

Abstract
The use of droplet digital PCR (ddPCR) for low-level DNA mutation detection in cancer, prenatal diagnosis, and infectious diseases is growing rapidly. However, although ddPCR has been implemented successfully for detection of rare mutations at pre-determined positions, no ddPCR adaptation for mutation scanning exists. Yet, frequently, clinically relevant mutations reside on multiple sequence positions in tumor suppressor genes or complex hotspot mutations in oncogenes. Here, we describe a combination of coamplification at lower denaturation temperature PCR (COLD-PCR) with ddPCR that enables digital mutation scanning within approximately 50-bp sections of a target amplicon. Two FAM/HEX-labeled hydrolysis probes matching the wild-type sequence are used during ddPCR. The ratio of FAM/HEX-positive droplets is constant when wild-type amplicons are amplified but deviates when mutations anywhere under the FAM or HEX probes are present. To enhance the change in FAM/HEX ratio, we employed COLD-PCR cycling conditions that enrich mutation-containing amplicons anywhere on the sequence. We validated COLD-ddPCR on multiple mutations in TP53 and in EGFR using serial mutation dilutions and cell-free circulating DNA samples, and demonstrate detection down to approximately 0.2% to 1.2% mutation abundance. COLD-ddPCR enables a simple, rapid, and robust two-fluorophore detection method for the identification of multiple mutations during ddPCR and potentially can identify unknown DNA variants present in the target sequence.
In the era of personalized medicine, mutation detection methods that target mutations known to influence therapy response or clinical outcome are of great interest. Although real-time PCR methodologies have been described and are widely used for detecting mutations in clinical samples,1–4 interest in digital PCR5 is rising in view of the unique aspects of the technology and the emergence of commercial droplet digital PCR (ddPCR) platforms.5–8 ddPCR has been implemented in a variety of fields such as cancer biomarker and viral load detection, fetal screening, or library quantification for next-generation sequencing.9 One of the most common ddPCR applications is in the detection of known DNA variants present within a large excess of wild-type DNA, for instance in DNA from heterogeneous samples that harbor subclonal populations of mutated tumor cells.10
In ddPCR, the amplification reaction is compartmentalized into microscopic emulsion-based droplets containing at most a few target molecules per droplet. By segregating the interrogated sample, the effect of in-droplet target competition is reduced, which translates into increased assay discrimination and facile determination of wild-type versus mutant status.11 However, as currently applied, ddPCR can only be used to detect mutations at known sequence positions. ddPCR incorporates two reporter probes, one mutant-specific and one wild-type, because of the requirement to account for PCR-amplification variability among droplets. This approach, by design, allows only the detection of previously known mutations. In cancer, tumor suppressor genes such as TP53 harbor mutations that are scattered throughout the gene as opposed to oncogenes that usually carry mutations located in specific hotspots.12 Although mutation scanning methods based on amplicon fluorescent melting analysis following real-time PCR have been developed,13–16 monitoring fluorescent melting within individual droplets during ddPCR is not available at this time. Accordingly, a ddPCR approach that could be implemented in a mutation scanning format is desirable.
Here, we demonstrate that incorporation of coamplification at lower denaturation temperature PCR (COLD-PCR),17–21 within the ddPCR workflow in conjunction with two fluorescently labeled probes matching the wild-type amplicon, provides a simple and robust method for mutation scanning of target amplicons. COLD-PCR suppresses wild-type sequences and enables preferential amplification of mutation-containing droplets, for any mutation along the amplicon.21,22 Detecting changes to the ratio of COLD-PCR–enhanced signals caused by mutations anywhere within the probed region enables mutation scanning with high selectivity. This novel enhanced ratio of signal-based mutation scanning COLD-ddPCR enables a rapid method for the detection of mutations during ddPCR without prior knowledge of the specific DNA variant present in the target sequence. We demonstrate in this paper the application of COLD-ddPCR to the detection of multiple mutations present in TP53 exon 8, as well as for the T790M resistance mutation in EGFR exon 20 in DNA from mutated cell lines and cell-free circulating DNA (cfDNA) from clinical cancer samples.
Materials and Methods
Cell Lines and Clinical Samples
Missense mutations in several positions of the TP53 exon 8 and mutation p.T790M in EGFR exon 20 were assessed in this study. Human genomic DNA from cancer cell lines DU-145 (ATCC HTB-81D), HCC2218 (ATCC CRL-2343), and MDA-MB-231 (ATCC HTB-26D) was purchased from ATCC (Manassas, VA). Genomic DNA from commercial cell lines SW480 (ATCC no. CCL-228) and H1975 (ATCC no. CRL-5908) was extracted using the DNeasy Blood and Tissue kit (Qiagen, Valencia, CA) following the manufacturer's protocol (Table 1). Human genomic DNA (Promega, Madison, WI) was used as wild-type control DNA and for creating dilutions of gradually decreasing mutation abundances. All experiments were replicated at least three independent times for assessing the reproducibility of the results.
Table 1.
Summary of Cell Lines Used
| Target region | Cell line | Mutation (nt) | Mutation (aa) |
|---|---|---|---|
| EGFR exon 20 | H1975 | c.2369C>T | p.T790M |
| TP53 exon 8 | SW480 | c.818G>A | p.R273H |
| DU-145 | c.820G>T | p.V274F | |
| MDA-MB-231 | c.839G>A | p.R280K | |
| HCC2218 | c.847C>T | p.R283C |
aa, amino acid; nt, nucleotide.
To evaluate the efficacy of this assay in characterizing specimens from different origins, we analyzed a colorectal tumor sample, known to harbor a G>A (p.R273H) missense mutation present at a low frequency (approximately 1%). This mutation had been previously identified and validated using different methods [COLD-PCR sequencing, denaturing high-pressure liquid chromatography, restriction endonuclease-mediated selective PCR, and differential strand separation at critical temperature (Tc)16,23]. We also evaluated cfDNA isolated from patients with lung adenocarcinomas at different stages of disease progression and treatment, and a cfDNA sample from an esophageal cancer case. Mutations in these cfDNA samples had been previously identified using a ddPCR allele-specific approach with hydrolysis probes specific either to the wild-type or the mutant allele.24 Samples were obtained from patients after informed consent and Dana Farber-Cancer Institute Institutional Review Board approval (Table 2).
Table 2.
List of Clinical Samples Used for Validation Purposes
| Target region | Sample name | Type | Tumor/origin | Mutation (nt) | Mutation (aa) | Estimated frequency∗ |
|---|---|---|---|---|---|---|
| TP53 exon 8 | CT20 | gDNA | Colorectal cancer | c.818G>A | p.R273H | ∼1% |
| FC39–4 | cfDNA | Esophageal cancer | c.847C>T | p.R283C | ∼5% | |
| EGFR exon 20 | PT-04–09 | cfDNA | Lung adenocarcinoma | c.2369C>T | p.T790M | ∼0.1% |
| PT-21 | cfDNA | Lung adenocarcinoma | c.2369C>T | p.T790M | Wild type | |
| PT-04–11 | cfDNA | Lung adenocarcinoma | c.2369C>T | p.T790M | ∼2.1% | |
| PT-10–06 | cfDNA | Lung adenocarcinoma | c.2369C>T | p.T790M | ∼0.43% | |
| PT-07–16 | cfDNA | Lung adenocarcinoma | c.2369C>T | p.T790M | ∼2.03% | |
| PT-10–33 | cfDNA | Lung adenocarcinoma | c.2369C>T | p.T790M | ∼0.38% | |
| PT-04–17 | cfDNA | Lung adenocarcinoma | c.2369C>T | p.T790M | ∼4.08% |
Circulating cell-free tumor DNA samples (cfDNA).
aa, amino acid; DNA, genomic DNA; nt, nucleotide.
Mutation frequencies were estimated by previous analysis or conventional digital PCR using allele-specific probes.
Genomic and cfDNA Pre-Amplification Step
Gene-specific primers were used to pre-amplify selected targets from genomic or cfDNA (approximately 20 ng and approximately 10 ng, respectively) before performing ddPCR experiments. Twenty cycles of pre-amplification of EGFR exon 20 were done using the GoTaq Flexi DNA polymerase system (Promega) and 0.2 μmol/L forward and reverse primers. Inconsistent with previous studies, the forward primer was engineered to encompass the A/G single nucleotide polymorphism present within this amplicon to generate a uniform template for downstream analysis.25,26 The initial 30 cycles of pre-amplification of TP53 exon 8 were conducted using the Phusion polymerase system (New England Biolabs, Ipswich, MA) and 0.2 μmol/L forward and reverse primers (Tables 3 and 4). Amplified DNA was then diluted and used as a template for ddPCR experiments using nested primers (Table 3).
Table 3.
List of Primers and Probes Sequences Used in This Study
| Target region | PCR round | Primer and probe sequences | Size (bp) |
|---|---|---|---|
| TP53 exon 8 | Genomic DNA pre-amplification | F: 5′-GCTTCTCTTTTCCTATCCTG-3′ | 167 |
| R: 5′-CTTACCTCGCTTAGTGCT-3′ | |||
| Digital PCR | F: 5′-TGGTAATCTACTGGGACG-3′ | 87 | |
| R: 5′-CGGAGATTCTCTTCCTCT-3′ | |||
| Hydrolysis probes | 5′-FAM-TGGGAGAGACCGGCGCA-BHQ_1-3′∗ | ||
| 5′-HEX-TTTGAGGTGCGTGTTTGTGCC-BHQ_1-3′∗ | |||
| EGFR exon 20 | Genomic DNA pre-amplification | F: 5′-GCTGGGCATCTGCCTCACCTCCACCGTGCAACT-3′ | 91 |
| R: 5′-GTCTTTGTGTTCCCGGACATAG-3′ | |||
| Digital PCR | F: 5′-GCTGGGCATCTGCCTCA-3′ | 67 | |
| R: 5′-CAGGAGGCAGCCGAAGG-3′ | |||
| Hydrolysis probes | 5′-FAM-ATGAGTTGCACGGTGGA-BHQ_1-3′∗ | ||
| 5′-HEX-CTCATCACGCAGCTCATG-BHQ_1-3′∗ | |||
| 5′-FAM-CTCATCATGCAGCTCATG-BHQ_1-3′† | |||
| 5′-HEX-CTCATCACGCAGCTCATG-BHQ_1-3′† |
F, forward; R, reverse.
These hydrolysis probes had sequences complementary to the wild-type allele.
Hydrolysis probes specific to the mutant (FAM) or wild-type (HEX) allele used for precise quantification of mutational abundances.
Table 4.
Cycling Conditions Used
| Target region | PCR round | Cycling conditions | |
|---|---|---|---|
| TP53 exon 8 | Genomic DNA pre-amplification | Initial denaturation | 98°C for 30 seconds |
| Thermocycling: 30 cycles | 98°C for 10 seconds | ||
| 58°C for 20 seconds | |||
| 72°C for 10 seconds | |||
| Conventional Digital PCR | Initial denaturation | 95°C for 10 minutes | |
| Thermocycling: 40 cycles | 94°C for 30 seconds | ||
| 58°C for 60 seconds | |||
| Hold | 98°C for 10 minutes | ||
| Digital COLD-PCR | Initial denaturation | 95°C for 10 minutes | |
| Thermocycling: 5 cycles | 94°C for 30 seconds | ||
| 58°C for 60 seconds | |||
| Thermocycling: 40 cycles | 78°C (Tc) for 30 seconds | ||
| 58°C for 60 seconds | |||
| Hold | 98°C for 10 minutes | ||
| EGFR exon 20 | Genomic DNA pre-amplification | Initial denaturation | 95°C for 120 seconds |
| Thermocycling: 20 cycles | 95°C for 15 seconds | ||
| 55°C for 30 seconds | |||
| 72°C for 30 seconds | |||
| 72°C for 60 seconds | |||
| Conventional Digital PCR | Initial denaturation | 95°C for 10 minutes | |
| Thermocycling: 40 cycles | 94°C for 30 seconds | ||
| 52°C for 60 seconds | |||
| Hold | 98°C for 10 minutes | ||
| Digital COLD-PCR | Initial denaturation | 95°C for 10 minutes | |
| Thermocycling: 5 cycles | 94°C for 30 seconds | ||
| 52°C for 60 seconds | |||
| Thermocycling: 35 cycles | 79.9°C (Tc) for 30 seconds | ||
| 52°C for 60 seconds | |||
| Hold | 98°C for 10 minutes | ||
COLD-PCR, coamplification at lower denaturation temperature PCR.
ddPCR Using Ratio of Signals from Wild-Type Probes
To apply COLD-ddPCR approach, two TaqMan hydrolysis probes (FAM and HEX labeled, respectively) were designed to match the wild-type sequence and to bind to different regions on the target amplicon (Figure 1).
Figure 1.
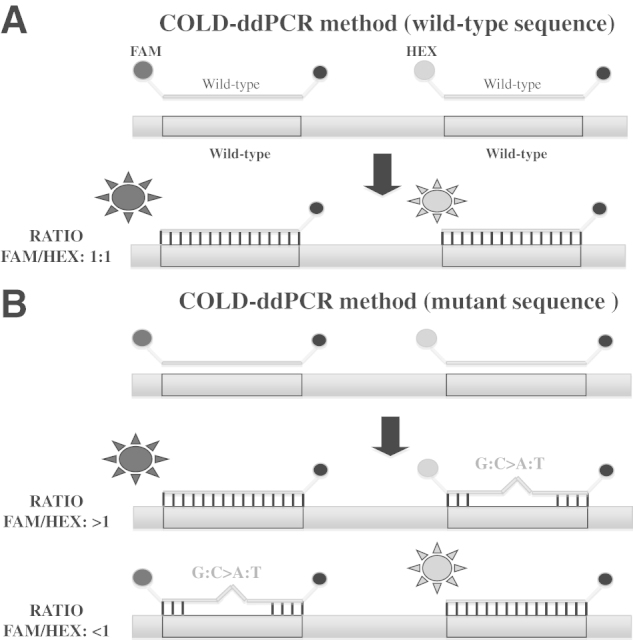
A: Signal generation during enhanced ratio of signals for mutation scanning coamplification at lower denaturation temperature–droplet digital PCR (COLD-ddPCR). The system incorporates two hydrolysis/reporter probes that are complementary to the wild-type sequence. When the interrogated DNA region does not contain mismatches that may prevent the reporter probes to hybridize, both probes emit similar signals, translated into a 1:1 FAM/HEX ratio. B: If there is a mutation (G:C>A:T or G:C>T:A) present under one of the probes, the mismatch will affect the probe-target DNA binding, reducing the overall fluorescence of that particular probe, producing a >1 or <1 FAM/HEX ratio. On the graph representing Taqman probes and DNA templates, the circles labeled on the left side of each probe are FAM (grey) or HEX (black). The circles labeled on the right side of each probe are quenchers (BHQ-1). The suns represent the release of FAM or HEX from the probe during COLD-ddPCR.
Conventional ddPCR Cycling Conditions
Conventional ddPCR reactions were performed following the manufacturer's indications (Bio-Rad Laboratories, Hercules CA). Amplifications were performed in a 20-μL volume containing 2× ddPCR supermix for probes (Bio-Rad Laboratories), 900 nmol/L forward and reverse primers (synthesized by Integrated DNA Technologies, Coralville, IA), 250 nmol/L FAM and HEX probes (Integrated DNA Technologies), and pre-amplified DNA template (using 1:4000 final dilution for the EGFR exon 20 experiments and 1:100,000 for the TP53 exon 8) (Table 3). We started from a pre-amplified template, given the nature (very low number of target copies) of the samples used for the validation stage (cfDNA). Droplets were then generated using the DG8 droplet generator cartridges by mixing the aqueous phase with 70 μL of droplet generation oil (DG; Bio-Rad Laboratories). Samples were transferred to a 96-well reaction plate and then sealed using the PX1 PCR plate sealer (Bio-Rad Laboratories) for 10 seconds at 180°C before thermal cycling. Cycling conditions for conventional ddPCR are summarized in Table 4.
COLD-ddPCR Principles and Cycling Conditions
In its simplest form, COLD-PCR (fast-COLD-PCR22) is performed by replacing the conventional denaturation temperature during the PCR cycling by a lower denaturation temperature. This results in a preferential amplification of mutations that decrease the melting temperature of the amplicon (G:C>A:T and G:C>T:A, or melting temperature–decreasing insertion-deletions). These mutation types comprise the majority (70% to 95%) of somatic mutations reported in breast, lung, gastric, colorectal, renal, ovarian, glioma, and melanoma cancers, as well as germline polymorphisms.27 In conventional ddPCR, once the DNA is compartmentalized, PCR amplification occurs simultaneously in every droplet. By contrast, COLD-ddPCR amplification takes place at a denaturation temperature lower than that of the denaturation temperature of the wild-type amplicon. As a result, amplification occurs preferentially within those droplets where the mutant allele is present, resulting in a mutant-biased amplification (Figure 2).
Figure 2.
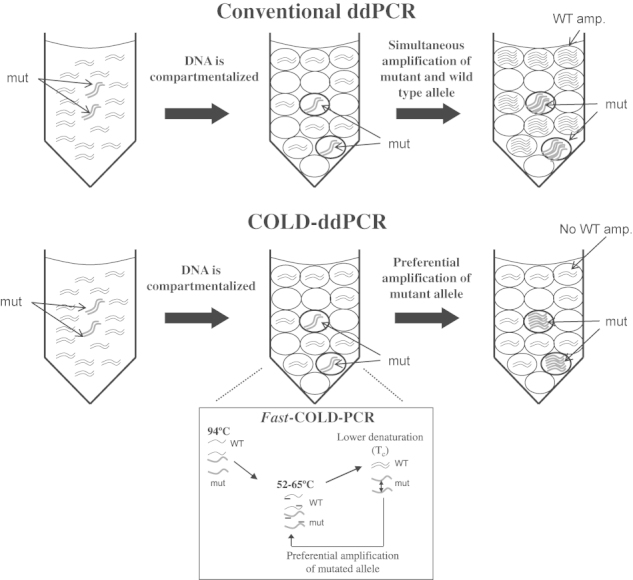
Diagram summarizes the principles of fast coamplification at lower denaturation temperature–droplet digital PCR (fast-COLD-ddPCR). Both mutant and wild-type alleles are compartmentalized into thousands of droplets before the thermal cycling step. In conventional ddPCR, both wild-type and mutant amplicons coamplify in different compartments. If fast-COLD-ddPCR is implemented during the thermal cycling inhibition of the wild-type amplicon, preferential amplification of mutation-containing droplets occurs. amp., amplification; mut, mutant; WT, wild type.
To determine the optimal critical denaturation temperature (Tc) for the COLD-ddPCR reactions, a gradient of Tc temperatures was tested using the Eppendorf Mastercycler (Eppendorf, Hamburg, Germany). Series of wild-type and 3% to 5% mutation-containing samples were used in ddPCR reactions using the temperature gradient feature of the thermal cycler. We first tested a broad temperature window and then narrowed it to a 1°C temperature window to find a suitable Tc (Supplemental Figures S1 and S2). As in bulk-solution COLD-PCR, the Tc chosen was the lowest temperature that generates substantial inhibition, yet reproducible amplification, for the wild-type sample.28 At this temperature, the FAM/HEX differences between wild-type and mutant samples were maximized.
Despite the wild-type inhibition, amplification still occurs in a fraction of wild-type DNA–containing droplets, hence providing positive signals in both HEX and FAM channels. The threshold in the QuantaSoft software (Bio-Rad Laboratories) is set such that for wild-type control samples, FAM/HEX = 1. The same threshold was then applied to all interrogated samples. Samples with the FAM/HEX ratio deviating substantially from 1 (P < 0.01) are considered mutant. ddPCR reactions were then analyzed and processed using the droplet reader and the QuantaSoft software version 1.3.2.0.
Allele-Specific ddPCR and Calculation of Fractional Abundance of Mutation
To validate mutational abundances in the cfDNA samples, two hydrolysis (TaqMan) probes, binding to the same DNA region, were designed for allele-specific ddPCR. Reactions were run under conventional PCR cycling conditions per manufacturer recommendations: FAM-labeled, complementary to the mutant allele, and HEX-labeled, complementary to the wild-type allele. Hydrolysis probes (Table 3) were synthesized by Integrated DNA Technologies.
Calculation of absolute number of positive events for a given channel (FAM or HEX), and the ratio and the fractional abundance of mutation for each sample were performed by the QuantaSoft software. The ratio was calculated as the number of copies per microliter obtained in the FAM channel (A), divided by copies per microliter in the HEX channel (B): A/B. The fractional abundance of mutant allele was obtained by dividing the number of copies per microliter of mutant allele (FAM channel) by the total copies per microliter of wild-type allele (HEX) plus mutant (FAM): [A/(A + B)]. The determination of number of target copies per droplet (number of copies of target molecule) was adjusted by the software to fit a Poisson distribution model with a 95% confidence level. The t-test was conducted to evaluate the statistical significance of the FAM/HEX ratio differences between wild-type and mutant alleles. Results with a two-tailed P value below 0.01 were considered significant.
Results
COLD-ddPCR Method for EGFR p.T790M Mutation
During COLD-ddPCR, the presence of a mutation-generated mismatch anywhere in the DNA sequence under a hydrolysis probe decreases the overall fluorescent signal generated from that probe compared to the signal contributed by the second probe that remains fully matched (Figure 1). Accordingly, the ratio of FAM/HEX signals either decreases relative to the wild type or increases, depending on whether there is a mutation under the FAM or HEX, respectively. This principle was first applied on a EGFR exon 20 amplicon where the p.T790M resistance-causing mutation lies.29
We initially used conventional PCR cycling conditions using serial dilutions of p.T790M mutant DNA into wild-type DNA. Under conventional cycling, the FAM-positive versus HEX-positive droplets changed by about approximately 10-fold in the presence of 100% p.T790M mutant sequence (Figure 3A). Serial dilutions of mutant into wild-type indicated that the lowest mutation dilution for which the FAM/HEX ratio is different from the wild-type samples is 3% (P < 0.01), given the SD of the signals from three replicates. Two-dimensional plots of the signals from droplets following ddPCR are depicted in Supplemental Figure S3 (one replicate of each sample). For wild-type samples, the vast majority of droplets generate both FAM- and HEX-positive signals, consistent with both TaqMan probes binding efficiently to the target sequence. A 100% mutant probe shifts the majority of droplets with positive signals toward FAM, whereas reducing the mutant abundance below 3% yields results statistically indistinguishable from the wild-type (t-test P > 0.01). Changing the threshold that separates FAM/HEX-positive from negative droplets in these two-dimensional plots changes the absolute number of droplets within each quadrangle but does not affect significantly the FAM/HEX-positive ratio or the lowest mutation abundance detectable (not shown).
Figure 3.

Absolute number of FAM-positive (dark gray) or HEX-positive (light gray) events (p.T790M region) after either conventional droplet digital PCR (ddPCR) (A) or coamplification at lower denaturation temperature–ddPCR (COLD-ddPCR) (B). The presence of the p.T790M mismatch under the HEX-labeled probe during COLD-ddPCR results in an increase in the absolute number of events for FAM, distinguishable from the wild-type sample down to mutational abundances of 5% to 10% or approximately 0.15% (P < 0.01) for conventional ddPCR or COLD-ddPCR, respectively. The error bars represent maximum and minimum number of positive events in each sample. WT, wild type.
By replacing conventional PCR with COLD-PCR cycling conditions, the overall number of droplets generating signals (positive events) for FAM and HEX are sharply reduced, consistent with inhibition of the wild-type amplification (Figure 3B). Furthermore, the shift in the ratio of FAM- to HEX-positive events is more pronounced, and the serial dilutions indicate that approximately 0.2% mutational abundance can be discriminated from wild type (with approximately 1.7× higher FAM/HEX ratio) (Figure 3B). Supplemental Figure S4 depicts two-dimensional plots of FAM versus HEX amplitude following COLD-ddPCR for wild-type, 5%, 1.25%, and 0.3% mutational abundance (one replicate of each sample). The sample with 0.3% p.T790M mutational abundance was distinguishable from that of the wild type according to t-test with two-tailed P < 0.01. The smear-like appearance is an unavoidable aspect of the technique; however, because the method preferentially inhibits the amplification of the wild-type amplicon, it does not affect the power of discrimination of the mutant amplicon. Experiments have been repeated multiple times, and these smears are reproducible between replicates. Changing by 10% to 20% the threshold that separates FAM/HEX-positive from negative droplets in these two-dimensional plots does not affect significantly the FAM/HEX-positive ratio (data not shown).
Finally, to compare the ratio-of-signals method using real-time PCR instead of ddPCR, we performed the same experiment using real-time PCR on an ECO thermocycler (Illumina, San Diego, CA). We calculated the FAM/HEX ratio of fluorescence signals at cycle 40, as used in ddPCR, and also plotted the cycle threshold values for each of the probes (FAM or HEX) (Supplemental Figure S5). The data indicate that the lowest mutation abundance detectable via real-time PCR is approximately 25% to 50%. In summary, the limits of detection using the present signal ratio–based approach with real-time PCR, ddPCR, and COLD-ddPCR are approximately 25% to 50%, approximately 5% to 10%, and approximately 0.2% to 0.3% respectively.
cfDNA samples isolated from plasma of patients with lung adenocarcinoma, harboring p.T790M mutations, were then examined. Mutational abundances for p.T790M for these clinical samples were independently derived using the established ddPCR approach, with FAM- and HEX-labeled allele-specific probes.24 Using the present approach, COLD-ddPCR, FAM/HEX ratios from clinical samples (as an average of three independent replicates) were distinguishable from the wild-type (seven independent replicates) with the exception of PT-04-09 (Figure 4). The latter sample had an estimated mutation abundance ≤0.1% using the allele-specific ddPCR approach.
Figure 4.

Application of coamplification at lower denaturation temperature–droplet digital PCR (COLD-ddPCR) FAM/HEX ratio method for detection of mutation p.T790M in cell-free circulating DNA samples. Serial dilution samples were also tested in parallel to enable assessment of mutational abundances. Numbers over the histograms indicate mutation abundances derived via independent quantification performed using allele-specific ddPCR for p.T790M as described.24 The error bars represent variations among three independent detections. WT, wild type.
Mutation Scanning Using COLD-ddPCR for Multiple TP53 Exon 8 Mutations
To validate the ability of COLD-ddPCR for mutation scanning and to examine multiple mutations across an amplicon, we developed a COLD-ddPCR assay directed to TP53 exon 8, where cancer-relevant mutations can be present in multiple positions. A FAM-labeled probe was designed over the target region harboring missense mutations p.R280K (MDA-MB-231) and p.R283C (HCC2218), and a HEX-labeled probe over missense mutations p.R273H (SW480) and p.V274F (DU-145). COLD-ddPCR experiments using serial dilutions of mutational abundances for cell lines with mutations under FAM or HEX probes, and their corresponding ratios, are plotted in Figure 5. FAM/HEX ratios are smaller or larger than the value of the wild type for mutations under the FAM probe or under the HEX probe, respectively. The lowest mutational abundance with a FAM/HEX ratio distinguishable from wild-type DNA is 0.6% to 1.25% p.V274F (t-test with two-tailed P < 0.01).
Figure 5.

Sensitivity of the coamplification at lower denaturation temperature–droplet digital PCR (COLD-ddPCR) FAM/HEX ratio method for the detection of mutations in TP53 exon 8. A and B: FAM/HEX ratio values for serial mutant dilutions (100%, 10%, 5%, 1.25%, 0.63%, and 0%) of missense mutation p.R280K (located under the FAM-labeled probe) and mutant dilutions (50%, 5%, 1.25%, 0.63%, and 0%) for p.V274F (located under the HEX-labeled probe). C and D: FAM/HEX ratio values for serial mutant dilutions (100%, 10%, 5%, 1.25%, 0.63%, and 0%) of missense mutation p.R273H (located under the HEX-labeled probe) and serial dilutions (100%, 10%, 5%, 1.25%, 0.63%, and 0%) of p.R283C (located under the FAM-labeled probe). The lowest distinguishable mutation abundances in these experiments were 0.6% to 1.25% (P < 0.01). On the graph representing Taqman probes and DNA templates, the circles labeled on the left side of each probe are FAM (grey) or HEX (black). The circles labeled on the right side of each probe are quenchers (BHQ-1). The suns represent the release of FAM or HEX from the probe during COLD-ddPCR. The error bars represent variations among three independent detections.
Two clinical samples known to contain low-level TP53 mutations on exon 8 were also analyzed via COLD-ddPCR. A colorectal cancer (CT20), containing a missense mutation p.R273H is depicted in Figure 6A. FAM/HEX ratio values for CT20 (1.54:1) were similar to those obtained for approximately 1% mutant abundance (1.58:1), in agreement with previous findings.23 And a plasma-cfDNA sample obtained from a radiation therapy patient containing a TP53 mutation (p.R283C) is depicted in Figure 6B. FAM/HEX values indicate that the estimated mutation abundance in that particular sample (as an average of six replicates) was about approximately 5%, in agreement with previous findings.23,30 Corresponding two-dimensional plots depicting signals from droplets following ddPCR are shown in Figure 6C. During COLD-ddPCR, cfDNA samples containing mutations are preferentially amplified, whereas the wild-type amplicon is inhibited. When the mutation overlaps the HEX probe, the FAM signal is increased (CT20), whereas the opposite occurs when the mutation overlaps the FAM probe (as in sample FC39-4).
Figure 6.

Detection of mutations in solid tumor and cell-free circulating DNA (cfDNA) samples by the coamplification at lower denaturation temperature–droplet digital PCR (COLD-ddPCR) FAM/HEX ratio method in TP53 exon 8. A: FAM/HEX ratio value for clinical sample CT20, harboring missense mutation p.R273H (overlapping the HEX-labeled probe). B: FAM/HEX ratio for a cfDNA sample, FC39-4 with the missense mutation p.R283C (overlapping the FAM-labeled probe). C: Two-dimensional plots for CT20 and FC39-4 samples obtained following COLD-ddPCR. The error bars represent variations among three independent detections. Ch1/2, channel 1/2.
Discussion
Sequencing remains the gold standard for identifying unknown DNA mutations, especially at the present time, when throughput and sensitivity issues for next-generation sequencing are gradually being addressed.31–33 Nevertheless, sequencing remains a relatively expensive option when a limited group of targets needs to be examined. Mutation scanning is an attractive alternative that balances well resources versus the information obtained.34 There are several methods for mutation scanning using PCR-based technology, including high-resolution melting,13,35 which provides the ability to rapidly pre-screen samples before sequencing.34 However, high-resolution melting cannot be performed on individual droplets, thus it has not been possible to combine high-resolution melting with ddPCR technology for mutation scanning. Because ddPCR is becoming widely used now that commercial platforms are available, a mutation scanning approach based on ddPCR is timely.
COLD-ddPCR is a novel combination of COLD-PCR and ddPCR technologies that enables mutation scanning over a target region as opposed to allele-specific ddPCR, which interrogates a single nucleotide. COLD-PCR cycling inhibits amplification of the wild-type amplicon. This inhibition reduces wild-type background signals and improves the limit for mutation detection.9
COLD-PCR suppresses wild-type DNA amplification for amplicons up to 200-bp long.21 As applied here, COLD-ddPCR interrogates the region covered by the two hydrolysis probes, ie, an approximately 50-bp region here. To interrogate additional sections of the amplicon, a second reaction addressing different regions could be performed. Alternatively, three or more hydrolysis probes labeled with different fluorophores could be used simultaneously, whose relative signal ratios would inform about the presence of a mutation. Although the present application used the simplest form of COLD-PCR (fast-COLD-PCR), COLD-ddPCR may potentially also be applicable with other forms of COLD-PCR, full-PCR16,36 or ICE-COLD-PCR,20,37 which enrich for all possible mutations within an amplicon.
A question pertains to how the COLD-ddPCR ratio method behaves in the simultaneous presence of two mutations on the same DNA molecule. The probability that there will be two neighboring mutations on the same DNA molecule (ie, not on different alleles or different tumor clones) is low, but not negligible. If the two mutations lie under the same TaqMan probe, then the ratio method will still be effective. If the mutations lie under different TaqMan probes, there could be a problem in their detection. However, during fast-COLD-ddPCR, the binding of the two TaqMan probes will likely be affected to a different extent from each of the two mutations, hence, still resulting in an altered signal ratio.
In summary we presented a process using a combination of COLD-PCR and digital PCR technologies that enables droplet-digital PCR to identify multiple mutations and potentially perform mutation scanning of short target regions by observing shifts to the ratio of fluorescent signals from two probes. This approach, enhanced ratio of signals for mutation scanning COLD-ddPCR, is able to scan for low-level mutations, with a limit of detection of approximately 0.2% to 0.6% mutant-to-wild-type ratio.
This method can be applied to clinical samples where multiple mutations along an amplicon can have clinical significance, such as TP53, codons 12 and G13 of KRAS (v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog), or mutations around V600E in BRAF (v-Raf murine sarcoma viral oncogene homolog B1) in cfDNA, urine, or other clinical samples where mutations can serve as biomarkers for prognostic, predictive, or therapy monitoring purposes.
Footnotes
Supported by National Cancer Institute grant numbers R21CA-155615 (G.M.M.), R21CA-175542 (G.M.M.), and R01CA135257 (P.A.J.).
The contents of this paper do not necessarily represent the official views of the National Cancer Institute or the NIH.
Disclosures: COLD-PCR is a technology owned by the Dana-Farber Cancer Institute and has been commercially licensed.
Supplemental Data
One-dimensional and two-dimensional plots for the Tc gradient of coamplification at lower denaturation temperature–droplet digital PCR (COLD-ddPCR) on wild-type (WT) of TP53 exon8. Ch1/2, channel 1/2.
One-dimensional and two-dimensional plots for the critical temperature (Tc) gradient of coamplification at lower denaturation temperature–droplet digital PCR (COLD-ddPCR) on 3.6% p.R273H mutation of TP53 exon8. Ch1/2, channel 1/2.
Two-dimensional plots of FAM versus HEX amplitude for wild-type, 100%, 6.25%, and 3.13% mutational abundance after conventional droplet digital PCR (ddPCR) (one replicate of each sample is shown). Ch1/2, channel 1/2.
Two-dimensional plots of FAM versus HEX amplitude for wild-type, 5%, 1.25%, and a 0.3% mutational abundance samples after coamplification at lower denaturation temperature–droplet digital PCR (COLD-ddPCR) (one replicate of each sample is shown). Ch1/2, channel 1/2.
A: Real-time PCR cycle threshold (CT) values for FAM and HEX (p.T790M region) after conventional PCR. B: The FAM/HEX ratio of fluorescence values at cycle 40 (same number of cycles used for conventional ddPCR experiments). Values have been normalized against the wild-type FAM/HEX ratio.
References
- 1.Bernard P.S., Wittwer C.T. Real-time PCR technology for cancer diagnostics. Clin Chem. 2002;48:1178–1185. [PubMed] [Google Scholar]
- 2.Tyagi S., Kramer F.R. Molecular beacons: probes that fluoresce upon hybridization. Nat Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 3.Li J., Wang F., Mamon H., Kulke M.H., Harris L., Maher E., Wang L., Makrigiorgos G.M. Antiprimer quenching-based real-time PCR and its application to the analysis of clinical cancer samples. Clin Chem. 2006;52:624–633. doi: 10.1373/clinchem.2005.063321. [DOI] [PubMed] [Google Scholar]
- 4.Amicarelli G., Shehi E., Makrigiorgos G.M., Adlerstein D. FLAG assay as a novel method for real-time signal generation during PCR: application to detection and genotyping of KRAS codon 12 mutations. Nucleic Acids Res. 2007;35:e131. doi: 10.1093/nar/gkm809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vogelstein B., Kinzler K.W. Digital PCR. Proc Natl Acad Sci U S A. 1999;96:9236–9241. doi: 10.1073/pnas.96.16.9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taly V., Pekin D., Benhaim L., Kotsopoulos S.K., Le Corre D., Li X., Atochin I., Link D.R., Griffiths A.D., Pallier K., Blons H., Bouche O., Landi B., Hutchison J.B., Laurent-Puig P. Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin Chem. 2013;59:1722–1731. doi: 10.1373/clinchem.2013.206359. [DOI] [PubMed] [Google Scholar]
- 7.Day E., Dear P.H., McCaughan F. Digital PCR strategies in the development and analysis of molecular biomarkers for personalized medicine. Methods. 2013;59:101–107. doi: 10.1016/j.ymeth.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 8.Hindson C.M., Chevillet J.R., Briggs H.A., Gallichotte E.N., Ruf I.K., Hindson B.J., Vessella R.L., Tewari M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat Methods. 2013;10:1003–1005. doi: 10.1038/nmeth.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huggett J.F., Foy C.A., Benes V., Emslie K., Garson J.A., Haynes R., Hellemans J., Kubista M., Mueller R.D., Nolan T., Pfaffl M.W., Shipley G.L., Vandesompele J., Wittwer C.T., Bustin S.A. The digital MIQE guidelines: Minimum Information for Publication of Quantitative Digital PCR Experiments. Clin Chem. 2013;59:892–902. doi: 10.1373/clinchem.2013.206375. [DOI] [PubMed] [Google Scholar]
- 10.Diehl F., Schmidt K., Choti M.A., Romans K., Goodman S., Li M., Thornton K., Agrawal N., Sokoll L., Szabo S.A., Kinzler K.W., Vogelstein B., Diaz L.A., Jr. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985–990. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hindson B.J., Ness K.D., Masquelier D.A., Belgrader P., Heredia N.J., Makarewicz A.J. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83:8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marsh D.J., Theodosopoulos G., Howell V., Richardson A.L., Benn D.E., Proos A.L., Eng C., Robinson B.G. Rapid mutation scanning of genes associated with familial cancer syndromes using denaturing high-performance liquid chromatography. Neoplasia. 2001;3:236–244. doi: 10.1038/sj.neo.7900154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wittwer C.T., Reed G.H., Gundry C.N., Vandersteen J.G., Pryor R.J. High-resolution genotyping by amplicon melting analysis using LCGreen. Clin Chem. 2003;49:853–860. doi: 10.1373/49.6.853. [DOI] [PubMed] [Google Scholar]
- 14.Li J., Berbeco R., Distel R.J., Janne P.A., Wang L., Makrigiorgos G.M. s-RT-MELT for rapid mutation scanning using enzymatic selection and real time DNA-melting: new potential for multiplex genetic analysis. Nucleic Acids Res. 2007;35:e84. doi: 10.1093/nar/gkm403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rice J.E., Reis A.H., Jr., Rice L.M., Carver-Brown R.K., Wangh L.J. Fluorescent signatures for variable DNA sequences. Nucleic Acids Res. 2012;40:e164. doi: 10.1093/nar/gks731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Milbury C.A., Li J., Makrigiorgos G.M. COLD-PCR-enhanced high-resolution melting enables rapid and selective identification of low-level unknown mutations. Clin Chem. 2009;55:2130–2143. doi: 10.1373/clinchem.2009.131029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carotenuto P., Roma C., Cozzolino S., Fenizia F., Rachiglio A.M., Tatangelo F., Iannaccone A., Baron L., Botti G., Normanno N. Detection of KRAS mutations in colorectal cancer with Fast COLD-PCR. Int J Oncol. 2012;40:378–384. doi: 10.3892/ijo.2011.1221. [DOI] [PubMed] [Google Scholar]
- 18.Pritchard C.C., Akagi L., Reddy P.L., Joseph L., Tait J.F. COLD-PCR enhanced melting curve analysis improves diagnostic accuracy for KRAS mutations in colorectal carcinoma. BMC Clin Pathol. 2010;10:6. doi: 10.1186/1472-6890-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kristensen L.S., Daugaard I.L., Christensen M., Hamilton-Dutoit S., Hager H., Hansen L.L. Increased sensitivity of KRAS mutation detection by high-resolution melting analysis of COLD-PCR products. Hum Mutat. 2010;31:1366–1373. doi: 10.1002/humu.21358. [DOI] [PubMed] [Google Scholar]
- 20.How Kit A., Mazaleyrat N., Daunay A., Nielsen H.M., Terris B., Tost J. Sensitive detection of KRAS mutations using enhanced-ice-COLD-PCR mutation enrichment and direct sequence identification. Hum Mutat. 2013;34:1568–1580. doi: 10.1002/humu.22427. [DOI] [PubMed] [Google Scholar]
- 21.Li J., Wang L., Mamon H., Kulke M.H., Berbeco R., Makrigiorgos G.M. Replacing PCR with COLD-PCR enriches variant DNA sequences and redefines the sensitivity of genetic testing. Nat Med. 2008;14:579–584. doi: 10.1038/nm1708. [DOI] [PubMed] [Google Scholar]
- 22.Li J., Milbury C.A., Li C., Makrigiorgos G.M. Two-round coamplification at lower denaturation temperature-PCR (COLD-PCR)-based Sanger sequencing identifies a novel spectrum of low-level mutations in lung adenocarcinoma. Hum Mutat. 2009;30:1583–1590. doi: 10.1002/humu.21112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guha M., Castellanos-Rizaldos E., Liu P., Mamon H., Makrigiorgos G.M. Differential strand separation at critical temperature: a minimally disruptive enrichment method for low-abundance unknown DNA mutations. Nucleic Acids Res. 2013;41:e50. doi: 10.1093/nar/gks1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oxnard G.R., Paweletz C.P., Kuang Y., Mach S.L., O'Connell A., Messineo M.M., Luke J.J., Butaney M., Kirschmeier P., Jackman D.M., Janne P.A. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res. 2014;20:1698–1705. doi: 10.1158/1078-0432.CCR-13-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guha M., Castellanos-Rizaldos E., Makrigiorgos G.M. DISSECT method using PNA-LNA clamp improves detection of T790m mutation. PLoS One. 2013;8:e67782. doi: 10.1371/journal.pone.0067782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greenman C., Stephens P., Smith R., Dalgliesh G.L., Hunter C., Bignell G. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galbiati S., Brisci A., Lalatta F., Seia M., Makrigiorgos G.M., Ferrari M., Cremonesi L. Full COLD-PCR protocol for noninvasive prenatal diagnosis of genetic diseases. Clin Chem. 2011;57:136–138. doi: 10.1373/clinchem.2010.155671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuang Y., Rogers A., Yeap B.Y., Wang L., Makrigiorgos M., Vetrand K., Thiede S., Distel R.J., Janne P.A. Noninvasive detection of EGFR T790M in gefitinib or erlotinib resistant non-small cell lung cancer. Clin Cancer Res. 2009;15:2630–2636. doi: 10.1158/1078-0432.CCR-08-2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Castellanos-Rizaldos E., Liu P., Milbury C.A., Guha M., Brisci A., Cremonesi L., Ferrari M., Mamon H., Makrigiorgos G.M. Temperature-tolerant COLD-PCR reduces temperature stringency and enables robust mutation enrichment. Clin Chem. 2012;58:1130–1138. doi: 10.1373/clinchem.2012.183095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Narayan A., Carriero N.J., Gettinger S.N., Kluytenaar J., Kozak K.R., Yock T.I., Muscato N.E., Ugarelli P., Decker R.H., Patel A.A. Ultrasensitive measurement of hotspot mutations in tumor DNA in blood using error-suppressed multiplexed deep sequencing. Cancer Res. 2012;72:3492–3498. doi: 10.1158/0008-5472.CAN-11-4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kinde I., Wu J., Papadopoulos N., Kinzler K.W., Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A. 2011;108:9530–9535. doi: 10.1073/pnas.1105422108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Milbury C.A., Correll M., Quackenbush J., Rubio R., Makrigiorgos G.M. COLD-PCR enrichment of rare cancer mutations prior to targeted amplicon resequencing. Clin Chem. 2012;58:580–589. doi: 10.1373/clinchem.2011.176198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Milbury C.A., Li J., Makrigiorgos G.M. PCR-based methods for the enrichment of minority alleles and mutations. Clin Chem. 2009;55:4632–4640. doi: 10.1373/clinchem.2008.113035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herrmann M.G., Durtschi J.D., Bromley L.K., Wittwer C.T., Voelkerding K.V. Amplicon DNA melting analysis for mutation scanning and genotyping: cross-platform comparison of instruments and dyes. Clin Chem. 2006;52:494–503. doi: 10.1373/clinchem.2005.063438. [DOI] [PubMed] [Google Scholar]
- 36.Delaney D., Diss T.C., Presneau N., Hing S., Berisha F., Idowu B.D., O'Donnell P., Skinner J.A., Tirabosco R., Flanagan A.M. GNAS1 mutations occur more commonly than previously thought in intramuscular myxoma. Mod Pathol. 2009;22:718–724. doi: 10.1038/modpathol.2009.32. [DOI] [PubMed] [Google Scholar]
- 37.Milbury C.A., Li J., Makrigiorgos G.M. Ice-COLD-PCR enables rapid amplification and robust enrichment for low-abundance unknown DNA mutations. Nucleic Acids Res. 2011;39:e2. doi: 10.1093/nar/gkq899. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
One-dimensional and two-dimensional plots for the Tc gradient of coamplification at lower denaturation temperature–droplet digital PCR (COLD-ddPCR) on wild-type (WT) of TP53 exon8. Ch1/2, channel 1/2.
One-dimensional and two-dimensional plots for the critical temperature (Tc) gradient of coamplification at lower denaturation temperature–droplet digital PCR (COLD-ddPCR) on 3.6% p.R273H mutation of TP53 exon8. Ch1/2, channel 1/2.
Two-dimensional plots of FAM versus HEX amplitude for wild-type, 100%, 6.25%, and 3.13% mutational abundance after conventional droplet digital PCR (ddPCR) (one replicate of each sample is shown). Ch1/2, channel 1/2.
Two-dimensional plots of FAM versus HEX amplitude for wild-type, 5%, 1.25%, and a 0.3% mutational abundance samples after coamplification at lower denaturation temperature–droplet digital PCR (COLD-ddPCR) (one replicate of each sample is shown). Ch1/2, channel 1/2.
A: Real-time PCR cycle threshold (CT) values for FAM and HEX (p.T790M region) after conventional PCR. B: The FAM/HEX ratio of fluorescence values at cycle 40 (same number of cycles used for conventional ddPCR experiments). Values have been normalized against the wild-type FAM/HEX ratio.