secretory pathway (Figure 2).
Figure 2. Quality control of glycoprotein folding in the endoplasmic reticulum, ERAD and sorting in the secretory pathway.

The glycan G3M9 transferred to proteins during N-glycosylation is immediately trimmed by glucosidases GI and GII. Monoglucosylated species generated by GII may interact with lectin/chaperones CNX or CRT, thus facilitating folding, preventing aggregation and providing a mechanism for ER retention of misfolded species. A second cleavage by GII liberates glycoproteins from the CNX/CRT anchor. This is a check point in the secretory pathway: if the proteins have acquired their native conformation, they can continue to transit through the secretory pathway to their final destination. If not yet properly folded, UGGT adds a glucose unit to allow another round of interactions between misfolded glycoproteins and lectin/chaperones. GII is also responsible for the removal of the glucose added by UGGT. Cycles of deglucosylation and reglucosylation catalyzed by the opposing activities of UGGT and GII continue until the glycoproteins acquire their native tertiary structure, thereby allowing their transit to their final destination. Misfolded/slow-folding species are characterized by ER mannosidase(s) (ERManI/EDEM)-catalyzed N-glycan demannosylation. OS-9 recognizes Manα 1,6Man on the trimmed C arm and facilitates entry of misfolded glycoproteins into the ERAD pathway where the misfolded glycoproteins exit the ER and are degraded by the proteosome in the cytosol. A decrease in N-glycan mannose content significantly diminishes in vivo GII-mediated deglucosylation rates but does not affect in vivo UGGT-mediated glucosylation, thus increasing the possibility of displaying monoglucosylated structures able to interact with CNX/CRT for longer time periods, and providing one more chance to escape from ERAD. If the final destination of a glycoprotein is the lysosome (as for acidic hydrolases), a M6P tag is added by UDP-N-acetylglucosamine:lysosomal enzyme GlcNAc-1-phosphotranfserase (PT) and the N-acetylglucosamine-1-phosphodiester α-N-acetylglucosaminidase (UCE) in the Golgi. M6P receptors (CD-MPR and CI-MPR) recognize this tag and concentrate these proteins in clatrin-coated vesicles that bud from trans Golgi network. MRH domain-containing proteins present in GIIβ subunit, OS-9, PT γ subunit and CD-MPR or CI-MPR are indicated.
If the folding process is too slow or fails, one or more mannose residues are removed by ER-resident mannosidases (residues i and k, Figure 1B). Mannose trimming generates ligands for two α1,6-linked mannose-binding lectins, OS-9 and XTP3-B. These lectins facilitate the transport of misfolded glycoproteins across the ER membrane for degradation [31–33]. However it was reported recently that unlike OS-9, XTP3-B did not enhance the degradation of misfolded glycoproteins but instead protected misfolded proteins bearing M9 glycan structures from degradation [34]. As it will be discussed below, GII activity is reduced upon reduction in mannose content in the B and C arms of the glycan while UGGT activity is not, thus providing an opportunity for slow-folding/misfolded glycoproteins to re-enter the CNX/CRT cycles [35, 36] (Figure 2). It is unknown whether the G2M9 structure triggers a signal, but the recent discovery of Malectin, a membrane protein that recognizes the disaccharide Glcα1,3Glc moiety of this glycan, suggests a possible function for this intermediate in the ER retention of G2M9-misfolded glycoproteins upon ER stress [37, 38]. Accordingly, it was recently shown that Malectin preferentially associates with misfolded glycoproteins and inhibits their secretion [39].
4) Glucosidase II
GII plays a key role in the quality control of glycoprotein folding within the ER. First, the removal of the middle glucose residue in G2M9-bearing glycoproteins catalyzed by GII’s first cleavage step generates monoglucosylated species able to interact with CNX and/or CRT; second, GII removal of the innermost glucose in G1M9-glycoproteins liberates their association with CNX/CRT allowing exiting from QC and finally, GII is also responsible for the removal of the glucose added by UGGT (Figure 2). GII is a soluble ER heterodimer whose subunits are tightly but non-covalently bound. The heterodimeric nature of GII was initially described by Helenius and co-workers when a 100–120 KDa polypeptidic chain from rat liver (GIIα) reported previously to bear the catalytic activity was unable to be separated from a smaller, uncharacterized polypeptide chain (GIIβ) using conditions close to denaturation [40]. GII heterodimeric composition was also demonstrated genetically using strains of Schizosaccharomyces pombe (a fission yeast that displays a QC mechanism similar to that of mammalian cells) lacking either GIIα or GIIβ coding genes. Microsomes purified from both strains were completely devoid of GII trimming activity in vitro toward the glycan G1M9, demonstrating that both subunits were required for GII full activity. In vivo studies showed, however, that although mutants lacking GIIα formed only G2M9 glycans bound to proteins after a 15 minute-labeling indicating that GII activity was completely abolished, small amounts of G1M9 were formed in the ER of mutants lacking GIIβ, thus confirming the catalytic role of GIIα subunit [41].
GIIα is conserved in divergent organisms including yeast, mammals, parasites and plants [40–44]. This subunit bears the (G/F)(L/I/V/M)WXDMNE) consensus sequence of the active site of family 31 glycosyl hydrolases. Mutation of aspartic acid or glutamic acid of this sequence completely abolishes the enzymatic activity of GII [45]. GII has a nearly neutral pH optimum, no cation requirements and is inhibited by 1-deoxynojirimycin (DNJ), castanospermine and bromoconduritol [46]. Recently, N-alkylated DNJ inhibitors of GII have been developed and evaluated for their antiviral effect in cells upon dengue virus infection [47]. It has been proposed that the kinetics of the first glucose cleavage mediated by GII (formation of G1M9) is much faster than the second glucose cleavage reaction (formation of M9) [46, 48]. This rate differential would increase the life-time of the monoglucosylated species able to interact with CNX and CRT, even in the absence of UGGT activity as occurring in a few organisms as S. cerevisiae [49]. Other work suggests, however, that the different kinetics of the first and second GII-trimming reactions may not be significative at the high protein concentrations present in the ER lumen [50]. Interestingly, GIIα subunit was recently proposed as a potential novel cancer biomarker as it was reported to be frequently overexpressed in human lung tumor tissues and exhibited a stress response similar to p53 [51].
a) Substrate specificity of GII
GII activity has been determined using two types of substrates: small artificial analogues such as p-nitro phenyl-α-D-glucopyranoside (pNPG) or N-glycans (G2M9, G1M9 and derivatives such as methotrexate (MTX)-linked glycans) [23, 40, 41, 52, 53]. GII heterodimer and isolated GIIα subunit activities toward pNPG were similar. However, the dramatically reduced activity of the isolated GIIα subunit toward N-glycans compared to the GIIα/GIIβ heterodimer prompted the study of the role of GIIβ subunit in N-glycan deglucosylation by GIIα [54, 55]. The roles of GIIβ subunit will be reviewed in the following sections.
Several years ago it was determined that the GII deglucosylation activity in vitro toward high mannose N-glycans decreased dramatically when the mannose content of the B- or C-arm of the glycan was reduced [35]. Later, it was shown that the order of rat liver GII deglucosylation initial velocities of MTX-conjugated substrates was G1M9-MTX ≥ G1M8B-MTX (lacking terminal mannose i of arm B, Figure 1A) > G1M8C-MTX (lacking terminal mannose k of arm C) > G1M7-MTX (lacking terminal mannose residues i, k of arms B and C, Figure 1A), suggesting that the outermost mannose of the C-arm of the glycan was involved in substrate recognition by GII [53]. More recently, the in vivo activity of GII was analyzed in S. pombe mutant cells in which the biosynthesis of the Dolichol-PP glycan was interfered by mutating one or more alg genes. The transferred glycans were G2M9, G2M7 (lacking mannoses j and k of arm C), G2M6 (lacking mannose i of arm B and mannoses j and k of arm C) or G2M5 (lacking mannoses h and i, of arm B and mannoses j and k, of arm C) in the different mutants (Figure 1A) [36]. The cells were pulse-labelled and the glycan pattern produced after 15 min in the presence of dithiotreitol (to avoid glycoprotein exiting from the ER) was analyzed. Measurement of the relative proportion of di-glucosylated, monoglucosylated and unglucosylated species (G2M9, G1M9 and M9; G2M7, G1M7 and M7; G2M6, G1M6 and M6; and G2M5, G1M5 and M5) showed that the in vivo activity of GII was progressively reduced as the mannose content of B and C arms decreased. Because in vivo GII activity toward M6 was observed to be even lower than that observed toward M7, this indicates that not only is the outermost mannose of arm C important for GII heterodimer activity, but also the outermost mannose of arm B, although to a less extent. Surprisingly, the in vivo activity of UGGT was not dependent on glycoprotein mannose content. These results suggested that upon demannosylation of slow folding glycoproteins, the relative activities of GII and UGGT would shift the equilibrium toward the monoglucosylated forms that are able to interact with CNX, giving yet another chance for slow-folding demannosylated proteins to enter the QC cycles and fold properly (Figures 1 and 2) [36].
b) Glucosidase II beta subunit
The study of GIIβ subunit has been the object of increasing interest as polycystic liver disease, an autosomal dominant disorder characterized by the appearance of fluid-filled cysts in the liver and an increased liver volume may develop in individuals bearing mutations in GIIβ gene. This gene is also known as hepatocystin, PRKCSH or protein kinase C substrate 80H-K [56–59]. It was postulated that defective quality control of proteins in patients with PRKCSH disrupts hepatic homeostasis. However, the precise molecular mechanisms of the outcome of the disease are not known. Recently it was shown that the knockdown of hepatocystin induces autophagy through a mammalian target of rapamycin (mTOR)-dependent pathway [60].
GIIβ subunit is a 50–70 kDa polypeptide non-covalently bound to GIIα subunit [40]. GIIβ bears a signal peptide sequence to deliver proteins into the ER and a canonical ER retention/retrieval signal HisAspGluLeu at its C-terminus [40, 41]. In some species it may contain one or two EF hand Ca++ binding domains and a glutamic acid-rich motif. Absolutely conserved among GIIβs is a C-terminal domain homologous to the mannose 6-phosphate receptor (MPR) lectin domain (MRH) [61]. Another GIIβ conserved domain (G2B domain) is involved in GIIα-GIIβ interaction [36, 62, 63] (see Figure 3). GIIβ has been suggested to be responsible for GIIα folding, maturation or stability in mammals, heterodimer localization in the ER (GIIα lacks any known ER retention signal), and/or enhancing GIIα N-glycan processing rates [40, 41, 54, 55, 63–67].
Figure 3. Proposed models for GIIβ MRH domain-mediated enhancement of N-glycan deglucosylation.

GII is a heterodimer composed of a GIIα catalytic and a GIIβ regulatory subunit. GIIβ enhances GII deglucosylation activity towards both G2M9 and G1M9 through its MRH domain. Upon binding mannose units in the B and/or C arms of the glycan, the GIIβ MRH domain presents bonds to be cleaved to the GIIα catalytic site (star). GIIβ also provides the retention/retrieval signal for proper ER localization of the heterodimer (−ValAspGluLeu (VDEL) in S. pombe). G2B domain is involved in GIIα-GIIβ interaction. Residues Gln-384, Arg-414, Glu-433, Tyr-439 (QREY) form the binding pocket that is the “signature motif” for MRH domain-containing proteins. There are two possible models for the role of Trp-409 in GII activity: In (A) mannose-binding essential residues Gln-384, Arg-414, Glu-433, Tyr-439 form a pocket which binds arm C of the glycan, while residue Trp-409 (W) interacts with arm B. This bidentate interaction allows the glucose-containing arm A to be juxtaposed to GIIα’s catalytic site. In (B) Trp-409 interacts with other regions of the β-subunit and influences its affinity for N-glycans. These models suggest that removal of mannoses by ER mannosidases will reduce both the binding of the glycan and GII activity.
i. Role of GIIβ on GIIα folding, maturation or activity
Although it was initially suggested that GIIβ was necessary for proper GIIα folding or stability [66, 67], work of Trombetta et al. showed that GIIβ subunit could be specifically proteolyzed from GII heterodimer without loosing activity toward the small substrate analogue pNPG [68]. A more recent work showed that the same GIIβ-proteolyzed preparation was unable to trim G2M9 or G1M9 physiological substrates of GII, demonstrating a key role of GIIβ subunit in N-glycan trimming [54]. Total cell extracts of mutant S. pombe cells lacking endogenous GIIβ were active towards pNPG, indicating that GIIβ is not required for GIIα folding or maturation. However, in vivo trimming of G2M9 and G1M9 of ER folding proteins was severely delayed in those mutants, thus confirming that GIIβ subunit was required for N-glycan trimming by GII [41, 54]. Accordingly, Wilkinson et al. showed that GIIα subunit was as stable alone as in the heterodimer in S. cerevisiae [63]. Watanabe et al. also demonstrated in vitro that GIIβ was not required for pNPG processing in Aspergillus oryzae [55]. In summary, all recent evidence leads to the conclusion that GIIβ is not involved in GIIα folding, maturation, stability or in the activity toward pNPG (at least in S. pombe, S. cerevisiae and in A. oryzae), but that it is involved in N-glycan trimming by GII.
ii. Role of GIIβ on GII localization within the ER
GII has been shown to be present in the ER lumen [69]. This localization correlates well with that of UGGT and CRT [70] also involved in QC. However, GIIα subunit lacks in its sequence any known ER localization signal. As GIIβ of fungal and mammalian cells bear the canonical C-terminal XAspGluLeu-like retention/retrieval sequence of ER resident soluble proteins, it was proposed that GIIβ is responsible for the heterodimer subcellular localization due to its tight interaction with GIIα [40, 41]. Removal of the HisAspGluLeu signal of human GIIβ increased GIIα secretion in COS7 cells [67]. In agreement with these studies, a reduced ER amount of GIIα was present in microsomal fractions of S. pombe cells lacking GIIβ. GIIα ER levels could be recovered to those of wild-type cells when the ValAspGluLeu C terminal signal present in GIIβ was added to GIIα [54]. Although there is a consensus opinion attributing to GIIβ the role of retaining GIIα catalytic subunit in the ER, one or more unknown ER localization mechanisms seem to be operating as well. For instance, a normal ER localization of GIIβ occurred in cells in which the GIIβ ValAspGluLeu C terminal sequence was occluded by a YFP tag [54]. In S. cerevisiae, GIIβ localized to the ER although it does not display any known retention/retrieval sequence [63]. Moreover, in S. pombe cells lacking GIIβ, GIIα content was still between 20–50 % of that of wild type cells, suggesting that also GIIα may bear another unknown ER localization signal [54].
iii. Role of GIIβ on N-glycan trimming
The newest role proposed for GIIβ subunit in the QC is that of being a lectin that enhances GIIα-mediated N-glycan trimming both in vivo and in vitro [54, 55, 63]. Work of Deprez et al. showed that the association of GII with a single-glycan-bearing protein was independent of its hydrolytic activity, but that trimming of the middle glucose by GII (residue n in Figure 1A) only occurred efficiently when a second glycan was present in the polypeptide chain. The authors proposed that the second glycan would bind to the MRH domain present in GIIβ subunit and this would change the conformation of the protein, thus activating GIIα [64]. Wilkinson et al. showed later that S. cerevisiae mutants lacking GIIβ subunit were defective in the processing of monoglucosylated glycans [63]. A more recent work by Stigliano et al. showed that S. pombe cells lacking GIIβ completely abolished in vitro trimming of glucose units from both G2M9 and G1M9. Moreover, microsomes of S. pombe cells in which GIIα content was normal (cells lacking both endogenous GIIα and GIIβ and expressing GIIα with a ValAspGluLeu retention signal) showed an activity towards pNPG similar to that of wild type cells while no in vitro activity was observed when N-glycans were used as substrates. Also, G2M9 and G1M9 trimming was severely delayed in vivo demonstrating that GIIβ is required for efficient N-glycan trimming by GII [54]. Similar conclusions were made by Watanabe et al. using microsomes of A. oryzae lacking GIIβ and assaying activity towards N-glycans in vitro [55]. Olson et al. were able to express and purify for the first time recombinant full length GIIβ subunit. This preparation was able to recover in trans the activity toward G1M9 of microsomes of S. pombe cells expressing only GIIα in the ER [71]. The observation that GIIβ is required to efficiently trim N-glycans and that it bears a MRH domain similar to that of MPRs prompted the authors to propose that the GIIβ MRH domain is responsible for N-glycan recognition by GII.
c) The role of GIIβ MRH domain on N-glycan recognition by GII and its influence in QC
Mannose 6-phosphate Receptor Homology (MRH) domains were first described in the two MPRs responsible for delivering lysosomal acid hydrolases from the trans Golgi network (TGN) to endosomal compartments in mammalian cells, but they are also present in a few resident proteins of the ER and Golgi: the GIIβ subunit of GII, the lectins OS-9 and XTP3-B (involved in ERAD), and the gamma subunit of the Golgi GlcNAc-1-phosphotransferase (generates the M6P signal recognized by MPRs) [61] (Figure 4). GIIβ, OS-9 and XTP3-B are ER resident proteins and GlcNAc-1-phosphotransferase and MPRs localize to the Golgi apparatus, with the MPRs recycling between TGN, endosomes and plasma membrane. Two of these proteins are associated in a complex with known enzymatic activity, GII and GlcNAc-1-phosphotransferase, whereas the others are lectins in the secretory pathway.
Figure 4. A) Schematic diagram of human MRH domain-containing proteins.

The location of the MRH domains (blue) is shown. The CI-MPR contains 15 contiguous MRH domains, with MRH domain 13 containing a 48-residue fibronectin type II (FnII) insert (gray). The CD-MPR and CI-MPR are type I integral membrane proteins and the location of the single transmembrane domain is shown by a vertical hatched bar. The number of amino acids in each protein, including the N-terminal signal sequence that is not shown, is indicated. The oligomeric state of the protein is listed (note: the oligomeric state of human OS-9 has not been established). GIIβ is the non-catalytic subunit of the heterodimeric glycosidase, glucosidase II, a resident protein of the ER. OS-9 and XTP3-B are also resident proteins of the ER. The γ-subunit of GlcNAc-1-phosphotransferase is the non-catalytic subunit of this hexameric glycosyltransferase localized to early Golgi compartments. CD-MPR and CI-MPR constitutively recycle between TGN, endosomes and plasma membrane. ER = endoplasmic reticulum, TGN = trans Golgi network. B) Structure-based sequence alignment. Structure-based sequence alignment of the MRH domains of bovine CD-MPR (A27068), domains 3, 5 and 9 of the bovine CI-MPR (A30788), human GII β subunit (CAA04006), human GlcNAc-1-phosphotransferase γ subunit (Q9UJJ9), the N- and C-terminal MRH domains of human XTP3-B (NP_056516), and human OS-9 (BAA24363). GII β subunit, OS-9, XTP3-B, and GlcNAc-1-phosphotransferase γ subunit contain all four essential residues for M6P recognition (Gln, Arg, Glu, and Tyr), which are shaded in red. The WW motif of OS-9 is boxed in orange. Y679, which is present in the binding pocket of domain 5, is boxed in blue. The residues known to bind the phosphate group in the CD-MPR (D103, N104, H105) and CI-MPR MRH domain 3 (S386) are boxed in red. The conserved tryptophan residue in loop C of GII β subunit is boxed in green. Residues in OS-9 predicted to prevent binding of the phosphate moiety of M6P (D182, L183) are boxed in green. The cysteine residues are shaded in yellow. The secondary structural elements of CD-MPR and OS-9 are shown, with dark blue arrows representing the β-strands and the green line representing an α-helix. Location of loops C and D are shown. PTγ = GlcNAc-1-phosphotransferase γ subunit, Erl1 = XTP3-B N-terminal MRH domain, Erl2 = XTP3-B C-terminal MRH domain
The observation that GIIβ bears an MRH domain lead to the hypothesis that the MRH domain mediates N-glycan recognition by GII [54, 55, 65, 72]. This hypothesis is supported by experiments showing that the N-glycan trimming rates carried out by GII in vitro [53, 54] and in vivo [36] were decreased significantly upon hydrolysis of mannose residues from the B and C arms of the N-glycan, indicating that GIIβ MRH domain acts as a lectin that binds mannose in arms B and/or C but not in arm A. Using frontal affinity chromatography, Hu et al. showed direct binding of the isolated recombinant human GIIβ MRH domain forming a tetramer to high mannose N-glycans coupled to 2-aminopyridine [72]. Stigliano et al. showed that MRH domain-containing proteins with mutations in the conserved amino acids known to be critical for the interaction between MPRs and mannose residues of lysosomal acid hydrolases (“MRH binding pocket signature motif” that includes amino acids Gln, Arg, Glu, Tyr, see Figures 6 and 7) [73, 74] abolished the G2M9 and G1M9 hydrolysis activity by GII in vitro and sharply decreased the deglucosylation of those glycans in vivo without affecting the GIIβ ER localization or GIIα-GIIβ interaction [54]. Olson et al. showed that the isolated MRH domain can compete with full-length GIIβ for N-glycan binding. Moreover, surface plasmon resonance analyses (SPR) of purified MRH domain mutated in those same amino acids proposed to be in the binding pocket showed a decreased binding affinity to high mannose glycoproteins [71].
Figure 6. Comparison of the structures of the MRH domains.
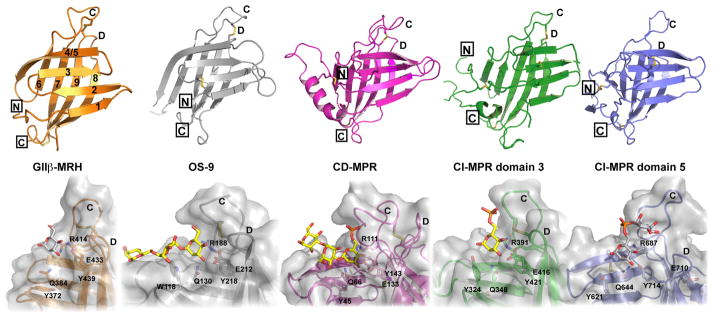
Top, Ribbon diagram of the MRH domains of S. pombe GIIβ (orange, solution structure, PDB ID: 2LVX), human OS-9 (gray, crystal structure, PDB ID: 3AIH), bovine CD-MPR (magenta, crystal structure, PDB ID: 1C39), bovine CI-MPR MRH domain 3 (green, crystal structure, PDB ID: 1SZO), bovine CI-MPR MRH domain 5 (blue, solution structure, PDB ID: 2KVB). Disulfide bridges are shown in yellow, N and C termini are boxed, β-strands are numbered from the N to C terminus, and loops C and D are labeled. Bottom, Close-up view of the carbohydrate binding sites are shown below the respective ribbon diagram. The four essential residues for mannose binding are shown along with the proposed linkage-sensing Tyr or Trp. Molecular surfaces are shown in gray over the ribbon diagram. Carbohydrate ligands are depicted as ball-and-stick. Structures solved in the presence of a bound ligand (Manα1,6Manα1,6Man for OS-9, pentamannosyl phosphate for CD-MPR, M6P for CI-MPR MRH domain 3) are shown in yellow. Modeled ligands are depicted in gray (mannose for GIIβ, methyl-M6P-GlcNAc for CI-MPR MRH domain 5) and are placed in the binding pocket based on superimposition of the four essential residues with a known MRH domain structure containing a bound ligand.
Figure 7. Close-up view of the carbohydrate binding pocket.

Overlay of the four essential residues (Gln, Arg, Glu, Tyr) of S. pombe GIIβ (orange, PDB ID: 2LVX), human OS-9 (gray, PDB ID: 3AIH), bovine CD-MPR (magenta, PDB ID: 1C39), bovine CI-MPR MRH domain 3 (green, PDB ID: 1SZO), bovine CI-MPR MRH domain 5 (blue, PDB ID: 2KVB). Disulfide bridges are shown in yellow and loops C and D are labeled.
These findings demonstrate that the MRH domain is indeed responsible for N-glycan recognition by GIIβ, and a model was proposed for GIIβ MRH domain enhancement of GIIα activity: upon binding mannose in the B and/or C arms of the glycan, the MRH domain positions the arm A of the glycan to be near the catalytic site of GIIα (Figure 3) [54]. This model was an alternative model to that previously proposed by Deprez et al. in which the basal activity of GIIα catalytic site bound to a glycan would be activated by a conformational change produced upon binding of a second glycan in the same glycoprotein to the GIIβ MRH domain [64]. The 3D structure of GIIβ MRH domain was solved by NMR spectroscopy and revealed that the amino acids that constitute the “signature motif” for MRH-containing proteins are positionally conserved in the binding pocket. The model for GIIβ enhancement of GIIα activity was refined and it was proposed that those amino acids bind to the C arm of the glycan [71]. The refined model and the structure of GIIβ MRH domain will be discussed below.
As GII activity in vivo progressively decreases upon demannosylation in B and/or C arms of slow folding/misfolded glycoproteins while UGGT does not, it was proposed that GII would act as a regulator of the permanence of such species in the ER [36]. In the absence of GIIβ, the sensitivity of GII toward N-glycan mannose content is lost. Moreover, when GIIβ subunits containing mutations in the key amino acids of MRH binding pocket are expressed to replace the endogenous GIIβ, the same loss of mannose sensitivity was observed, indicating that GIIβ subunit’s MRH domain was responsible for sensing the mannose content of N-glycans. This observation confers GIIβ MRH domain a key role in quality control of glycoprotein folding in the ER [36] (Figure 2).
5) Other MRH domain-containing proteins in the secretory pathway
As mentioned above, in addition to the β-subunit of glucosidase II, two other resident ER proteins with MRH domains participate in the quality control of glycoprotein folding, OS-9 and XTP3-B. OS-9 and XTP3-B differ from GIIβ in that they are not a regulatory subunit of a glycosidase. Rather, they serve as lectin sensors in an ubiquitin ligase complex to screen glycoproteins for entry into a retro-translocation process across the ER membrane that ultimately leads to their proteasomal degradation in the cytosol. Several proteins with MRH domains are also present in post-ER compartments and serve complementary roles: the γ-subunit of GlcNAc-1-phosphotransferase which generates a M6P tag on newly synthesized acid hydrolases in the Golgi and the recycling receptors, CD-MPR and CI-MPR, that route M6P-tagged acid hydrolases to the lysosome (Figure 4).
a) OS-9
The ER resident protein, OS-9, was originally identified as a protein upregulated in human osteosarcomas [75]. The human OS9 gene (alternate notation is ERLEC2) encodes a 667-residue glycoprotein and has a single MRH domain near its N-terminus (Figure 4A). Unlike the yeast ortholog, Yos9p, mammalian OS-9 does not contain an ER retention signal. Yos9 is a dimer, and the ~170-residue dimerization domain, which is adjacent to the C terminus of the MRH domain, has been crystallized [76].
Much of the studies have been conducted on either the human or S. cerevisiae form of this ER lectin, with many, but not all, of the results being consistent between both species. OS-9 associates with a HRD1-SEL1L ubiquitin ligase complex, although the glycan dependence of OS-9’s interaction with SEL1L remains controversial [31, 33, 77]. OS-9 functions late in the quality control process of glycoprotein folding and plays a key role in determining whether a glycoprotein is targeted for proteasomal degradation (Figure 2). The MRH domain is essential for this process as it recognizes specific N-glycans on misfolded glycoproteins (reviewed in [78]). Both OS-9 and Yos9p bind truncated N-glycans containing a terminal Manα1,6Man sequence on arm C (residue k, Figure 1B and Figure 5) which serves as a glycan degradation signal [28, 31, 79]. However, Yos9p, but not OS-9, binds to a Man3GlcNAc2 glycan containing a terminal Manα1,6Man sequence [80]. Taken together, these studies indicate that entry of misfolded glycoproteins into ERAD requires trimming of the outermost α1,2-linked mannose residue on arm C, with OS-9’s MRH domain serving as the specific carbohydrate sensor.
Figure 5. Summary of glycan specificity of the MRH domain-containing proteins.

Top panel, Binding of glucosidase II β-subunit (GIIβ), OS-9, and XTP3-B to high mannose-type glycans is shown. The glycan binding specificity of XTP3-B remains unresolved, with recent studies indicating a preference for M9 glycan (asterisk-labeled bar) wheras other groups report binding to M5, M6 and M7 glycans. GlcNAc-1-phosphotransferase γ subunit’s ability to interact with specific glycan structures has not yet been determined. Bottom panel, Binding of MPRs to phosphorylated glycans. The CD-MPR and each of the three carbohydate binding domains of the CI-MPR recognize different populations of phosphorylated glycans. For simplicity, only the M6P (P) containing glycans are shown, with the phosphorylated mannose residue highlighted. M6P-GlcNAc-containing glycans have the identical structure, except with a phosphodiester at the analogous position as the phosphomonoester. Domains 1–3 and 9 preferentially bind M6P-containing glycans (grey bars), whereas domain 5 binds only M6P-GlcNAc-containing glycans (open bar). Both MPRs are unable to bind a M7 glycan containing a phosphomannosyl residue on the B (α1,3 linked mannose h) or C (α1,6 linked mannose j) arm. Residues are labeled as in Figure 1A: glucose (blue triangles), mannose (green circles) and GlcNAc (blue squares).
OS-9 may play multiple roles in ERAD. Yos9p has been reported to bind misfolded polypeptides [81]. In addition, recent studies have shown that Yos9p functions to retain misfolded proteins in the ER independent of the glycan degradation signal, even when proteasomal degradation is inhibited [82]. Although more studies are needed to fully understand OS-9’s role(s) in the ER, these studies implicate OS-9 in multiple steps of the ERAD process.
b) XTP3-B
XTP3-B, or Erlectin, is an ER luminal protein. The human ERLEC1 gene encodes a 483-residue glycoprotein that contains two MRH domains. Unlike the CI-MPR, the two MRH domains are not contiguous but are separated by 96 residues of unknown function (Figure 4A). Early studies revealed a difference in the activity of the two MRH domains. Only the C-terminal MRH domain was able to bind Kremen2, a co-receptor involved in early Xenopus development [83]. Subsequent studies indicate that human XTP3-B’s C-terminal MRH domain functions in ERAD by recognizing N-glycans on SEL1L of the HRD1-SEL1L ubiquitin ligase complex [77, 84]. Consistent with the earlier report, only the C-terminal MRH domain was active and was shown to bind glycans containing a terminal Manα1,6Man sequence on arm C [84] (Figure 5). The common feature of binding truncated N-glycans containing a terminal Manα1,6Man sequence suggests that XTP3-B and OS-9 have similar functions in ERAD.
A recent report by Fujimori et al. offers a different view of XTP3-B’s function and glycan specificity [34]. Consistent with previous studies, endogenous XTP3-B was found associated with the HRD1-SEL1L membrane-bound ubiquitin ligase complex. However, XTP3-B’s lectin activity was required for its interaction with an ERAD substrate, not SEL1L. Frontal affinity chromatography analyses demonstrated interaction with a M9 glycan (Figure 5), but not with a known substrate for OS-9 (M7 isomer A lacking mannose residues i and k, see Figure 1B). XTP3-B inhibited the degradation of M9-containing glycoproteins as opposed to OS-9 which enhances the degradation of misfolded glycoproteins. Taken together, these studies implicate a protective, rather than degradative-promoting, role for XTP3-B due to its ability to prevent the premature degradation of newly synthesized glycoproteins bearing M9 glycans [34]. Clearly more studies are needed to rectify these apparent discrepancies in the function and glycan specificity of XTP3-B. In addition, XTP3-B and OS-9 may have a broader role in quality control in that they have been shown to interact with nonglycosylated proteins [85].
c) GlcNAc-1-Phosphotransferase γ-Subunit and lysosomal enzyme targeting
Lysosomes carry out the degradation and recycling of macromolecules that are delivered to them via endocytic, phagocytic, and autophagic pathways. The activity of lysosomes is dependent upon the acquisition of a robust army of hydrolytic enzymes: these acidified organelles contain over 60 soluble acid hydrolases that degrade a diverse array of proteins, lipids, and glycans [86]. The selective delivery of newly synthesized acid hydrolases to lysosomes involves modification of their N-glycans with a unique tag, M6P. In the cis Golgi, UDP-N-acetylglucosamine:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase (GlcNAc-1-phosphotransferase; EC 2.7.8.17) adds GlcNAc-1-phosphate to the C-6 hydroxyl group of selected mannose residues (5 out of the 9 mannoses can become phosphorylated) to form a phosphodiester, M6P-GlcNAc [87–90]. Following this modification, N-acetylglucosamine-1-phosphodiester α-N-acetylglucosaminidase (EC 3.1.4.45), may act to remove GlcNAc in the TGN to reveal a phosphomonoester, M6P, that serves as a high affinity ligand for the CD-MPRs and CI-MPRs (see below) [91–95].
GlcNAc-1-phosphotransferase acts as the initial filter in this process by distinguishing the ~60 acid hydrolases from among the 1,000’s of proteins traveling through the secretory pathway and marking their N-glycans with M6P-GlcNAc. GlcNAc-1-phosphotransferase is an α2β2γ2 heterohexamer produced by two genes. GNPTAB encodes a 1256-residue α/β precursor that is a type III membrane protein with its N and C termini facing the cytosol [96]. The site-1 protease in the Golgi catalyzes the cleavage of the precursor between residues Lys928 and Asp929 to generate the mature membrane proteins (a type II α-subunit and a type I β-subunit), and this processing event is required for its optimum activity [97, 98]. The α/β subunits contain the catalytic activity and binding site for lysosomal enzymes [90, 96]. Recent studies have shown that the DNA methyltransferase-associated protein (DMAP) domain of the α subunit acts to specifically recognize acid hydrolases [99], a recognition event that was previously shown to involve conformation-dependent regions containing two or more lysine residues that must have a defined orientation relative to each other and to the N-glycan chain [100–103].
The GNPTG gene encodes the non-catalytic 305-residue γ subunit. The γ subunit is a soluble glycoprotein that contains a single MRH domain near its N-terminus (Figure 4A) and can form disulfide-linked dimers [104]. The γ subunit appears to regulate the activity of GlcNAc-1-phosphotransferase, including analyses of a patient with a mutation in the GNPTG gene [105] and studies of human macrophages, which lack M6P on their proteins, in which the γ subunit undergoes proteolysis and fails to form a complex with the α/β subunits [106]. In a recent study by Qian et al., in vivo and in vitro studies were used to evaluate the role of the γ subunit. In mice lacking the γ subunit, approximately one third of the acid hydrolases were phosphorylated at levels similar to that observed in wild-type animals while the remainder required the γ subunit for optimal phosphorylation [107]. These studies demonstrate that the γ subunit enhances the activity of the α/β subunits toward a subset of the enzymes. Further analyses showed that, although GlcNAc-1-phosphotransferase can add one or two GlcNAc-P residues to a high mannose glycan on an acid hydrolase, the addition of the second GlcNAc-P is stimulated by the presence of the γ subunit [107]. It is interesting to note that the order of addition is not random: the first GlcNAc-P is almost always added to a mannose on the 6′ arm (i.e., arm C) of the glycan whereas the second GlcNAc-P is added to a mannose on the 3′ arm (i.e., arm A or arm B) of the glycan [108]. Because the MRH domain of the γ subunit contains the four residues critical for carbohydrate recognition (Figure 4B and see below), the notion that the γ-subunit binds high mannose N-glycans and orients the glycan near the catalytic α/β subunits has been proposed [107] and may resemble the GIIβ MRH domain-mediated enhancement of GIIα activity in GII. This hypothesis awaits validation because binding of the γ subunit to high mannose glycans has not been directly demonstrated.
Given the key role GlcNAc-1-phosphotransferase plays in the biogenesis of lysosomes, it is not surprising that it is critical for normal human development as mutations in the GNPTAB gene cause the pediatric lysosomal storage disease mucolipidosis II (MLII, or I-cell disease) and MLIII alpha/beta, whereas mutations in the GNPTG gene cause a milder form of mucolipidosis, MLIII gamma. MLII patients typically die prior to their teenage years from cardiovascular dysfunction as a result of the progressive storage of undigested material in the lysosomes of fibroblasts found in heart valves, endocardium, myocardium, and the perivascular region [109]. Unlike the majority of lysosomal storage diseases that affect a single enzyme in a catabolic pathway, cells from MLII/III patients secrete multiple newly synthesized acid hydrolases because the enzymes are unable to be modified with M6P and be recognized by MPRs.
d) The P-type lectins, CD-MPR and CI-MPR
The P-type lectins, CD-MPR and CI-MPR, are among the most extensively characterized lectins in the secretory pathway (for reviews see [110–112]). The MPRs are the only two members of the P-type lectin family and are unique in their ability to bind phosphomannosyl residues. The only known function of the CD-MPR is targeting newly synthesized acid hydrolases to the lysosome, whereas the CI-MPR is a multifunctional protein that interacts with several different ligands, including M6P-tagged acid hydrolases as well as the non-M6P-containing proteins insulin-like growth factor 2 (IGF-2) [113, 114], plasminogen [115, 116], and urokinase-type plasminogen activator receptor [117, 118]. Transgenic mice lacking the CD-MPR are viable [119]. In contrast, CI-MPR-deficient mice have high serum levels of IGF-2 and the oversized embryos usually die perinatally, a phenotype that can be partially rescued in mice lacking both CI-MPR and IGF-2 [120]. Analyses of fibroblasts derived from mice lacking a single MPR or both MPRs indicate that CD-MPR and CI-MPR complement each other in their acid hydrolase targeting capabilities, as expression of one MPR cannot completely compensate for the absence of the other [121].
MPRs are dimeric type I membrane proteins. CD-MPR encoded by the M6PR gene has an ~160-residue extracytoplasmic region containing a single MRH domain [122, 123]. In contrast, CI-MPR encoded by the IGF2R gene contains a large, ~2300-residue extracytoplasmic region with 15 contiguous MRH domains, three of which bind carbohydrate (domains 3, 5 and 9) (reviewed in [110, 114]). The 13th MRH domain of the CI-MPR differs from all other MRH domains characterized to date in that it has an inserted sequence: a 48-residue fibronectin type II repeat located near the C-terminal region of the domain [113, 124, 125] (Figure 4A). MPRs recycle between endosomes, TGN, and the plasma membrane during their lifetime (reviewed in [126]). MPRs carry newly synthesized M6P-tagged acid hydrolases from the TGN to late endosomal compartments. Optimal ligand binding by these receptors occurs at pH ~6.4, with dissociation from their cargo occurring in the acidic environment of the late endosome [127–132]. The soluble acid hydrolases are packaged into clathrin-coated vesicles that bud from the TGN, shed their coat and fuse with late endosomes. The acid hydrolases enter into lysosomes, whereas the receptors return to the TGN to acquire more cargo or move to the plasma membrane where the CI-MPR, but not the CD-MPR, can endocytose exogenous ligands.
MPRs recognize a heterogeneous population of soluble acid hydrolases in the TGN. This diversity arises from: 1) the number (>50) of different acid hydrolases that exist in monomeric or oligomeric forms, 2) the number and spacial positioning of N-glycans on each hydrolytic enzyme, 3) the presence of one or two phosphomannosyl residues that can be a phosphomonoester (M6P) or a phosphodiester (M6P-GlcNAc), 4) the location of the phosphomannosyl residue at one of five different positions in the glycan chain, and 5) the size of the phosphorylated N-glycan that can vary in its mannose content [87, 88, 133, 134]. To understand how MPRs can efficiently transport this diverse population of acid hydrolases, SPR analyses using acid hydrolases containing phosphomonoesters or phosphodiesters and a solid phase binding assay using a novel phosphorylated glycan array were performed with truncated forms of the MPRs [135–137]. These studies showed that the CD-MPR and MRH domains 3, 5, and 9 of the CI-MPR each recognize a distinct repertoire of phosphorylated glycans [136, 137] (Figure 5). A major difference between the MPRs is that the CI-MPR, but not the CD-MPR, can bind phosphodiester-containing glycans (Figure 5) [128, 136]. Both MRH domains 3 and 9 of the CI-MPR recognize phosphomonoesters. MRH domain 9 interacts with glycan structures M6 to M9 bearing one or two phosphomonoesters (Figure 5). In contrast to MRH domain 9 and the CD-MPR, MRH domain 3 is unable to bind glycans containing a single phosphate on the terminal α1,2-linked mannose of arm C (residue k in Figure 1A; Figure 5). Unlike MRH domains 3 and 9, MRH domain 5 of the CI-MPR recognizes a wide range of phosphodiester-containing glycans, and no significant binding to glycans containing phosphomonoesters is observed (Figure 5) [135, 137]. Internalization of M6P-GlcNAc-containing, but not M6P-containing, acid hydrolases was observed in fibroblasts expressing a mutant CI-MPR that lacked functional M6P binding sites in MRH domains 3 and 9, indicating that MRH domain 5 functions in the context of the full-length receptor to deliver phosphodiester-containing enzymes to the lysosome [138]. Furthermore, the observation that transgenic mice expressing only M6P-GlcNAc-containing acid hydrolases have a normal phenotype [139] indicates that the CI-MPR, due to its unique capacity to bind M6P-GlcNAc-containing ligands, rescues these mice from the severe ML-II-like phenotype observed in mice deficient in GlcNAc-1-phosphotransferase activity. The unique glycan specificities of these three MRH domains allows the CI-MPR to bind a structurally diverse population of phosphorylated N-glycans found on acid hydrolases, and supports previous in vitro [132, 140] and in vivo [121, 141–143] studies which show that the CI-MPR is more efficient than the CD-MPR in delivering acid hydrolases to lysosomes.
6) Structure of the MRH domains of GIIβ, OS-9 and MPRs
a) MRH Fold
The three-dimensional structures of the extracytoplasmic MRH domain of the CD-MPR [144, 145] and eight (domains 1, 2, 3, 5, 11, 12, 13, 14) out of the 15 MRH domains of the CI-MPR are known and have the same fold [138, 146–150]. The crystal structure of human OS-9 MRH domain [151] and the recent solution structure of S. pombe GIIβ MRH domain [71] demonstrate the conservation of the MRH fold in proteins that do not bind phosphorylated mannose residues (Figure 6, top panel). The MRH fold is a flattened nine-stranded β-barrel comprising two antiparallel β-sheets (β1-β4 and β5-β9, with β9 interjecting between β7 and β8) oriented orthogonally over each other. GIIβ and OS-9 have a compact β-barrel structure due to their lack of ~20 amino acids present at the N-terminus of the CD-MPR and CI-MPR domain that form either an α-helix or two to three short β-strands. GIIβ MRH domain contains only two disulfide bonds in contrast to the three (OS-9, CD-MPR, CI-MPR domain 5) or four (CI-MPR domains 3 and 9) disulfide bonds found in the other glycan-binding MRH domains.
b) Conservation of a signature motif for mannose Binding
The CD-MPR was the first MRH domain to be crystallized and its three-dimensional structure solved in the presence of M6P revealed the presence of four residues, Gln, Arg, Glu, and Tyr, that contact the 2-, 3-, and 4-hydroxyl groups of the mannose ring and now serve as a signature motif for mannose recognition by an MRH domain [144, 145]. Structure-based sequence alignments of the MRH domains of the MPRs, GIIβ, OS-9, XTP3-B and γ subunit of GlcNAc-1-phosphotransferase show all four signature motif residues (Gln, Arg, Glu, and Tyr) are found in only three (domains 3, 5, and 9) out of the fifteen MRH domains of the CI-MPR, which are the only MRH domains of the CI-MPR known to have carbohydrate binding activity, as well as in GIIβ, OS-9, both MRH domains of XTP3-B, and the γ subunit of GlcNAc-1-phosphotransferase (Figure 4B) [152]. Mutagenesis studies of these residues verified their essential role by showing that replacement of any of the signature motif residues results in: 1) a dramatic decrease in binding affinity of the MPRs for acid hydrolases [73, 74, 135, 153], 2) inhibition of GII activity [54, 71, 72], 3) inhibition of glycan binding and degradation of ERAD substrates by OS-9 [31, 154] 4) inhibition of XTP3-B’s interaction with an ERAD substrate [34] or component of the ubiquitin ligase complex [77].
c) Carbohydrate binding pockets
The architecture of the MRH domain binding pocket has adapted to accommodate its preferred carbohydrate ligand. Although the length, positioning and residues of loops C and D differ between the MRH domains, these regions, along with the nearly identical position of the four essential residues (Gln, Arg, Glu, and Tyr) (Figure 7), play an important role in ligand binding (Figure 6, bottom panel). In contrast to the residues that interact with the mannose ring, the residues involved in binding the phosphate moiety by the MRH domains of the MPRs are not conserved (Figure 4B).
CD-MPR is ideally designed to bind a phosphomonoester. The longer length of loop D plus the position of loop C results in a binding pocket that in the CD-MPR (Figure 6) tightly surrounds the phosphate moiety, leaving no room in the pocket for a GlcNAc moiety of a phosphodiester. Only the CD-MPR requires divalent cations for optimal ligand binding [74, 127, 155]. A conserved aspartic acid residue (Asp103) in loop C of CD-MPR confers cation-dependence of ligand binding [74]. These mutagenesis studies indicate that a Mn2+ cation in the binding pocket shields the negative charge of Asp103, thereby permitting high affinity binding of the CD-MPR to the phosphate moiety of M6P [144, 145]. In contrast, MRH domain 5 of CI-MPR has a short D loop and lacks a disulfide bond that tethers loops C and D together in the other MRH domains (Figure 6). The resulting shape of the pocket, along with a characteristic Tyr residue (Figure 4B) responsible for M6P-GlcNAc recognition, allows for the unique architecture that allows CI-MPR’s MRH domain 5 to accommodate a GlcNAc residue. The openness of the pocket of CI-MPR’s MRH domain 3 is intermediate between MRH domain 5 and CD-MPR, and consistent with the observation that MRH domain 3 can recognize M6P-methyl phosphodiester, but not the more bulky M6P-GlcNAc [131].
Because OS-9 is found in the ER, it is not surprising that its binding pocket is optimized to interact with high mannose-type glycans. Asp182 and Leu183, which are found in loop C of OS-9 (Figure 4B) and are missing in the other binding pockets, interact with the terminal mannose ring (residue j in Figure 1A) [151]. It is predicted that the negatively charged Asp182 would electrostatically repel the negatively charged phosphate group of M6P, thereby preventing OS-9 from interacting with phosphorylated glycans. Furthermore, when M6P is modeled into the binding pocket of OS-9, the phosphate moiety cannot fit into the binding pocket. In contrast to Tyr residues found in the other MRH domains (Figures 4B and 6), OS-9 has a Trp residue (Trp118) that is proposed to confer the Manα1,6Man linkage specificity observed for this lectin.
GIIβ exhibits the most shallow binding pocket of the MRH domains (Figure 6, bottom panel). This finding is consistent with GIIβ’s ability to recognize glycoproteins containing either high mannose, phosphomonoesters or M6P-GlcNAc diesters with similar affinity [71]. Thus, S. pombe GIIβ differs from human OS-9 which binds mannose but not M6P [79]. It should be noted that the current solution structure of GIIβ was obtained in the absence of ligand [71], and therefore additional structural studies are needed to determine whether the positioning of loops C and/or D will change significantly in the presence of a bound glycan. Unique to GIIβ, a conserved Trp residue (Trp409) is present in loop C (Figure 4B) that is required for optimal GII activity [71]. Two models have been proposed for the influence of Trp409 on GII activity (Figure 3). In the first model (Figure 3A), the mannose-binding essential residues Gln384, Arg414, Glu433, Tyr439 form a pocket which binds arm C of the glycan, while residue Trp409 interacts with arm B. This bidentate interaction allows the glucose-containing arm A to be juxtaposed to GIIα’s catalytic site. In the second model (Figure 3B), Trp409 interacts with other regions of the α or β-subunit, thereby influencing its affinity for N-glycans. These models imply that removal of mannoses by ER mannosidases will reduce both binding of the glycan and GII activity [36, 71]. Additional studies are needed to discriminate between these two models.
d) MRH domain interactions stabilize the carbohydrate binding pocket
The crystal structure of the dimeric CD-MPR reveals that the dimer interface is extensive and consists of the two β5–β9 sheets [144]. The CD-MPR differs from other lectins in that it is dynamic and its quaternary structure undergoes significant changes upon phosphomannosyl binding [156–158]. Importantly, these studies show that the binding pocket is stabilized by intersubunit salt bridges, including those between loop D of one subunit and the N-terminal α-helix of the other subunit. MRH domain 3 of the CI-MPR is stabilized by very different interactions. The crystal structure of the CI-MPR’s N-terminal three MRH domains reveals a compact structure, with the three domains contacting each other mainly through their loops and linker regions [148, 149], rather than via their β sheets as observed for the CD-MPR. These contacts stabilize the carbohydrate binding pocket of MRH domain 3, as evidenced by a construct encoding MRH domain 3 alone that is unable to interact with phosphorylated acid hydrolases with high affinity [159]. Together these studies suggest that MRH domains 1–3 form a compact, structural unit within the full-length CI-MPR. A construct encoding CI-MPR’s MRH domain 5 alone binds phosphodiesters with a lower affinity than a construct encoding MRH domains 5–9 [137], suggesting that other MRH domains interact and stabilize the carbohydrate binding site within MRH domain 5.
The MRH domain of GIIβ, OS-9, and γ subunit of GlcNAc-1-phosphotransferase exists as a single MRH domain within a significantly larger polypeptide, whereas XTP3-B houses two non-contiguous MRH domains separated by ~95 residues (Figure 4A). Additional studies are needed to determine if these MRH domains require specific interactions with other regions of the protein to generate a high affinity carbohydrate binding site. Although Yos9p is a dimer, its dimerization region maps to a site other than its MRH domain [76]. This dimerization domain of Yos9p is located adjacent to the C-terminus of the MRH domain and exhibits an αβ-roll architecture. Because the MRH domain of OS-9 and the dimerization domain Yos9p were crystallized independent of each other, it is not know whether a flexible linker joins these two domains of the protein together as beads on a string or facilitates extensive interactions between these two domains tightly in the intact protein.
7) Perspectives
In recent years, much progress has been made in elucidating the role of N-glycans in regulating the quality control of glycoprotein folding in the ER and in targeting newly synthesized acid hydrolases to lysosomes. However, the complexity and heterogeneity of the substrates involved and the highly dynamic nature of these processes leave many important questions unanswered. Did the MRH fold first appear in resident ER proteins as recognition modules for nascent peptides bearing high mannose-type glycan chains, and subsequently evolve to accommodate new glycan modifications (i.e., phosphorylation) in the Golgi? How does the MRH domain of GIIβ enhance glucosidase II’s activity? Does the MRH domain of the γ subunit of GlcNAc-1-phosphotransferase enhance the catalytic activity of GlcNAc-1-phosphotransferase toward a select subset of acid hydrolases? Because structural studies of XTP3-B and GlcNAc-1-phosphotransferase are lacking, the prediction that their MRH domains adopt the MRH fold requires validation. Is the lack of lectin activity of XTP3-B’s N-terminal MRH domain due to the presence of a stretch of ~45 highly charged residues (i.e., Lys and Glu) not found in other carbohydrate-binding MRH domains? Rather than bind carbohydrate, can XTP3-B’s N-terminal MRH domain serve as a protein-protein interaction domain similar to the CI-MPR’s MRH domain 11 that binds the polypeptide IGF-2? How are the three carbohydrate-binding MRH domains of the CI-MPR oriented with respect to each other in the dimeric structure of the receptor? How flexible is the linker region between each of the 15 contiguous MRH domains of the CI-MPR, and can this flexibility impact multivalent interactions with a heterogeneous population of acid hydrolases by altering the spatial arrangement of the three carbohydrate-binding MRH domains? Clearly, MRH domains play a key role in the recognition of specific N-glycan structures and define the fate of glycoproteins in the secretory pathway. However, additional studies are required to fully elucidate the functional roles of the MRH domains in this diverse family of MRH domain-containing proteins in the secretory pathway.
Acknowledgments
We would like to thank Dr. Linda Olson for generating figures 6 and 7. This work was supported by National Research Council (Argentina) grant PIP-N824 and Agencia Nacional de Promoción Científica y Tecnológica (ANPCYT) grant PICT-2012-1504 (to C.D.), National Institutes of Health grant R01DK042667 (to N.M.D.) and Mizutani Foundation for Glycosciences grant 10-0056 (to N.M.D.). CD is a Career Investigator of the National Research Council (Argentina).
Abbreviations used
The abbreviations used are
- CD-MPR
cation-dependent mannose 6-phosphate receptor
- CI-MPR
cation-independent mannose 6-phosphate receptor
- CNX
calnexin
- CRT
calreticulin
- ER
endoplasmic reticulum
- ERAD
ER associated degradation
- GI
glucosidase I
- GII
glucosidase II
- IGF-2
insulin-like growth factor 2
- M6P
mannose 6-phosphate
- MPR
mannose 6-phosphate receptor
- MRH
mannose 6-phosphate receptor homology domain
- pNGP
p-nitrophenyl α-D-glucopyranoside
- QC
quality control of glycoprotein folding in the ER
- SPR
surface plasmon resonance
- UGGT
UDP-Glc:glycoprotein glucosyltransferase
N-glycan abbreviations used are
- G3M9
Glc3Man9GlcNAc2
- G2M9
Glc2Man9GlcNAc2
- G1M9
Glc1Man9GlcNAc2
- M9
Man9GlcNAc2
- G1M8
Glc1Man8GlcNAc2
- M8
Man8GlcNAc2
- M7
Man7GlcNAc2
- M6
Man6GlcNAc2
Footnotes
CONFLICT OF INTERESTS. None declared
References
- 1.D’Alessio C, Caramelo JJ, Parodi AJ. UDP-Glc:Glycoprotein glucosyltransferase-glucosidase II, the ying-yang of the er quality control. Semin Cell Dev Biol. 2010;21(5):491–9. doi: 10.1016/j.semcdb.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annual Review of Biochemistry. 1985;54:631–64. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- 3.Aebi M, Bernasconi R, Clerc S, Molinari M. N-glycan structures: Recognition and processing in the er. Trends Biochem Sci. 2010;35(2):74–82. doi: 10.1016/j.tibs.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the swiss-prot database. Biochim Biophys Acta. 1999;1473(1):4–8. doi: 10.1016/s0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- 5.Nilsson IM, von Heijne G. Determination of the distance between the oligosaccharyltransferase active site and the endoplasmic reticulum membrane. J Biol Chem. 1993;268(8):5798–801. [PubMed] [Google Scholar]
- 6.Ruiz-Canada C, Kelleher DJ, Gilmore R. Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian ost isoforms. Cell. 2009;136(2):272–83. doi: 10.1016/j.cell.2008.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen MM, Bartlett AI, Nerenberg PS, Friel CT, Hackenberger CP, Stultz CM, Radford SE, Imperiali B. Perturbing the folding energy landscape of the bacterial immunity protein Im7 by site-specific N-linked glycosylation. Proc Natl Acad Sci U S A. 2010;107(52):22528–33. doi: 10.1073/pnas.1015356107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parodi AJ. Protein glucosylation and its role in protein folding. Annu Rev Biochem. 2000;69:69–93. doi: 10.1146/annurev.biochem.69.1.69. [DOI] [PubMed] [Google Scholar]
- 9.Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–49. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 10.Li E, Tabas I, Kornfeld S. The synthesis of complex-type oligosaccharides. I. Structure of the lipid-linked oligosaccharide precursor of the complex-type oligosaccharides of the vesicular stomatitis virus G protein. Journal of Biological Chemistry. 1978;253(21):7762–70. [PubMed] [Google Scholar]
- 11.Aebi M. N-linked protein glycosylation in the ER. Biochim Biophys Acta. 2013;1833(11):2430–7. doi: 10.1016/j.bbamcr.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 12.Parodi AJ, Martin-Barrientos J, Engel JC. Glycoprotein assembly in leishmania mexicana. Biochem Biophys Res Commun. 1984;118(1):1–7. doi: 10.1016/0006-291x(84)91058-1. [DOI] [PubMed] [Google Scholar]
- 13.Parodi AJ. N-glycosylation in trypanosomatid protozoa. Glycobiology. 1993;3(3):193–9. doi: 10.1093/glycob/3.3.193. [DOI] [PubMed] [Google Scholar]
- 14.Petrescu AJ, Milac AL, Petrescu SM, Dwek RA, Wormald MR. Statistical analysis of the protein environment of N-glycosylation sites: Implications for occupancy, structure, and folding. Glycobiology. 2004;14(2):103–14. doi: 10.1093/glycob/cwh008. [DOI] [PubMed] [Google Scholar]
- 15.Sato C, Kim JH, Abe Y, Saito K, Yokoyama S, Kohda D. Characterization of the N-oligosaccharides attached to the atypical asn-x-cys sequence of recombinant human epidermal growth factor receptor. J Biochem. 2000;127(1):65–72. doi: 10.1093/oxfordjournals.jbchem.a022585. [DOI] [PubMed] [Google Scholar]
- 16.Chi YH, Koo YD, Dai SY, Ahn JE, Yun DJ, Lee SY, Zhu-Salzman K. N-glycosylation at non-canonical Asn-X-Cys sequence of an insect recombinant cathepsin b-like counter-defense protein. Comp Biochem Physiol B Biochem Mol Biol. 2010;156(1):40–7. doi: 10.1016/j.cbpb.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 17.Matsui T, Takita E, Sato T, Kinjo S, Aizawa M, Sugiura Y, Hamabata T, Sawada K, Kato K. N-glycosylation at noncanonical Asn-X-Cys sequences in plant cells. Glycobiology. 2011;21(8):994–9. doi: 10.1093/glycob/cwq198. [DOI] [PubMed] [Google Scholar]
- 18.Mohorko E, Glockshuber R, Aebi M. Oligosaccharyltransferase: The central enzyme of N-linked protein glycosylation. J Inherit Metab Dis. 2011;34(4):869–78. doi: 10.1007/s10545-011-9337-1. [DOI] [PubMed] [Google Scholar]
- 19.Ballou L, Gopal P, Krummel B, Tammi M, Ballou CE. A mutation that prevents glucosylation of the lipid-linked oligosaccharide precursor leads to underglycosylation of secreted yeast invertase. Proc Natl Acad Sci U S A. 1986;83(10):3081–5. doi: 10.1073/pnas.83.10.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freeze HH. Congenital disorders of glycosylation: CDG-I, CDG-II and beyond. Curr Mol Med. 2007;7(4):389–96. doi: 10.2174/156652407780831548. [DOI] [PubMed] [Google Scholar]
- 21.Nasab FP, Schulz BL, Gamarro F, Parodi AJ, Aebi M. All in one: Leishmania major STT3 proteins substitute for the whole oligosaccharyltransferase complex in Saccharomyces cerevisiae. Mol Biol Cell. 2008;19(9):3758–68. doi: 10.1091/mbc.E08-05-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shailubhai K, Pukazhenthi BS, Saxena ES, Varma GM, Vijay IK. Glucosidase I, a transmembrane endoplasmic reticular glycoprotein with a luminal catalytic domain. J Biol Chem. 1991;266(25):16587–93. [PubMed] [Google Scholar]
- 23.Ugalde RA, Staneloni RJ, Leloir LF. Microsomal glucosidases acting on the saccharide moiety of the glucose-containing dolichyl diphosphate oligosaccharide. Biochem Biophys Res Commun. 1979;91(3):1174–81. doi: 10.1016/0006-291x(79)92003-5. [DOI] [PubMed] [Google Scholar]
- 24.Trombetta SE, Bosch M, Parodi AJ. Glucosylation of glycoproteins by mammalian, plant, fungal, and trypanosomatid protozoa microsomal membranes. Biochemistry. 1989;28(20):8108–16. doi: 10.1021/bi00446a022. [DOI] [PubMed] [Google Scholar]
- 25.Trombetta SE, Parodi AJ. Purification to apparent homogeneity and partial characterization of rat liver UDP-glucose:Glycoprotein glucosyltransferase. J Biol Chem. 1992;267(13):9236–40. [PubMed] [Google Scholar]
- 26.Sousa MC, Ferrero-Garcia MA, Parodi AJ. Recognition of the oligosaccharide and protein moieties of glycoproteins by the UDP-Glc:Glycoprotein glucosyltransferase. Biochemistry. 1992;31(1):97–105. doi: 10.1021/bi00116a015. [DOI] [PubMed] [Google Scholar]
- 27.Herscovics A, Romero PA, Tremblay LO. The specificity of the yeast and human class I ER alpha 1,2-mannosidases involved in er quality control is not as strict previously reported. Glycobiology. 2002;12(4):14G–15G. [PubMed] [Google Scholar]
- 28.Clerc S, Hirsch C, Oggier DM, Deprez P, Jakob C, Sommer T, Aebi M. Htm1 protein generates the N-glycan signal for glycoprotein degradation in the endoplasmic reticulum. J Cell Biol. 2009;184(1):159–72. doi: 10.1083/jcb.200809198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lederkremer GZ. Glycoprotein folding, quality control and ER-associated degradation. Curr Opin Struct Biol. 2009;19(5):515–23. doi: 10.1016/j.sbi.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 30.Caramelo JJ, Castro OA, Alonso LG, De Prat-Gay G, Parodi AJ. UDP-Glc:Glycoprotein glucosyltransferase recognizes structured and solvent accessible hydrophobic patches in molten globule-like folding intermediates. Proc Natl Acad Sci U S A. 2003;100(1):86–91. doi: 10.1073/pnas.262661199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hosokawa N, Kamiya Y, Kamiya D, Kato K, Nagata K. Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J Biol Chem. 2009;284(25):17061–8. doi: 10.1074/jbc.M809725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hosokawa N, Wada I, Nagasawa K, Moriyama T, Okawa K, Nagata K. Human XTP3-b forms an endoplasmic reticulum quality control scaffold with the hrd1-sel1l ubiquitin ligase complex and bip. J Biol Chem. 2008;283(30):20914–24. doi: 10.1074/jbc.M709336200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bernasconi R, Pertel T, Luban J, Molinari M. A dual task for the xbp1-responsive OS-9 variants in the mammalian endoplasmic reticulum: Inhibiting secretion of misfolded protein conformers and enhancing their disposal. J Biol Chem. 2008;283(24):16446–54. doi: 10.1074/jbc.M802272200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujimori T, Kamiya Y, Nagata K, Kato K, Hosokawa N. Endoplasmic reticulum lectin XTP3-b inhibits endoplasmic reticulum-associated degradation of a misfolded alpha1-antitrypsin variant. FEBS J. 2013 doi: 10.1111/febs.12157. in press. [DOI] [PubMed] [Google Scholar]
- 35.Grinna LS, Robbins PW. Substrate specificities of rat liver microsomal glucosidases which process glycoproteins. J Biol Chem. 1980;255(6):2255–8. [PubMed] [Google Scholar]
- 36.Stigliano ID, Alculumbre SG, Labriola CA, Parodi AJ, D’Alessio C. Glucosidase II and N-glycan mannose content regulate the half-lives of monoglucosylated species in vivo. Mol Biol Cell. 2011;22(11):1810–23. doi: 10.1091/mbc.E11-01-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schallus T, Jaeckh C, Feher K, Palma AS, Liu Y, Simpson JC, Mackeen M, Stier G, Gibson TJ, Feizi T, Pieler T, Muhle-Goll C. Malectin: A novel carbohydrate-binding protein of the endoplasmic reticulum and a candidate player in the early steps of protein N-glycosylation. Mol Biol Cell. 2008;19(8):3404–14. doi: 10.1091/mbc.E08-04-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galli C, Bernasconi R, Solda T, Calanca V, Molinari M. Malectin participates in a backup glycoprotein quality control pathway in the mammalian er. PLoS One. 2011;6(1):e16304. doi: 10.1371/journal.pone.0016304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qin SY, Hu D, Matsumoto K, Takeda K, Matsumoto N, Yamaguchi Y, Yamamoto K. Malectin forms a complex with ribophorin i for enhanced association with misfolded glycoproteins. J Biol Chem. 2012;287(45):38080–9. doi: 10.1074/jbc.M112.394288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trombetta ES, Simons JF, Helenius A. Endoplasmic reticulum glucosidase II is composed of a catalytic subunit, conserved from yeast to mammals, and a tightly bound noncatalytic HDEL-containing subunit. J Biol Chem. 1996;271(44):27509–16. doi: 10.1074/jbc.271.44.27509. [DOI] [PubMed] [Google Scholar]
- 41.D’Alessio C, Fernandez F, Trombetta ES, Parodi AJ. Genetic evidence for the heterodimeric structure of glucosidase II. The effect of disrupting the subunit-encoding genes on glycoprotein folding. J Biol Chem. 1999;274(36):25899–25905. doi: 10.1074/jbc.274.36.25899. [DOI] [PubMed] [Google Scholar]
- 42.Jones DC, Mehlert A, Guther ML, Ferguson MA. Deletion of the glucosidase ii gene in trypanosoma brucei reveals novel N-glycosylation mechanisms in the biosynthesis of variant surface glycoprotein. J Biol Chem. 2005;280(43):35929–42. doi: 10.1074/jbc.M509130200. [DOI] [PubMed] [Google Scholar]
- 43.Arendt CW, Ostergaard HL. Identification of the CD45-associated 116-kda and 80-kda proteins as the alpha- and beta-subunits of alpha-glucosidaseII. J Biol Chem. 1997;272(20):13117–25. doi: 10.1074/jbc.272.20.13117. [DOI] [PubMed] [Google Scholar]
- 44.Soussilane P, D’Alessio C, Paccalet T, Fitchette AC, Parodi AJ, Williamson R, Plasson C, Faye L, Gomord V. N-glycan trimming by glucosidase II is essential for arabidopsis development. Glycoconj J. 2009;26(5):597–607. doi: 10.1007/s10719-008-9201-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feng J, Romaniouk AV, Samal SK, Vijay IK. Processing enzyme glucosidase ii: Proposed catalytic residues and developmental regulation during the ontogeny of the mouse mammary gland. Glycobiology. 2004;14(10):909–21. doi: 10.1093/glycob/cwh110. [DOI] [PubMed] [Google Scholar]
- 46.Alonso JM, Santa-Cecilia A, Calvo P. Effect of bromoconduritol on glucosidase ii from rat liver. A new kinetic model for the binding and hydrolysis of the substrate. Eur J Biochem. 1993;215(1):37–42. doi: 10.1111/j.1432-1033.1993.tb18004.x. [DOI] [PubMed] [Google Scholar]
- 47.Yu W, Gill T, Wang L, Du Y, Ye H, Qu X, Guo JT, Cuconati A, Zhao K, Block TM, Xu X, Chang J. Design, synthesis, and biological evaluation of N-alkylated deoxynojirimycin (DNJ) derivatives for the treatment of dengue virus infection. J Med Chem. 2012;55(13):6061–75. doi: 10.1021/jm300171v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cumpstey I, Butters TD, Tennant-Eyles RJ, Fairbanks AJ, France RR, Wormald MR. Synthesis of fluorescence-labelled disaccharide substrates of glucosidase II. Carbohydr Res. 2003;338(19):1937–49. doi: 10.1016/s0008-6215(03)00255-6. [DOI] [PubMed] [Google Scholar]
- 49.Fernandez FS, Trombetta SE, Hellman U, Parodi AJ. Purification to homogeneity of udp-glucose:Glycoprotein glucosyltransferase from schizosaccharomyces pombe and apparent absence of the enzyme from saccharomyces cerevisiae. J Biol Chem. 1994;269(48):30701–6. [PubMed] [Google Scholar]
- 50.Totani K, Ihara Y, Matsuo I, Ito Y. Effects of macromolecular crowding on glycoprotein processing enzymes. J Am Chem Soc. 2008;130(6):2101–2107. doi: 10.1021/ja077570k. [DOI] [PubMed] [Google Scholar]
- 51.Suradej B, Pata S, Kasinrerk W, Cressey R. Glucosidase II exhibits similarity to the p53 tumor suppressor in regards to structure and behavior in response to stress signals: A potential novel cancer biomarker. Oncol Rep. 2013;30(5):2511–9. doi: 10.3892/or.2013.2721. [DOI] [PubMed] [Google Scholar]
- 52.Herscovics A, Jelinek-Kelly S. A rapid method for assay of glycosidases involved in glycoprotein biosynthesis. Anal Biochem. 1987;166(1):85–9. doi: 10.1016/0003-2697(87)90550-1. [DOI] [PubMed] [Google Scholar]
- 53.Totani K, Ihara Y, Matsuo I, Ito Y. Substrate specificity analysis of endoplasmic reticulum glucosidase ii using synthetic high mannose-type glycans. J Biol Chem. 2006;281(42):31502–8. doi: 10.1074/jbc.M605457200. [DOI] [PubMed] [Google Scholar]
- 54.Stigliano ID, Caramelo JJ, Labriola CA, Parodi AJ, D’Alessio C. Glucosidase II beta subunit modulates N-glycan trimming in fission yeasts and mammals. Mol Biol Cell. 2009;20(17):3974–3984. doi: 10.1091/mbc.E09-04-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Watanabe T, Totani K, Matsuo I, Maruyama J, Kitamoto K, Ito Y. Genetic analysis of glucosidase II beta-subunit in trimming of high-mannose-type glycans. Glycobiology. 2009;19(8):834–40. doi: 10.1093/glycob/cwp061. [DOI] [PubMed] [Google Scholar]
- 56.Li A, Davila S, Furu L, Qian Q, Tian X, Kamath PS, King BF, Torres VE, Somlo S. Mutations in prkcsh cause isolated autosomal dominant polycystic liver disease. Am J Hum Genet. 2003;72(3):691–703. doi: 10.1086/368295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Drenth JP, te Morsche RH, Smink R, Bonifacino JS, Jansen JB. Germline mutations in prkcsh are associated with autosomal dominant polycystic liver disease. Nat Genet. 2003;33(3):345–7. doi: 10.1038/ng1104. [DOI] [PubMed] [Google Scholar]
- 58.Drenth JP, Martina JA, van de Kerkhof R, Bonifacino JS, Jansen JB. Polycystic liver disease is a disorder of cotranslational protein processing. Trends Mol Med. 2005;11(1):37–42. doi: 10.1016/j.molmed.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 59.Janssen MJ, Waanders E, Woudenberg J, Lefeber DJ, Drenth JP. Congenital disorders of glycosylation in hepatology: The example of polycystic liver disease. J Hepatol. 2010;52(3):432–40. doi: 10.1016/j.jhep.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 60.Yang J, Zhao Y, Ma K, Jiang FJ, Liao W, Zhang P, Zhou J, Tu B, Wang L, Kampinga HH, Xie Z, Zhu WG. Deficiency of hepatocystin induces autophagy through an mtor-dependent pathway. Autophagy. 2011;7(7):748–59. doi: 10.4161/auto.7.7.15822. [DOI] [PubMed] [Google Scholar]
- 61.Munro S. The mrh domain suggests a shared ancestry for the mannose 6-phosphate receptors and other N-glycan-recognising proteins. Curr Biol. 2001;11(13):R499–501. doi: 10.1016/s0960-9822(01)00302-5. [DOI] [PubMed] [Google Scholar]
- 62.Arendt CW, Ostergaard HL. Two distinct domains of the beta-subunit of glucosidase II interact with the catalytic alpha-subunit. Glycobiology. 2000;10(5):487–92. doi: 10.1093/glycob/10.5.487. [DOI] [PubMed] [Google Scholar]
- 63.Wilkinson BM, Purswani J, Stirling CJ. Yeast gtb1 encodes a subunit of glucosidase II required for glycoprotein processing in the endoplasmic reticulum. J Biol Chem. 2006;281(10):6325–33. doi: 10.1074/jbc.M510455200. [DOI] [PubMed] [Google Scholar]
- 64.Deprez P, Gautschi M, Helenius A. More than one glycan is needed for er glucosidase II to allow entry of glycoproteins into the calnexin/calreticulin cycle. Mol Cell. 2005;19(2):183–195. doi: 10.1016/j.molcel.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 65.Quinn RP, Mahoney SJ, Wilkinson BM, Thornton DJ, Stirling CJ. A novel role for gtb1p in glucose trimming of N-linked glycans. Glycobiology. 2009;19(12):1408–1416. doi: 10.1093/glycob/cwp087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Treml K, Meimaroglou D, Hentges A, Bause E. The alpha- and beta-subunits are required for expression of catalytic activity in the hetero-dimeric glucosidase ii complex from human liver. Glycobiology. 2000;10(5):493–502. doi: 10.1093/glycob/10.5.493. [DOI] [PubMed] [Google Scholar]
- 67.Pelletier MF, Marcil A, Sevigny G, Jakob CA, Tessier DC, Chevet E, Menard R, Bergeron JJ, Thomas DY. The heterodimeric structure of glucosidase ii is required for its activity, solubility, and localization in vivo. Glycobiology. 2000;10(8):815–27. doi: 10.1093/glycob/10.8.815. [DOI] [PubMed] [Google Scholar]
- 68.Trombetta ES, Fleming KG, Helenius A. Quaternary and domain structure of glycoprotein processing glucosidase II. Biochemistry. 2001;40(35):10717–22. doi: 10.1021/bi010629u. [DOI] [PubMed] [Google Scholar]
- 69.Lucocq JM, Brada D, Roth J. Immunolocalization of the oligosaccharide trimming enzyme glucosidase ii. J Cell Biol. 1986;102(6):2137–46. doi: 10.1083/jcb.102.6.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zuber C, Fan JY, Guhl B, Parodi A, Fessler JH, Parker C, Roth J. Immunolocalization of udp-glucose:Glycoprotein glucosyltransferase indicates involvement of pre-golgi intermediates in protein quality control. Proc Natl Acad Sci U S A. 2001;98(19):10710–5. doi: 10.1073/pnas.191359198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Olson LJ, Orsi R, Alculumbre SG, Peterson FC, Stigliano ID, Parodi AJ, D’Alessio C, Dahms NM. Structure of the lectin mannose 6-phosphate receptor homology (mrh) domain of glucosidase ii, an enzyme that regulates glycoprotein folding quality control in the endoplasmic reticulum. J Biol Chem. 2013;288(23):16460–75. doi: 10.1074/jbc.M113.450239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hu D, Kamiya Y, Totani K, Kamiya D, Kawasaki N, Yamaguchi D, Matsuo I, Matsumoto N, Ito Y, Kato K, Yamamoto K. Sugar-binding activity of the mrh domain in the er alpha-glucosidase II beta subunit is important for efficient glucose trimming. Glycobiology. 2009;19(10):1127–35. doi: 10.1093/glycob/cwp104. [DOI] [PubMed] [Google Scholar]
- 73.Olson LJ, Hancock MK, Dix D, Kim JJP, Dahms NM. Mutational analysis of the binding site residues of the bovine cation-dependent mannose 6-phosphate receptor. Journal of Biological Chemistry. 1999;274(52):36905–36911. doi: 10.1074/jbc.274.52.36905. [DOI] [PubMed] [Google Scholar]
- 74.Sun G, Zhao H, Kalyanaraman B, Dahms NM. Identification of residues essential for carbohydrate recognition and cation dependence of the 46-kda mannose 6-phosphate receptor. Glycobiology. 2005;15(11):1136–1149. doi: 10.1093/glycob/cwi098. [DOI] [PubMed] [Google Scholar]
- 75.Kimura Y, Nakazawa M, Yamada M. Cloning and characterization of three isoforms of os-9 cdna and expression of the os-9 gene in various human tumor cell lines. J Biochem (Tokyo) 1998;123(5):876–882. doi: 10.1093/oxfordjournals.jbchem.a022019. [DOI] [PubMed] [Google Scholar]
- 76.Hanna J, Schutz A, Zimmermann F, Behlke J, Sommer T, Heinemann U. Structural and biochemical basis of yos9 protein dimerization and possible contribution to self-association of 3-hydroxy-3-methylglutaryl-coenzyme a reductase degradation ubiquitin-ligase complex. J Biol Chem. 2012;287(11):8633–40. doi: 10.1074/jbc.M111.317644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Christianson JC, Shaler TA, Tyler RE, Kopito RR. Os-9 and grp94 deliver mutant alpha1-antitrypsin to the hrd1? Sel1l ubiquitin ligase complex for ERAD. Nature Cell Biology. 2008;10(3):272–282. doi: 10.1038/ncb1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hosokawa N, Kamiya Y, Kato K. The role of MRH domain-containing lectins in erad. Glycobiology. 2010;20(6):651–60. doi: 10.1093/glycob/cwq013. [DOI] [PubMed] [Google Scholar]
- 79.Mikami K, Yamaguchi D, Tateno H, Hu D, Qin SY, Kawasaki N, Yamada M, Matsumoto N, Hirabayashi J, Ito Y, Yamamoto K. The sugar-binding ability of human OS-9 and its involvement in er-associated degradation. Glycobiology. 2010;20(3):310–21. doi: 10.1093/glycob/cwp175. [DOI] [PubMed] [Google Scholar]
- 80.Quan EM, Kamiya Y, Kamiya D, Denic V, Weibezahn J, Kato K, Weissman JS. Defining the glycan destruction signal for endoplasmic reticulum-associated degradation. Mol Cell. 2008;32(6):870–7. doi: 10.1016/j.molcel.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bhamidipati A, Denic V, Quan EM, Weissman JS. Exploration of the topological requirements of erad identifies Yos9p as a lectin sensor of misfolded glycoproteins in the er lumen. Molecular Cell. 2005;19(6):741–751. doi: 10.1016/j.molcel.2005.07.027. [DOI] [PubMed] [Google Scholar]
- 82.Izawa T, Nagai H, Endo T, Nishikawa S. Yos9p and Hrd1p mediate er retention of misfolded proteins for er-associated degradation. Mol Biol Cell. 2012;23(7):1283–93. doi: 10.1091/mbc.E11-08-0722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cruciat CM, Hassler C, Niehrs C. The MRH protein erlectin is a member of the endoplasmic reticulum synexpression group and functions in N-glycan recognition. Journal of Biological Chemistry. 2006;281(18):12986–12993. doi: 10.1074/jbc.M511872200. [DOI] [PubMed] [Google Scholar]
- 84.Yamaguchi D, Hu D, Matsumoto N, Yamamoto K. Human xtp3-b binds to alpha1-antitrypsin variant null(hong kong) via the C-terminal MRH domain in a glycan-dependent manner. Glycobiology. 2010;20(3):348–55. doi: 10.1093/glycob/cwp182. [DOI] [PubMed] [Google Scholar]
- 85.Shenkman M, Groisman B, Ron E, Avezov E, Hendershot LM, Lederkremer GZ. A shared endoplasmic reticulum-associated degradation pathway involving the EDEM1 protein for glycosylated and nonglycosylated proteins. J Biol Chem. 2013;288(4):2167–78. doi: 10.1074/jbc.M112.438275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Holtzman E, editor. Lysosomes. Plenum; New York: 1989. [Google Scholar]
- 87.Hasilik A, Klein U, Waheed A, Strecker G, von Figura K. Phosphorylated oligosaccharides in lysosomal enzymes: Identification of alpha-N-acetylglucosamine(1)phospho(6)mannose diester groups. Proceedings of the National Academy of Sciences of the United States of America. 1980;77(12):7074–7078. doi: 10.1073/pnas.77.12.7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Varki A, Kornfeld S. Structural studies of phosphorylated high mannose-type oligosaccharides. Journal of Biological Chemistry. 1980;255(22):10847–10858. [PubMed] [Google Scholar]
- 89.Bao M, Booth JL, Elmendorf BJ, Canfield WM. Bovine UDP-N-acetylglucosamine:Lysosomal-enzyme N-acetylglucosamine-1-phosphotransferase. I. Purification and subunit structure. Journal of Biological Chemistry. 1996;271(49):31437–45. doi: 10.1074/jbc.271.49.31437. [DOI] [PubMed] [Google Scholar]
- 90.Kudo M, Bao M, D’Souza A, Ying F, Pan H, Roe BA, Canfield WM. The alpha- and beta-subunits of the human UDP-N-acetylglucosamine:Lysosomal enzyme phosphotransferase are encoded by a single cdna. Journal of Biological Chemistry. 2005;280(43):36141–36149. doi: 10.1074/jbc.M509008200. [DOI] [PubMed] [Google Scholar]
- 91.Waheed A, Hasilik A, von Figura K. Processing of the phosphorylated recognition marker in lysosomal enzymes. Characterization and partial purification of a microsomal alpha-N-acetylglucosaminyl phosphodiesterase. Journal of Biological Chemistry. 1981;256(11):5717–21. [PubMed] [Google Scholar]
- 92.Varki A, Sherman W, Kornfeld S. Demonstration of the enzymatic mechanisms of alpha-N-acetyl-d-glucosamine-1-phosphodiester N-acetylglucosaminidase (formerly called alpha-N-acetylglucosaminylphosphodiesterase) and lysosomal alpha-N-acetylglucosaminidase. Archives of Biochemistry & Biophysics. 1983;222(1):145–149. doi: 10.1016/0003-9861(83)90511-8. [DOI] [PubMed] [Google Scholar]
- 93.Kornfeld R, Bao M, Brewer K, Noll C, Canfield W. Molecular cloning and functional expression of two splice forms of human N-acetylglucosamine-1-phosphodiester alpha-N- acetylglucosaminidase. J Biol Chem. 1999;274(46):32778–32785. doi: 10.1074/jbc.274.46.32778. [DOI] [PubMed] [Google Scholar]
- 94.Rohrer J, Kornfeld R. Lysosomal hydrolase mannose 6-phosphate uncovering enzyme resides in the trans-golgi network. Mol Biol Cell. 2001;12(6):1623–31. doi: 10.1091/mbc.12.6.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Do H, Lee WS, Ghosh P, Hollowell T, Canfield W, Kornfeld S. Human mannose 6-phosphate-uncovering enzyme is synthesized as a proenzyme that is activated by the endoprotease furin. J Biol Chem. 2002;277(33):29737–44. doi: 10.1074/jbc.M202369200. [DOI] [PubMed] [Google Scholar]
- 96.Tiede S, Storch S, Lubke T, Henrissat B, Bargal R, Raas-Rothschild A, Braulke T. Mucolipidosis II is caused by mutations in gnpta encoding the alpha/beta glcnac-1-phosphotransferase. Nat Med. 2005;11(10):1109–12. doi: 10.1038/nm1305. [DOI] [PubMed] [Google Scholar]
- 97.Kudo M, Canfield WM. Structural requirements for efficient processing and activation of recombinant human udp-N-acetylglucosamine:Lysosomal-enzyme-N-acetylglucosamine-1-phosphotransferase. Journal of Biological Chemistry. 2006;281(17):11761–11768. doi: 10.1074/jbc.M513717200. [DOI] [PubMed] [Google Scholar]
- 98.Marschner K, Kollmann K, Schweizer M, Braulke T, Pohl S. A key enzyme in the biogenesis of lysosomes is a protease that regulates cholesterol metabolism. Science. 2011;333(6038):87–90. doi: 10.1126/science.1205677. [DOI] [PubMed] [Google Scholar]
- 99.Qian Y, Flanagan-Steet H, van Meel E, Steet R, Kornfeld SA. The dmap interaction domain of UDP-GlcNAc:Lysosomal enzyme N-acetylglucosamine-1-phosphotransferase is a substrate recognition module. Proc Natl Acad Sci U S A. 2013;110(25):10246–51. doi: 10.1073/pnas.1308453110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Reitman ML, Kornfeld S. UDP-N-acetylglucosamine:Glycoprotein N-acetylglucosamine-1-phosphotransferase. Proposed enzyme for the phosphorylation of the high mannose oligosaccharide units of lysosomal enzymes. Journal of Biological Chemistry. 1981;256(9):4275–81. [PubMed] [Google Scholar]
- 101.Lang L, Reitman M, Tang J, Roberts RM, Kornfeld S. Lysosomal enzyme phosphorylation. Recognition of a protein-dependent determinant allows specific phosphorylation of oligosaccharides present on lysosomal enzymes. Journal of Biological Chemistry. 1984;259(23):14663–14671. [PubMed] [Google Scholar]
- 102.Warner JB, Thalhauser C, Tao K, Sahagian GG. Role of N-linked oligosaccharide flexibility in mannose phosphorylation of lysosomal enzyme cathepsin l. J Biol Chem. 2002;277(44):41897–905. doi: 10.1074/jbc.M203097200. [DOI] [PubMed] [Google Scholar]
- 103.Steet R, Lee WS, Kornfeld S. Identification of the minimal lysosomal enzyme recognition domain in cathepsin d. Journal of Biological Chemistry. 2005;280(39):33318–33323. doi: 10.1074/jbc.M505994200. [DOI] [PubMed] [Google Scholar]
- 104.Raas-Rothschild A, Cormier-Daire V, Bao M, Genin E, Salomon R, Brewer K, Zeigler M, Mandel H, Toth S, Roe B, Munnich A, Canfield WM. Molecular basis of variant pseudo-hurler polydystrophy (mucolipidosis iiic) [see comments] J Clin Invest. 2000;105(5):673–681. doi: 10.1172/JCI5826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pohl S, Tiede S, Castrichini M, Cantz M, Gieselmann V, Braulke T. Compensatory expression of human N-acetylglucosaminyl-1-phosphotransferase subunits in mucolipidosis type iii gamma. Biochim Biophys Acta. 2009;1792(3):221–5. doi: 10.1016/j.bbadis.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 106.Pohl S, Tiede S, Marschner K, Encarnacao M, Castrichini M, Kollmann K, Muschol N, Ullrich K, Muller-Loennies S, Braulke T. Proteolytic processing of the gamma-subunit is associated with the failure to form GlcNAc-1-phosphotransferase complexes and mannose 6-phosphate residues on lysosomal enzymes in human macrophages. J Biol Chem. 2010;285(31):23936–44. doi: 10.1074/jbc.M110.129684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Qian Y, Lee I, Lee WS, Qian M, Kudo M, Canfield WM, Lobel P, Kornfeld S. Functions of the alpha, beta, and gamma subunits of UDP-GlcNAc:Lysosomal enzyme N-acetylglucosamine-1-phosphotransferase. J Biol Chem. 2010;285(5):3360–3370. doi: 10.1074/jbc.M109.068650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lazzarino DA, Gabel CA. Biosynthesis of the mannose 6-phosphate recognition marker in transport-impaired mouse lymphoma cells. Demonstration of a two-step phosphorylation. J Biol Chem. 1988;263(21):10118–10126. [PubMed] [Google Scholar]
- 109.Kornfeld S, Sly WS. In: I cell disease and pseudo-hurler polydystrophy: Disorders of lysosomal enzyme phosphorylation and localization. Scriver CR, et al., editors. III. McGraw Hill; 2001. pp. 3469–3482. [Google Scholar]
- 110.Kim JJ, Olson LJ, Dahms NM. Carbohydrate recognition by the mannose-6-phosphate receptors. Curr Opin Struct Biol. 2009;19(5):534–542. doi: 10.1016/j.sbi.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nadimpalli SK, Amancha PK. Evolution of mannose 6-phosphate receptors (MPR300 and 46): Lysosomal enzyme sorting proteins. Curr Protein Pept Sci. 2010;11(1):68–90. doi: 10.2174/138920310790274644. [DOI] [PubMed] [Google Scholar]
- 112.Coutinho MF, Prata MJ, Alves S. Mannose-6-phosphate pathway: A review on its role in lysosomal function and dysfunction. Mol Genet Metab. 2012;105(4):542–50. doi: 10.1016/j.ymgme.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 113.Morgan DO, Edman JC, Standring DN, Fried VA, Smith MC, Roth RA, Rutter WJ. Insulin-like growth factor II receptor as a multifunctional binding protein. Nature. 1987;329(6137):301–7. doi: 10.1038/329301a0. [DOI] [PubMed] [Google Scholar]
- 114.Brown J, Jones EY, Forbes BE. Keeping IGF-II under control: Lessons from the igf-ii-igf2r crystal structure. Trends Biochem Sci. 2009;34(12):612–619. doi: 10.1016/j.tibs.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 115.Godar S, Horejsi V, Weidle UH, Binder BR, Hansmann C, Stockinger H. M6p/igfii-receptor complexes urokinase receptor and plasminogen for activation of transforming growth factor-beta1. Eur J Immunol. 1999;29(3):1004–1013. doi: 10.1002/(SICI)1521-4141(199903)29:03<1004::AID-IMMU1004>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 116.Leksa V, Pfisterer K, Ondrovicova G, Binder B, Lakatosova S, Donner C, Schiller HB, Zwirzitz A, Mrvova K, Pevala V, Kutejova E, Stockinger H. Dissecting mannose 6-phosphate-insulin-like growth factor 2 receptor complexes that control activation and uptake of plasminogen in cells. J Biol Chem. 2012;287(27):22450–62. doi: 10.1074/jbc.M112.339663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nykjaer A, Christensen EI, Vorum H, Hager H, Petersen CM, Roigaard H, Min HY, Vilhardt F, Moller LB, Kornfeld S, Gliemann J. Mannose 6-phosphate/insulin-like growth factor-II receptor targets the urokinase receptor to lysosomes via a novel binding interaction. J Cell Biol. 1998;141(3):815–828. doi: 10.1083/jcb.141.3.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kreiling JL, Byrd JC, Deisz RJ, Mizukami IF, Todd RF, 3rd, MacDonald RG. Binding of urokinase-type plasminogen activator receptor (uPAR) to the mannose 6-phosphate/insulin-like growth factor II receptor: Contrasting interactions of full-length and soluble forms of uPAR. J Biol Chem. 2003;27:27. doi: 10.1074/jbc.M302249200. [DOI] [PubMed] [Google Scholar]
- 119.Koster A, Saftig P, Matzner U, von Figura K, Peters C, Pohlmann R. Targeted disruption of the m(r) 46,000 mannose 6-phosphate receptor gene in mice results in misrouting of lysosomal proteins. EMBO Journal. 1993;12(13):5219–5223. doi: 10.1002/j.1460-2075.1993.tb06217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ludwig T, Eggenschwiler J, Fisher P, D’Ercole AJ, Davenport ML, Efstratiadis A. Mouse mutants lacking the type 2 IGF receptor (IGF2R) are rescued from perinatal lethality in igf2 and igf1r null backgrounds. Dev Biol. 1996;177(2):517–535. doi: 10.1006/dbio.1996.0182. [DOI] [PubMed] [Google Scholar]
- 121.Sohar I, Sleat D, Gong Liu C, Ludwig T, Lobel P. Mouse mutants lacking the cation-independent mannose 6- phosphate/insulin-like growth factor II receptor are impaired in lysosomal enzyme transport: Comparison of cation-independent and cation- dependent mannose 6-phosphate receptor-deficient mice. Biochem J. 1998;330(Pt 2):903–908. doi: 10.1042/bj3300903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dahms NM, Lobel P, Breitmeyer J, Chirgwin JM, Kornfeld S. 46 kd mannose 6-phosphate receptor: Cloning, expression, and homology to the 215 kd mannose 6-phosphate receptor. Cell. 1987;50(2):181–192. doi: 10.1016/0092-8674(87)90214-5. [DOI] [PubMed] [Google Scholar]
- 123.Pohlmann R, Nagel G, Schmidt B, Stein M, Lorkowski G, Krentler C, Cully J, Meyer HE, Grzeschik KH, Mersmann G, Hasilik A, von Figura K. Cloning of a cDNA encoding the human cation-dependent mannose 6- phosphate-specific receptor. Proc Natl Acad Sci U S A. 1987;84(16):5575–5579. doi: 10.1073/pnas.84.16.5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lobel P, Dahms NM, Breitmeyer J, Chirgwin JM, Kornfeld S. Cloning of the bovine 215-kda cation-independent mannose 6-phosphate receptor. Proceedings of the National Academy of Sciences of the United States of America. 1987;84(8):2233–2237. doi: 10.1073/pnas.84.8.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lobel P, Dahms NM, Kornfeld S. Cloning and sequence analysis of the cation-independent mannose 6- phosphate receptor. J Biol Chem. 1988;263(5):2563–2570. [PubMed] [Google Scholar]
- 126.Braulke T, Bonifacino JS. Sorting of lysosomal proteins. Biochim Biophys Acta. 2009;1793(4):605–14. doi: 10.1016/j.bbamcr.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 127.Tong PY, Kornfeld S. Ligand interactions of the cation-dependent mannose 6-phosphate receptor. Comparison with the cation-independent mannose 6-phosphate receptor. Journal of Biological Chemistry. 1989;264(14):7970–7975. [PubMed] [Google Scholar]
- 128.Hoflack B, Kornfeld S. Lysosomal enzyme binding to mouse p388d1 macrophage membranes lacking the 215-kda mannose 6-phosphate receptor: Evidence for the existence of a second mannose 6-phosphate receptor. Proceedings of the National Academy of Sciences of the United States of America. 1985;82(13):4428–4432. doi: 10.1073/pnas.82.13.4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Distler JJ, Patel R, Jourdian GW. Immobilization and assay of low-molecular-weight phosphomannosyl receptor in multiwell plates. Analytical Biochemistry. 1987;166(1):65–71. doi: 10.1016/0003-2697(87)90546-x. [DOI] [PubMed] [Google Scholar]
- 130.Watanabe H, Grubb JH, Sly WS. The overexpressed human 46-kda mannose 6-phosphate receptor mediates endocytosis and sorting of beta-glucuronidase. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(20):8036–8040. doi: 10.1073/pnas.87.20.8036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Marron-Terada PG, Hancock MK, Haskins DJ, Dahms NM. Recognition of dictyostelium discoideum lysosomal enzymes is conferred by the amino-terminal carbohydrate binding site of the insulin-like growth factor II/mannose 6-phosphate receptor. Biochemistry. 2000;39(9):2243–2253. doi: 10.1021/bi992226o. [DOI] [PubMed] [Google Scholar]
- 132.Hoflack B, Fujimoto K, Kornfeld S. The interaction of phosphorylated oligosaccharides and lysosomal enzymes with bovine liver cation-dependent mannose 6-phosphate receptor. Journal of Biological Chemistry. 1987;262(1):123–129. [PubMed] [Google Scholar]
- 133.Tabas I, Kornfeld S. Biosynthetic intermediates of beta-glucuronidase contain high mannose oligosaccharides with blocked phosphate residues. Journal of Biological Chemistry. 1980;255(14):6633–9. [PubMed] [Google Scholar]
- 134.Varki A, Kornfeld S. The spectrum of anionic oligosaccharides released by endo-beta-N-acetylglucosaminidase h from glycoproteins. Structural studies and interactions with the phosphomannosyl receptor. Journal of Biological Chemistry. 1983;258(5):2808–2818. [PubMed] [Google Scholar]
- 135.Chavez CA, Bohnsack RN, Kudo M, Gotschall RR, Canfield WM, Dahms NM. Domain 5 of the cation-independent mannose 6-phosphate receptor preferentially binds phosphodiesters (mannose 6-phosphate N-acetylglucosamine ester) Biochemistry. 2007;46(44):12604–12617. doi: 10.1021/bi7011806. [DOI] [PubMed] [Google Scholar]
- 136.Song X, Lasanajak Y, Olson LJ, Boonen M, Dahms NM, Kornfeld S, Cummings RD, Smith DF. Glycan microarray analysis of p-type lectins reveals distinct phosphomannose glycan recognition. J Biol Chem. 2009;284(50):35201–35214. doi: 10.1074/jbc.M109.056119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bohnsack RN, Song X, Olson LJ, Kudo M, Gotschall RR, Canfield WM, Cummings RD, Smith DF, Dahms NM. Cation-independent mannose 6-phosphate receptor: A composite of distinct phosphomannosyl binding sites. J Biol Chem. 2009;284:35215–35226. doi: 10.1074/jbc.M109.056184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Olson LJ, Peterson FC, Castonguay A, Bohnsack RN, Kudo M, Gotschall RR, Canfield WM, Volkman BF, Dahms NM. Structural basis for recognition of phosphodiester-containing lysosomal enzymes by the cation-independent mannose 6-phosphate receptor. Proc Natl Acad Sci USA. 2010;107(28):12493–8. doi: 10.1073/pnas.1004232107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Boonen M, Vogel P, Platt KA, Dahms N, Kornfeld S. Mice lacking mannose 6-phosphate uncovering enzyme activity have a milder phenotype than mice deficient for N-acetylglucosamine-1-phosphotransferase activity. Mol Biol Cell. 2009;20(20):4381–4389. doi: 10.1091/mbc.E09-05-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sleat DE, Lobel P. Ligand binding specificities of the two mannose 6-phosphate receptors. Journal of Biological Chemistry. 1997;272(2):731–738. doi: 10.1074/jbc.272.2.731. [DOI] [PubMed] [Google Scholar]
- 141.Ludwig T, Munier-Lehmann H, Bauer U, Hollinshead M, Ovitt C, Lobel P, Hoflack B. Differential sorting of lysosomal enzymes in mannose 6-phosphate receptor-deficient fibroblasts. EMBO Journal. 1994;13(15):3430–3437. doi: 10.1002/j.1460-2075.1994.tb06648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Pohlmann R, Boeker MW, von Figura K. The two mannose 6-phosphate receptors transport distinct complements of lysosomal proteins. Journal of Biological Chemistry. 1995;270(45):27311–27318. doi: 10.1074/jbc.270.45.27311. [DOI] [PubMed] [Google Scholar]
- 143.Kasper D, Dittmer F, von Figura K, Pohlmann R. Neither type of mannose 6-phosphate receptor is sufficient for targeting of lysosomal enzymes along intracellular routes. Journal of Cell Biology. 1996;134(3):615–23. doi: 10.1083/jcb.134.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Roberts DL, Weix DJ, Dahms NM, Kim JJP. Molecular basis of lysosomal enzyme recognition: Three-dimensional structure of the cation-dependent mannose 6-phosphate receptor. Cell. 1998;93(4):639–648. doi: 10.1016/s0092-8674(00)81192-7. [DOI] [PubMed] [Google Scholar]
- 145.Olson LJ, Zhang J, Lee YC, Dahms NM, Kim JJP. Structural basis for recognition of phosphorylated high mannose oligosaccharides by the cation-dependent mannose 6-phosphate receptor. Journal of Biological Chemistry. 1999;274(42):29889–29896. doi: 10.1074/jbc.274.42.29889. [DOI] [PubMed] [Google Scholar]
- 146.Brown J, Esnouf RM, Jones MA, Linnell J, Harlos K, Hassan AB, Jones EY. Structure of a functional igf2r fragment determined from the anomalous scattering of sulfur. Embo J. 2002;21(5):1054–1062. doi: 10.1093/emboj/21.5.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Uson I, Schmidt B, von Bulow R, Grimme S, von Figura K, Dauter M, Rajashankar KR, Dauter Z, Sheldrick GM. Locating the anomalous scatterer substructures in halide and sulfur phasing. Acta Crystallographica Section D-Biological Crystallography. 2003;59(Pt 1):57–66. doi: 10.1107/s090744490201884x. [DOI] [PubMed] [Google Scholar]
- 148.Olson LJ, Yammani RD, Dahms NM, Kim JJ. Structure of uPAR, plasminogen, and sugar-binding sites of the 300 kda mannose 6-phosphate receptor. EMBO J. 2004;23(10):2019–28. doi: 10.1038/sj.emboj.7600215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Olson LJ, Dahms NM, Kim JJ. The N-terminal carbohydrate recognition site of the cation-independent mannose 6-phosphate receptor. Journal of Biological Chemistry. 2004;279(32):34000–9. doi: 10.1074/jbc.M404588200. [DOI] [PubMed] [Google Scholar]
- 150.Brown J, Delaine C, Zaccheo OJ, Siebold C, Gilbert RJ, van Boxel G, Denley A, Wallace JC, Hassan AB, Forbes BE, Jones EY. Structure and functional analysis of the igf-ii/igf2r interaction. EMBO J. 2008;27(1):265–276. doi: 10.1038/sj.emboj.7601938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Satoh T, Chen Y, Hu D, Hanashima S, Yamamoto K, Yamaguchi Y. Structural basis for oligosaccharide recognition of misfolded glycoproteins by OS-9 in ER-associated degradation. Mol Cell. 2010;40(6):905–16. doi: 10.1016/j.molcel.2010.11.017. [DOI] [PubMed] [Google Scholar]
- 152.Castonguay AC, Olson LJ, Dahms NM. Mannose 6-phosphate receptor homology (MRH) domain-containing lectins in the secretory pathway. Biochim Biophys Acta. 2011;1810(9):815–826. doi: 10.1016/j.bbagen.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hancock MK, Haskins DJ, Sun G, Dahms NM. Identification of residues essential for carbohydrate recognition by the insulin-like growth factor ii/mannose 6-phosphate receptor. J Biol Chem. 2002;277:11255–11264. doi: 10.1074/jbc.M109855200. [DOI] [PubMed] [Google Scholar]
- 154.Szathmary R, Bielmann R, Nita-Lazar M, Burda P, Jakob CA. Yos9 protein is essential for degradation of misfolded glycoproteins and may function as lectin in ERAD. Molecular Cell. 2005;19(6):765–775. doi: 10.1016/j.molcel.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 155.Hoflack B, Kornfeld S. Purification and characterization of a cation-dependent mannose 6- phosphate receptor from murine p388d1 macrophages and bovine liver. J Biol Chem. 1985;260(22):12008–12014. [PubMed] [Google Scholar]
- 156.Olson LJ, Zhang J, Dahms NM, Kim JJP. Twists and turns of the CD-MPR: Ligand-bound versus ligand-free receptor. J Biol Chem. 2002;277(12):10156–10161. doi: 10.1074/jbc.M112230200. [DOI] [PubMed] [Google Scholar]
- 157.Olson LJ, Hindsgaul O, Dahms NM, Kim JJ. Structural insights into the mechanism of ph-dependent ligand binding and release by the cation-dependent mannose 6-phosphate receptor. J Biol Chem. 2008;283(15):10124–10134. doi: 10.1074/jbc.M708994200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Olson LJ, Sun G, Bohnsack RN, Peterson FC, Dahms NM, Kim JJ. Intermonomer interactions are essential for lysosomal enzyme binding by the cation-dependent mannose 6-phosphate receptor. Biochemistry. 2010;49(1):236–246. doi: 10.1021/bi901725x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hancock MK, Yammani RD, Dahms NM. Localization of the carbohydrate recognition sites of the insulin-like growth factor II/mannose 6-phosphate receptor to domains 3 and 9 of the extracytoplasmic region. J Biol Chem. 2002;277(49):47205–47212. doi: 10.1074/jbc.M208534200. [DOI] [PubMed] [Google Scholar]