

Abstract
The asymmetric total syntheses of the α-benzylidene-γ-butyrolactone natural products megacerotonic acid and shimobashiric acid A have been accomplished in nine and 11 steps, respectively, from simple, commercially available starting materials. The key step for each synthesis is the (arene)RuCl(monosulfonamide)-catalyzed dynamic kinetic resolution-asymmetric transfer hydrogenation (DKR-ATH) of racemic α,δ-diketo-β-aryl esters to establish the absolute stereochemistry. Intramolecular diastereoselective Dieckmann cyclization forms the lactone core, and ketone reduction/alcohol elimination installs the α-arylidene.
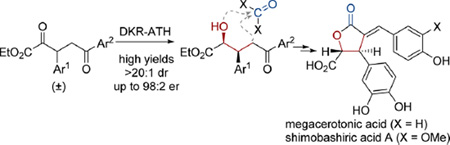
Natural products containing an α-benzylidene-γ-butyrolactone1 elicit a wide variety of biological responses, including antiviral,2 anticancer,3 antifungal,4 and anti-inflammatory activities.5 To date, megacerotonic acid6 1a and shimobashiric acid A7 1b are the only known members of this natural product class that contain both C3-aryl and C4-carboxylic acid substituents (Scheme 1).8 A racemic total synthesis of megacerotonic acid has been reported,9 but its biological activity has not been investigated. The purpose of this communication is to report the first asymmetric total syntheses of megacerotonic acid and shimobashiric acid A via a route that should be amenable to the preparation of unnatural congeners.
Scheme 1.

Retrosynthetic Analysis
Megacerotonic acid was isolated by Takeda and co-workers from Megaceros f lagellaris in 1990, and no investigation of its biological activity data has been reported to date.6 At the time of its isolation, megacerotonic acid was the only known γ-butyrolactone natural product to contain the C2-arylidene-C3-aryl-C4-carboxylic acid substitution pattern. Papin and coworkers9 carried out a racemic synthesis, unambiguously establishing its structure. The only other natural γ-butyrolactone known with this substituent array, shimobashiric acid A, was isolated by Murata and co-workers in 2012 from Keiskea japonica after its extracts showed hyaluronidase inhibitory activity.7 Although a number of other natural products isolated from these extracts showed biological activity, the isolation of 1b in a small quantity (1.2 mg) precluded the investigation of its biological activity.
Our laboratory recently reported facile access to enantioenriched γ-butyrolactones containing the required C3-aryl and C4-carboxylate functionality,10 potentially providing rapid entry to structures 1a and 1b. Approaching the retrosynthetic analysis of these natural products (Scheme 1), we envisioned Heck coupling of α-methylene lactone 2 with the appropriate haloarene (Ar2−X), allowing late-stage installation of a variety of benzylidene substituents and providing a modular approach to this class of natural products. Synthetic access to 2 would be achieved by a known three-step procedure utilizing ruthenium catalyzed dynamic kinetic resolution asymmetric transfer hydrogenation (DKR-ATH) of racemic β-aryl-α-keto esters followed by in situ diastereoselective lactonization.10 Racemic β-aryl-α-keto esters 3 can be prepared in a single step using a previously reported glyoxylate Stetter addition.
Initial investigation into this proposed synthetic pathway focused on the optimization of a route to 1a as we believed the same pathway would also be applicable to 1b (Scheme 2). DKR-ATH of β-aryl-α-keto ester 3 with concomitant lactonization smoothly provided γ-butyrolactone 4 in high diastereo- and enantioselectivity following a single recrystallization. Bromomethylation using dibromomethane followed by LiCl-promoted Krapcho dealkoxycarbonylation/elimination11 provided α-methylene lactone 2 for investigation of the projected Heck coupling.
Scheme 2.

Preliminary Route
Extant methodologies for Heck couplings that deliver exocyclic alkenes are generally limited to β-unsubstituted- or β-alkyl-α-methylidene lactones.12 Kim and co-workers reported the only example of Heck coupling using β-aryl-α-methylidene lactones, and formation of the butenolide was heavily favored over the desired exocyclic alkene.13 In our investigations, optimization of this coupling protocol provided arylidene 6a in only 19% yield (Scheme 3).14 Additionally, methylidene 2 was unreactive toward cross-metathesis with styrenes.15
Scheme 3.

Access to 6a via Heck Coupling
Consequently, we turned our attention to an alternative route from lactone 4, which would allow late stage installation of different benzylidene substituents (Scheme 4). Krapcho dealkoxycarbonylation of lactone 4 provided lactone 7 in good yield.16 Aldol reaction of lactone 7 with p-anisaldehyde followed by acid catalyzed elimination provided 6a.17
Scheme 4.

Krapcho Dealkoxycarbonylation/Aldol/Elimination Sequence
This modified route provided key intermediate 6a in a disappointing three step overall yield of 20%; however, the viability of alcohol elimination prompted us to consider a new retrosynthetic analysis for alcohol 8a (Scheme 5). In this proposed sequence, access to 8a would be provided by reduction of β-keto lactone 9, which would arise from (4 + 1)-annulation between a synthetic equivalent of 10 and a phosgene surrogate (11). DKR-ATH of α,δ-diketo-β-aryl ester 12 would establish the absolute stereochemistry.10
Scheme 5.

Revised Retrosynthetic Analysis
α,δ-Diketo-β-aryl ester 12a was prepared via N-heterocyclic carbene-catalyzed Stetter reaction of ethyl glyoxylate with enone 13a10 and was directly submitted to (arene)RuCl-(monosulfonamide) catalyzed DKR-ATH, providing alcohol 14a in excellent yield with high diastereo- and enantioselectivity (Scheme 6A).10 Installation of the requisite activated carbonyl functionality was achieved with 1,1′-carbonyldiimidazole to give tricarbonyl 15a.18 Selective enolate formation with potassium bis(trimethylsilyl)amide (KHMDS) led to a diastereoselective Dieckmann cyclization that delivered 9a. A subsequent ketone reduction to alcohol 8a was achieved with Pd/C and H2.19 Dehydration to enone 6a (Scheme 6B) was promoted by polymer supported perfluorosulfonic acid (Nafion SAC-13). In our hands, the reported ester epimerization and hydrolysis9 of 6a using lithium methoxide/methanol resulted in a number of undesired byproducts. Switching to sodium tert-butoxide in tert-amyl alcohol smoothly provided acid 16a as a 14:1 mixture of chromatographically inseparable diastereomers. Boron tribromide-mediated demethylation and purification by reverse phase HPLC completed the first asymmetric total synthesis of megacerotonic acid 1a in nine steps and 11% overall yield from commercially available materials.
Scheme 6.

Synthetic Route to Megacerotonic Acid and Shimobashiric Acid A
The route to 1b was identical to 1a through the synthesis of alcohol 8b (Scheme 6A). When 8b was submitted to the elimination conditions optimized for 8a (Scheme 6B), enone 6b was not formed, and only partial Boc deprotection was observed, even at elevated temperatures (100 °C). We sought a solution to this problem that would obviate the necessity of electron rich arenes and found that the Burgess reagent met this need, providing enone 6b as well as less electron rich α-benzylidene-γ-butyrolactones 6c and 6d, prepared via the same synthetic route (Scheme 7).20
Scheme 7.

Eliminations with the Burgess Reagent
tert-Butoxycarbonyl removal, epimerization at C4, and hydrolysis provided 16b in good overall yield as a 14:1 mixture of chromatographically inseparable diastereomers. Acetal deprotection and purification by reverse phase HPLC provided shimobashiric acid A 1b in 11 steps and 5% overall yield from commercially available material.21
In conclusion, we have reported the first asymmetric total syntheses of structurally unique α-benzylidene-γ-butyrolactones megacerotonic acid and shimobashiric acid A via a route amenable to the synthesis of analogues. This route utilized DKR-ATH to establish the absolute stereochemistry in excellent enantio- and diastereoselectivity, and diastereoselective Dieckmann cyclization formed the lactone core in high overall yield. Selective ketone reduction and subsequent elimination formed the α-benzylidene with excellent E:Z selectivity. Epimerization at C4, hydrolysis, and phenol deprotection concluded the first asymmetric total syntheses of megacerotonic acid and shimobashiric acid A. Screening of the synthesized natural products and analogues thereof for relevant biological activity is currently underway and will be reported in due course.
Supplementary Material
ACKNOWLEDGMENTS
The project described was supported by Award No. R01 GM084927 from the National Institute of General Medical Sciences. S.W.K. acknowledges a National Science Foundation Graduate Research Fellowship. The authors thank Professor Marcey Waters, Brendan Peacor, and Nicholas Pinkin (UNC) for use of and assistance with reverse phase HPLC systems, and Professor Michael Crimmins (UNC) for helpful discussions. A natural sample of shimobashiric acid A was generously provided by Professor Toshihiro Murata (Tohoku Pharmaceutical University).
Footnotes
ASSOCIATED CONTENT
Experimental procedures, characterization, and spectral data for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.Kitson RRA, Millemaggi A, Taylor RJK. Angew. Chem. Int. Ed. 2009;48:9426. doi: 10.1002/anie.200903108. [DOI] [PubMed] [Google Scholar]
- 2.(a) Tuchinda P, Kornsakulkarn J, Pohmakotr M, Kongsaeree P, Prabpai S, Yoosook C, Kasisit J, Napaswad C, Sophasan S, Reutrakul V. J. Nat. Prod. 2008;71:655. doi: 10.1021/np7007347. [DOI] [PubMed] [Google Scholar]; (b) Wen C-C, Kuo Y-H, Jan J-T, Liang P-H, Wang S-Y, Liu H-G, Lee C-K, Chang S-T, Kuo C-J, Lee S-S, Hou C-C, Hsiao P-W, Chien S-C, Shyur L-F, Yang N-S. J. Med. Chem. 2007;50:4087. doi: 10.1021/jm070295s. [DOI] [PubMed] [Google Scholar]; (c) Baroudy B, Archambault J. 2006/088824 A2. WO. 2006 Aug 24;
- 3. Mansoor TA, Borralho PM, Luo X, Mulhovod S, Rodrigues CMP, Ferreira M-JU. Phytomedicine. 2013;20:923. doi: 10.1016/j.phymed.2013.03.026. Ho P-J, Chou C-K, Kuo Y-H, Tu L-C, Yeh S-F. Life Sci. 2007;80:493. doi: 10.1016/j.lfs.2006.10.017. Dall’Acqua S, Viola G, Piacente S, Cappelletti EM, Innocenti G. J. Nat. Prod. 2004;67:1588. doi: 10.1021/np040046w. Chang S-T, Wang DS-Y, Wua C-L, Shiah S-G, Kuo Y-H, Chang C-J. Phytochemistry. 2000;55:227. doi: 10.1016/s0031-9422(00)00275-2. (e) Natural product derivatives: Ding Y-H, Fan H-X, Long J, Zhang Q, Chen Y. Bioorg. Med. Chem. Lett. 2013;23:6087. doi: 10.1016/j.bmcl.2013.09.028.
- 4.Hwang E-I, Yun B-S, Kim Y-K, Kwon B-M, Kim H-G, Lee H-B, Jeong W-J, Kim S-U. J. Antibiot. 2000;53:903. doi: 10.7164/antibiotics.53.903. [DOI] [PubMed] [Google Scholar]
- 5.Cho JY, Park J, Kim PS, Yoo ES, Baik KU, Park MH. Biol. Pharm. Bull. 2001;24:167. doi: 10.1248/bpb.24.167. [DOI] [PubMed] [Google Scholar]
- 6.Takeda R, Hasegawa J, Shinozaki M. Tetrahedron Lett. 1990;31:4159. [Google Scholar]; Takeda R, Hasegawa J, Shinozaki M. Proc. Phytochem. Soc. Eur. 1990;29:201. [Google Scholar]
- 7.Murata T, Miyase T, Yoshizaki F. Chem. Pharm. Bull. 2012;60:121. doi: 10.1248/cpb.60.121. [DOI] [PubMed] [Google Scholar]
- 8.For α-benzylidene-γ-butyrolactone natural products with C3-aryl-C4-ester substitution (two cases) see ref 1. For α-benzylidene-γ-butyrolactone natural products with C3-aryl-C4-methanol substitution (two cases) see: Chung Y-M, Wang H-C, El-Shazly M, Leu Y-L, Cheng M-C, Lee C-L, Fang-Rong Chang F-R, Wu Y-C. J. Agric. Food Chem. 2011;59:9219. doi: 10.1021/jf202119m.
- 9.(a) Brown E, Dhal R, Papin N. Tetrahedron. 1995;51:13061. [Google Scholar]; (b) Papin N, Dhal R, Brown E. Nat. Prod. Lett. 1994;4:303. [Google Scholar]
- 10.(a) Steward KM, Corbett MT, Goodman CG, Johnson JS. J. Am. Chem. Soc. 2012;134:20197. doi: 10.1021/ja3102709. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Steward KM, Gentry EC, Johnson JS. J. Am. Chem. Soc. 2012;134:7329. doi: 10.1021/ja3027136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bukowsa M, Kolodziejek W, Prejzner J. Polym. J. Chem. 1990;64:573. [Google Scholar]
- 12.(a) Belovodskii AV, Shults EE, Shakirov MM, Bagryanskaya IY, Gatilov YV, Tolstikov GA. Russ. J. Org. Chem. 2010;46:1719. [Google Scholar]; (b) Han C, Barrios FJ, V Riofski MV, David A, Colby DA. J. Org. Chem. 2009;74:7176. doi: 10.1021/jo901533e. [DOI] [PubMed] [Google Scholar]; (c) Arcadi A, Chiarini M, Marinelli F, Berente Z, Kollàr L. Eur. J. Org. Chem. 2001:3165. doi: 10.1021/ol9912130. [DOI] [PubMed] [Google Scholar]; (d) Arcadi A, Chiarini M, Marinelli F, Berente Z, Kollàr L. Org. Lett. 2000;2:69. doi: 10.1021/ol9912130. [DOI] [PubMed] [Google Scholar]
- 13.Kim SH, Kim KH, Lee HJ, Kim JN. Tetrahedron Lett. 2013;54:329. [Google Scholar]
- 14.Under the conditions reported, butenolide formation was not observed and the mass balance resides in a product incorporating two units of 2.
- 15.(a) Raju R, Allen LJ, Le T, Taylor CD, Howell AR. Org. Lett. 2007;9:1699. doi: 10.1021/ol070574+. [DOI] [PubMed] [Google Scholar]; (b) Moï1se J, Arseniyadis S, Cossy J. Org. Lett. 2007;9:1695. doi: 10.1021/ol0703940. [DOI] [PubMed] [Google Scholar]
- 16.Krapcho AP. Synthesis. 1982:893. [Google Scholar]
- 17.Mulzer J, Chucholowski A. Angew. Chem. 1986;98:632. [Google Scholar]
- 18.Armstrong A, Li W. N,N′-Carbonyldiimidazole. e-EROS Encyclopedia of Reagents for Organic Synthesis. 2007 [Google Scholar]
- 19.Pohmakotr M, Sampaongoen L, Issaree A, Tuchinda P, Reutrakul V. Tetrahedron Lett. 2003;44:6717. [Google Scholar]
- 20.(a) Khapli S, Dey S, Mal D. J. Indian Inst. Sci. 2001;81:461. [Google Scholar]; (b) Burgess EM, Penton HR, Jr, Taylor EA. J. Org. Chem. 1973;38:26. [Google Scholar]; (c) Atkins GM, Burgess EM. J. Am. Chem. Soc. 1968;90:4744. [Google Scholar]
- 21.The lower overall yield obtained for shimobashiric acid A in comparison to megacerotonic acid is a consequence of the Claisen– Schmidt condensation used in the synthesis of 13b (46% yield).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.