

In Chicago’s Field Museum of Natural History, there is an exhibit highlighting the evolutionary relationships between carnivorans. Glass cases lining two of the four walls of this room display specimens of feliforms, cats and their relatives. The other two walls are devoted to the caniforms, the dogs. In some future Museum of the Natural History of the Macromolecule, one might expect to encounter a similar room in the membrane protein wing. On one side one would find the channels, which form continuous pores in membranes allowing passive movement of ions and solutes down their concentration gradients, and on another side transporters that use the chemical energy of coupled reactions to move solutes against their concentration gradients. A close inspection of this exhibit would reveal that, unlike for the dogs and cats, there is no separation in the display cases between these two classes of membrane proteins. As one strolls past the ATP-binding cassette (ABC) transporters, a superfamily with thousands of members from archaea to mammals, one would arrive at the ABCC family. Members of this family range from ATP hydrolysis–driven transporters (like MRP1), to ion channel modulators (like SUR1 and SUR2), to an ABC protein that is a bona fide ion channel. This molecular dog–cat is known as the cystic fibrosis transmembrane conductance regulator, or CFTR. Because it is a true ion channel, passing tens of millions of Cl− ions per second down their concentration gradient, it is accessible to the tools of electrophysiology. By measuring Cl− currents, CFTR can be studied down to the single-molecule level and with high temporal precision. This is scarcely possible for other ABC proteins. In the April issue of the JGP, Chaves and Gadsby (2015) capitalize on CFTR’s accessibility to electrophysiological assays to probe changes in channel structure that are strongly coupled to the opening and closing of the pore. Their findings confirm an important feature of the CFTR gating cycle and increase our understanding of CFTR as well as those ABC proteins that can’t speak for themselves.
The natural history of CFTR
CFTR is perhaps best known for its dysfunction. It is the product of the gene mutated in patients with cystic fibrosis, a fatal disease primarily affecting the lungs. Reduced Cl− secretion as a result of poorly functional or absent CFTR in the airway epithelia produces abnormally thick mucus. Thus, patients experience frequent bacterial infections, inflammation, and ultimately destruction of lung tissue in addition to many other symptoms related to CFTR’s function in other organs and systems (Ashcroft, 2000).
Structurally, CFTR is a typical ABC exporter built from two six-helix transmembrane domains and two nucleotide-binding domains (NBDs; Fig. 1 A). These modules are connected in a single polypeptide with each transmembrane domain followed by an NBD, and the two halves separated by a regulatory insert after NBD1. This R domain is the site of extensive PKA phosphorylation, which increases the channel open probability (Csanády et al., 2005). CFTR’s transporter cousins have two gates, allowing for the alternating access to the substrate-binding core required to move a solute against its concentration gradient. In CFTR, one of these gates is absent or diminished such that opening of the other gate allows Cl− to flow freely down its electrochemical gradient.
Figure 1.
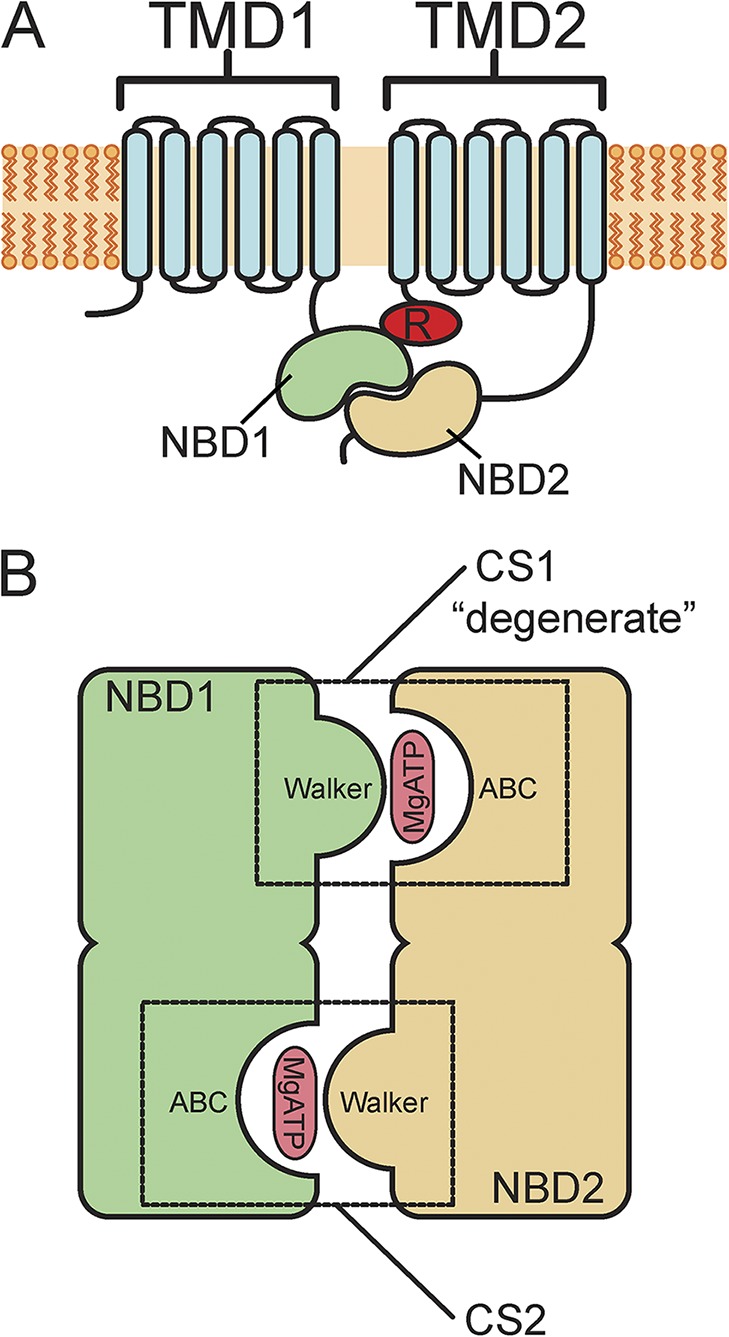
Schematic structure of CFTR and its NBDs. (A) Membrane topology of CFTR channels. CFTR consists of a single polypeptide chain with intracellular amino and carboxyl termini. There are two transmembrane domains (TMD1 and TMD2) and two NBDs (NBD1 and NBD2). There is also a regulatory domain (R) between NBD1 and TMD2. (B) Schematic of the NBDs (NBD1 and NBD2). NBD1 and NBD2 form a pseudo-twofold dimer with MgATP bound in between. The two composite nucleotide-binding sites (CS1 and CS2) are formed in part from the Walker A and B motifs (Walker) of one NBD and the ABC signature sequence (ABC) of the other. CS1, formed from the Walker groups of NBD1 and the ABC signature sequence of NBD2, is labeled “degenerate,” as it is catalytically incompetent.
The NBDs each consist of two subdomains (ter Beek et al., 2014). The amino-terminal portion is a RecA-like domain as found in other P-loop ATPases, e.g., the F1-ATPase. This domain contains the Walker A and B motifs, which are important for binding the phosphate groups of ATP and for ATP hydrolysis. A carboxyl-terminal helical domain contains the ABC signature sequence, which helps coordinate the γ-phosphate of ATP. In the available crystal structure of NBD1 of CFTR, the domain is monomeric. However, it is generally accepted that in the presence of nucleotide, NBD1 and NBD2 form a head-to-tail dimer with a pseudo-twofold symmetry (Fig. 1 B; Lewis et al., 2004). In the dimer, each NBD contributes to each ATP-binding site. Composite site 1 (CS1) is formed by the Walker A and B motifs of NBD1 and the signature sequence of NBD2. Composite site 2 (CS2) is formed in a similar fashion from the Walker motifs of NBD2 and the signature sequence of NBD1. In many ABC family proteins, both composite sites catalyze the hydrolysis of ATP in the presence of Mg+. However, in the ABCC family (and certain other transporters), CS1 contains modifications in certain key residues that prevent this “degenerate” site from hydrolyzing ATP. As a consequence, this site can stably bind nucleotides, probably through several channel gating cycles (Basso et al., 2003; Tsai et al., 2010).
Evidence that the NBDs form such dimers comes from several sources. Numerous crystal structures of isolated NBDs and NBDs as part of full ABC transporters have been solved. Most of these structures contain head-to-tail NBD dimers in the presence of bound nucleotide. Although there is no direct structural evidence that the NBDs of CFTR dimerize, there are many biochemical and electrophysiological studies that suggest that not only do such dimers exist, but dimer formation is coupled to channel opening. Cysteines inserted into several locations in the NBDs of CFTR can be cross-linked with bifunctional sulfhydryl-modifying reagents. The distance dependence and geometry of cross-linking is consistent with head-to-tail dimer formation, and the likelihood of cross-linking is increased with PKA phosphorylation, which favors channel opening (Mense et al., 2006). Furthermore, artificially creating dimerized NBDs by disulfide-bonding cysteines inserted into the NBD2 Walker A and the NBD1 signature sequence (both in CS2) opens CFTR channels in the absence of ATP. Finally, mutant cycle analysis demonstrated that residues along the dimer interface between NBD1 and NBD2 are thermodynamically coupled (and therefore likely to directly interact) when channels are open (Vergani et al., 2005).
Separation of the NBDs
The observation that dimerization is coupled to channel opening leads to an attractive mechanical model for CFTR gating based in part on the structure of the prokaryotic transporter Sav1866 (Dawson and Locher, 2006). In this structure, dimerization of the NBDs apparently tugs on the transmembrane domains so that they are in an outward-open conformation, which may be analogous to the open state of CFTR (Fig. 2). Channel closure would be expected to involve the separation of the NBDs. Is there any evidence that this is the case? At the single-channel level, binding of MgATP leads to a rate-limiting conformational change that initiates an open burst (Vergani et al., 2003). The burst is terminated by hydrolysis of ATP at CS2 and presumably release of ADP plus Pi from the binding site so that MgATP can be reloaded (Csanády et al., 2010). Release and reloading of nucleotides must involve some degree of separation of the NBDs, but open questions remain. Do the NBDs separate every time the channel closes? How big is the separation between them?
Figure 2.

Three possibilities for the arrangement of the NBDs. The structures of three full-length ABC transporters highlight possible arrangements of the NBDs. Sav1866 (Protein Data Bank [PDB] accession no. 2ONJ) is shown in a nucleotide-bound state. The transmembrane domains are in an outside-open conformation, and the NBDs are arranged in a tight head-to-tail dimer. TM287/288 is shown in the AMP-PNP–bound structure (PDB accession no. 3QF4), which is very similar in arrangement to the apo structure. The transmembrane domains are in an inside-open conformation, and the NBD dimer is partially open, but the NBDs have not completely separated. P-glycoprotein is shown without nucleotide (PDB accession no. 4M2T). Not only is the dimer interface broken, but the NBDs have separated entirely.
Available crystal structures offer several tantalizing proposals to answer the latter question (Fig. 2). Structures of P-glycoprotein and the MsbA transporter in the inward-facing conformation show that their NBDs are separated by very large distances (∼35 and >50 Å, respectively; Ward et al., 2007; Li et al., 2014). However, recent structures of TM287/288 bound to the poorly hydrolyzable nucleotide AMP-PNP and in the apo state tell a different story. In the absence of nucleotide, a cleft forms between the NBDs, allowing access to the nucleotide-binding sites (Hohl et al., 2014). Presumably, this separation between the NBDs pulls the transmembrane domain into the inward-facing conformation. A fascinating feature of these structures is that the NBDs are still in contact, burying around 832 Å2 of surface between them. Although the distance between the NBDs is more modest than in P-glycoprotein or MsbA, there is enough room for nucleotide exchange at either composite site. This conformation was confirmed using double electron–electron resonance (DEER), demonstrating that the interaction between the NBDs is meaningful, and not just a rare state captured in the crystal. Like CFTR, TM287/288 has both a functional and a degenerate nucleotide-binding site. AMP-PNP binding to the RecA-like domain of the degenerate site did not greatly alter the conformation of the protein relative to the apo state (Hohl et al., 2012). It is tempting to speculate that the AMP-PNP–bound structure may be analogous to a state of CFTR in which CS1 remains bound to MgATP, while the NBDs separate to allow nucleotide exchange at CS2.
How fast?
To address the how and when of NBD motions, Chaves and Gadsby (2015) studied the state-dependent accessibility of cysteines inserted into the NBD of an adequately cysteine-less channel. By probing the functional consequences of modifying these cysteines with various iterations of those old workhorses, the MTS reagents, they were able to probe the molecular structure of the NBDs in intact, functioning channels.
In the absence of ATP, when the channels were closed and the NBD sandwich was presumably open and waiting for nucleotides, the authors could readily modify cysteines inserted into the signature sequences of both composite sites. This confirmed the idea that the NBD dimer interface opened up in the absence of ATP. Positively charged MTSET, neutral MTSACE, and negatively charged MTSES were all able to severely impair channel opening after modifying the nucleotide-binding sites, indicating that the interference was primarily steric, presumably blocking the NBD dimer from comfortably reforming.
CFTR channels with the same binding-site cysteines were also exposed to MTS reagents in the presence of MgATP, with exciting results. In the presence of MgATP, reaction of cysteines in either composite site resulted in decay of the macroscopic current. At high concentrations of MTS reagents, the limiting rate of current decay was the same as the rate of channel closure subsequent to removal of MgATP. The macroscopic current decay after nucleotide removal is independent of the MgATP concentration and depends only on the rate of ATP hydrolysis at CS2. If the MTS reagents reacted very rapidly upon channel closure and the separation of the NBDs, i.e., before channels had a chance to reopen, it would be expected that current decay in the presence of MTS and MgATP would follow the same time course as channel closure in the absence of MgATP. Based on these results, it can be concluded that pore closure was tightly coupled to separation of the NBDs and that the NBDs must separate far enough to provide nearly unimpeded access to small modifying reagents and, by extension, ATP.
This similarity in the time constants of current decay from MTS modification and MgATP withdrawal could have been fortuitous. To provide further evidence that the modification rates were limited by separation of the NBD dimer upon channel closure, the authors took advantage of a mutation (K1250R) in the Walker A motif of CS2. This mutation impedes hydrolysis of MgATP, so that channel opening bursts are instead terminated by nucleotide dissociation, thus prolonging the length of the open burst. This can be seen in macroscopic current records as a lengthening of the time constant of current decay after removal of ATP (Carson et al., 1995; Gunderson and Kopito, 1995). In the K1250R background, modification of cysteines in the signature sequences of both composite sites occurred at a much slower rate. As for channels with the hydrolysis-competent K1250 background, the saturating modification rate by MTS reagents was very similar to the rate of current decay upon ATP withdrawal, i.e., channel closure. Therefore, it can be concluded that the rate of modification was limited by the rate of opening of the cleft between the NBDs when CFTR was in the closed state.
How much?
It is clear that the NBD dimer opens when CFTR is closed, but by how much? Obviously, it opens enough to let in MgATP, so probing with MTSET, MTACE, and MTSES offers little extra information regarding the size of the opening. To probe this further, Chaves and Gadsby (2015) also tested the accessibility of cysteines in the two composite sites to larger modifying reagents, including MTS-glucose and MTS-biotin (both comparable in size to ATP), and the larger and more conformationally challenged MTS-rhodamine. All of these compounds freely modified cysteines in the signature motifs of both composite sites when the channels were closed. There was a small decrease in the reaction rate of MTS-rhodamine at CS1 relative to MTSACE, and this may have been the result of somewhat restricted access of this compound to the target cysteine. Sadly, there is not an unlimited variety of MTS reagents that are practical and readily available to the electrophysiologist. The next larger MTS reagent that the authors used was the basketball that is avidin bound to MTS-biotin. At ∼45 Å wide and ∼58 Å across, it was excluded from the cleft between the NBDs, even when channels were closed. Therefore, cleft opening must be at least 8 Å at both composite sites (the narrowest dimension of MTS-rhodamine), but less than 45 Å.
The future
In terms of the models of NBD separation described above, this result eliminates the very large separation between NBDs observed in the MsbA structure, but does not differentiate between models for domain separation based on the P-glycoprotein structure or the TM287/288 structures in the presence or absence of bound nucleotide (Fig. 2). If a suitable variety of MTS reagents does not become available, perhaps future studies might use DEER to determine the distribution of distances between key points on the NBDs in the presence or absence of nucleotide. Fluorescence-based tools like FRET could also be used to monitor the separation between the NBDs and, like using MTS probes, FRET studies can be performed in intact functioning channels, such that a structural state can be directly correlated with a functional state.
The findings of Chaves and Gadsby (2015) offer structural insight into the normal workings of this critical ion channel. This sort of mechanistic insight may help in the design of compounds that can fix broken channels and alleviate certain forms of cystic fibrosis. Indeed, one such drug, ivacaftor, is used clinically to treat patients with certain mutations in the CFTR gene that lead to poorly gating channels. The drug potentiates CFTR in an apparently ATP-independent way, raising the intriguing possibility that it may artificially stabilize the NBD dimer interface (Eckford et al., 2012). Outside of the CFTR realm, the structural information that can be gleaned by interrogating an ABC protein at the single-molecule level and with fast time resolution can lead to new hypotheses and testable predictions for the ABC field at large. Regardless, this strange phenomenon of a transporter that has been made over as an ion channel has certainly earned CFTR a place in future museums.
Acknowledgments
The author would like to acknowledge Professor Frances Ashcroft and Dr. Natascia Vedovato for frequent discussions regarding NBDs.
The author is supported by a grant from the European Union (ERC Advanced grant 322620 to Professor Ashcroft).
Elizabeth M. Adler served as editor.
References
- Ashcroft F.M. 2000. Ion Channels and Disease: Channelopathies. Academic Press, San Diego: 481 pp. [Google Scholar]
- Basso C., Vergani P., Nairn A.C., and Gadsby D.C.. 2003. Prolonged nonhydrolytic interaction of nucleotide with CFTR’s NH2-terminal nucleotide binding domain and its role in channel gating. J. Gen. Physiol. 122:333–348. 10.1085/jgp.200308798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson M.R., Travis S.M., and Welsh M.J.. 1995. The two nucleotide-binding domains of cystic fibrosis transmembrane conductance regulator (CFTR) have distinct functions in controlling channel activity. J. Biol. Chem. 270:1711–1717. 10.1074/jbc.270.4.1711 [DOI] [PubMed] [Google Scholar]
- Chaves L.A.P., and Gadsby D.C.. 2015. Cysteine accessibility probes timing and extent of NBD separation along the dimer interface in gating CFTR channels. J. Gen. Physiol. 145:261–283. 10.1085/jgp.201411347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csanády L., Chan K.W., Nairn A.C., and Gadsby D.C.. 2005. Functional roles of nonconserved structural segments in CFTR’s NH2-terminal nucleotide binding domain. J. Gen. Physiol. 125:43–55. 10.1085/jgp.200409174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csanády L., Vergani P., and Gadsby D.C.. 2010. Strict coupling between CFTR’s catalytic cycle and gating of its Cl− ion pore revealed by distributions of open channel burst durations. Proc. Natl. Acad. Sci. USA. 107:1241–1246. 10.1073/pnas.0911061107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson R.J., and Locher K.P.. 2006. Structure of a bacterial multidrug ABC transporter. Nature. 443:180–185. 10.1038/nature05155 [DOI] [PubMed] [Google Scholar]
- Eckford P.D., Li C., Ramjeesingh M., and Bear C.E.. 2012. Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J. Biol. Chem. 287:36639–36649. 10.1074/jbc.M112.393637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunderson K.L., and Kopito R.R.. 1995. Conformational states of CFTR associated with channel gating: The role ATP binding and hydrolysis. Cell. 82:231–239. 10.1016/0092-8674(95)90310-0 [DOI] [PubMed] [Google Scholar]
- Hohl M., Briand C., Grütter M.G., and Seeger M.A.. 2012. Crystal structure of a heterodimeric ABC transporter in its inward-facing conformation. Nat. Struct. Mol. Biol. 19:395–402. 10.1038/nsmb.2267 [DOI] [PubMed] [Google Scholar]
- Hohl M., Hürlimann L.M., Böhm S., Schöppe J., Grütter M.G., Bordignon E., and Seeger M.A.. 2014. Structural basis for allosteric cross-talk between the asymmetric nucleotide binding sites of a heterodimeric ABC exporter. Proc. Natl. Acad. Sci. USA. 111:11025–11030. 10.1073/pnas.1400485111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis H.A., Buchanan S.G., Burley S.K., Conners K., Dickey M., Dorwart M., Fowler R., Gao X., Guggino W.B., Hendrickson W.A., et al. 2004. Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 23:282–293. 10.1038/sj.emboj.7600040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Jaimes K.F., and Aller S.G.. 2014. Refined structures of mouse P-glycoprotein. Protein Sci. 23:34–46. 10.1002/pro.2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mense M., Vergani P., White D.M., Altberg G., Nairn A.C., and Gadsby D.C.. 2006. In vivo phosphorylation of CFTR promotes formation of a nucleotide-binding domain heterodimer. EMBO J. 25:4728–4739. 10.1038/sj.emboj.7601373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Beek J., Guskov A., and Slotboom D.J.. 2014. Structural diversity of ABC transporters. J. Gen. Physiol. 143:419–435. 10.1085/jgp.201411164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai M.F., Li M., and Hwang T.C.. 2010. Stable ATP binding mediated by a partial NBD dimer of the CFTR chloride channel. J. Gen. Physiol. 135:399–414. 10.1085/jgp.201010399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani P., Nairn A.C., and Gadsby D.C.. 2003. On the mechanism of MgATP-dependent gating of CFTR Cl− channels. J. Gen. Physiol. 121:17–36. 10.1085/jgp.20028673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani P., Lockless S.W., Nairn A.C., and Gadsby D.C.. 2005. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature. 433:876–880. 10.1038/nature03313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward A., Reyes C.L., Yu J., Roth C.B., and Chang G.. 2007. Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc. Natl. Acad. Sci. USA. 104:19005–19010. 10.1073/pnas.0709388104 [DOI] [PMC free article] [PubMed] [Google Scholar]