

Abstract
Neurological disorders comprise a variety of complex diseases in the central nervous system, which can be roughly classified as neurodegenerative diseases and psychiatric disorders. The basic and translational research of neurological disorders has been hindered by the difficulty in accessing the pathological center (i.e., the brain) in live patients. The rapid advancement of sequencing and array technologies has made it possible to investigate the disease mechanism and biomarkers from a systems perspective. In this review, recent progresses in the discovery of novel risk genes, treatment targets and peripheral biomarkers employing genomic technologies will be discussed. Our major focus will be on two of the most heavily investigated neurological disorders, namely Alzheimer’s disease and autism spectrum disorder.
Keywords: Brain transcriptome, Genome-wide association study, Whole exome sequencing, Epigenome profiling, Biomarker
Introduction
Neurological disorders include a wide spectrum of diseases in the central nervous system (CNS). Up till now, hundreds of neurological disorders have been classified, with symptoms varying from cognitive dysfunction to manic behavior or depression [1]. Due to the complex nature of this group of diseases, it is difficult to identify the mechanisms using conventional methodologies, where only small pathways around specific target genes are investigated. The advent of systems biology approaches has made it possible to study these complex problems from the whole-genome perspective. In the recent years, genomic technologies have been increasingly applied to the investigation of neurological disorders [2]. Exciting discoveries have thus emerged including novel risk genes, peripheral biomarkers and treatment targets. For the convenience of the limited space, we will mainly focus on two of the most studied neurological disorders, Alzheimer’s disease (AD) and autism spectrum disorder (ASD).
AD is a major form of neurodegenerative diseases [3]. AD starts from memory loss and cognitive deficit in the early stage and gradually evolves into severe dementia in the late stage. The pathological hallmarks of AD include extracellular deposit of amyloid plaques and intra-neuronal neurofibrillary tangles (NFT). Although the disease-causing mutations have been identified for the familial early-onset AD (FEOAD), the genetic landscape has been perplexing for the late-onset AD (LOAD) that constitutes ∼95% of all AD patients [4]. The prevailing hypothesis for the disease mechanism of AD has been primarily based on the studies of FEOAD, which advocates the central role of amyloid-β (Aβ) in the chain of events leading to neuronal death and cognitive and behavioral symptoms. However, Aβ-based interventions have not been successful in the clinical trials so far [5]. Due to the lack of effective treatment for curing or slowing down AD, it becomes imperative to search for novel risk genes and drug targets, as well as biomarkers for early diagnosis.
Autism spectrum disorder is a neurodevelopmental disorder characterized by social and communication deficit as well as stereotyped and repetitive behaviors [6]. According to a recent survey, 1 in 68 US children has ASD. In contrast to AD, the disease onset for ASD starts from 3 years of age to early childhood. The gender ratio is approximately 4:1 disfavoring boys. Like other psychiatric disorders, there are no clear pathological hallmarks for ASD [1]. It is believed that brain wiring is altered in ASD children, although the exact interplay between gene and environment has not been clarified. In terms of the genetic factors, some types of ASD may be caused by rare mutations, while others may be due to the combination of common variations [7]. The genetic alterations in ASD are also more complex than those in AD, which include copy number variation, insertion, deletion and single nucleotide polymorphism (SNP). In addition to the genetic and environmental factors, prenatal and perinatal factors may also contribute to the development of ASD.
Genomic studies of neurological disorders involve the investigation of the genome, transcriptome and epigenome (Figure 1). There are two types of technologies available for genomic studies, including sequencing and various array platforms. For the investigation of genomic variation, the samples generally come from peripheral blood, although saliva has also been used. For the investigation of transcriptome, brain tissue is the most studied since it is more relevant to the disease mechanism. The peripheral blood and cerebrospinal fluid (CSF) have also been investigated, mostly for the discovery of novel biomarkers. These three tissues have also been utilized in the investigation of epigenomic alteration. In addition, skin fibroblast has been increasingly used in induced pluripotent stem cell (iPSC) technologies. Recent advances in these fields will be summarized in the following sections.
Figure 1.
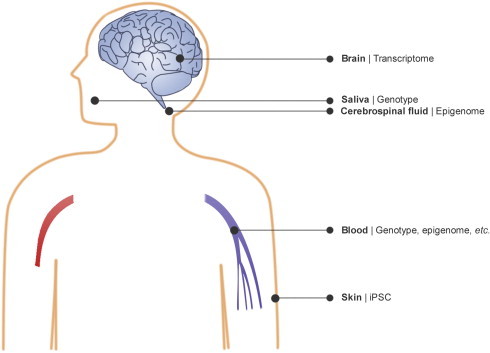
Application of genomic technologies to the investigation of neurological disorders High quality brain tissues can be used in transcriptome study, in addition to the examination of pathological hallmarks. Samples for genotyping and whole exome/genome sequencing generally come from peripheral blood or saliva. Transcriptome and epigenome profiling can also be performed on peripheral blood and cerebrospinal fluid in addition to other biomarker studies. Skin fibroblasts can be reprogrammed or trans-differentiated into neurons for comprehensive analysis of the dysfunctional network in patients.
Brain transcriptome studies
Since the disease mechanism for most of the neurological disorders is still under debate, it is necessary to conduct investigation from a systems perspective. In brain transcriptome studies, information regarding gene expression at the whole genome level can be extracted, and the dysregulation of gene expression in a disease condition can be revealed by comparing the gene expression with that from the matched healthy controls. Microarray platforms have been the main workhorse for brain transcriptome studies due to the mature technology and low cost. Sequencing technology has been increasingly used since 2008, but generally limited to small sample size due to the high cost. Although it is extremely challenging to collect relevant brain tissues for transcriptome studies considering the stringent requirement of short post mortem delay, dozens of brain transcriptome studies have already been performed and much of the original data have been released to the public [8,9].
Aberrations in the control of gene expression might contribute to the initiation and progression of AD [10] and other neurological disorders. In a recent work, Zhang et al. conducted a brain transcriptome study employing hundreds of brain samples covering three distinct brain regions, namely prefrontal cortex, primary visual cortex and cerebellum [11]. Based on the comprehensive analysis of the gene co-expression network, functional modules were identified, especially those with gain-of-function or loss-of-function in AD, compared to the control group. TYROBP was identified as one of the most important causal factors. Most importantly, this study provided to the public a rare resource of genome-scale measurement including both genome and transcriptome on a large cohort of AD patients and elderly controls.
In addition, several small-scale studies on AD have been conducted in recent years. A transcriptome study of astrocytes in the aging brain provided information regarding the correlation between APOE genotype and AD pathology [12]. In another study, distinct regions of AD brain were examined by RNA-seq, and the alternative splicing and promoter usage of APOE was found to be correlated with AD progression [10]. A network model integrating the AD proteome and transcriptome revealed key proteins, protein interactions, and the regulation between genes and their protein products in the disease pathology [13].
Compared to AD transcriptome studies, much fewer ASD transcriptome studies have been conducted due to inaccessibility of brain samples from young ASD patients. In a study by Geschwind and coworkers, the dysregulation of gene expression in the autistic brains was investigated [14]. The difference in gene expression between the frontal cortex and temporal cortex in normal brains diminished in autistic children. Critical modules were revealed by gene co-expression analysis, including a neuronal module and an immune and glia module. Based on comparison with risk genes from genome-wide association studies (GWAS), the neuronal module was highly enriched in risk genes, whereas the immune and glia module was not. These data suggest that genes in the neuronal module are likely causal factors while those in the immune and glia module merely reflect the secondary response during disease development.
GWAS studies
Brain transcriptome is generally considered as a consequence of the disease progression, because most of the brain samples come from the late stage of the disease. Discovery of the causal genetic factors requires the examination of genetic variations in patients. Since the human genome became available a decade ago, genome-wide association study gradually became the predominant method for the discovery of risk genes/variations, as compared to the traditional target gene/loci approach. Several SNP array platforms have been developed and constantly improved through the past decade. The general analytical procedure consists of genotyping, quality control, imputation, association test and meta-analysis.
In addition to APOE, recent large-scale GWAS studies on LOAD have identified nine other genes/loci, including CR1, BIN1, CLU, PICALM, MS4A4/MS4A6E, CD2AP, CD33, EPHA1 and ABCA7 [15]. For example, in the work by Lambert and his colleagues [16], two loci showed evidence of association with AD in two independent sample pools [16]: one within CLU (also called APOJ) on chromosome 8 (rs11136000, P = 7.5 × 10−9) and the other within CR1 on chromosome 1 (rs6656401, P = 3.7 × 10−9). In another work, Harold et al. undertook a two-stage GWAS study involving over 16,000 individuals including AD patients and controls [17]. In the first stage, two loci with significant P value included CLU (rs11136000, P = 1.4 × 10−9) and PICALM (rs3851179, P = 1.9 × 10−8). These associations were recapitulated in stage 2, producing compelling evidence for association with AD in the combined dataset. A three-stage design was adopted in a recent work [18] and several novel risk loci were discovered. These include MS4A4A (rs4938933; P = 8.2 × 10−12), CD2AP (rs9349407; P = 8.6 × 10−9), EPHA1 (rs11767557; P = 6.0 × 10−10) and CD33 (rs3865444; P = 1.6 × 10−9). The same group also replicated previous associations at CR1, CLU, BIN1 and PICALM. Similarly, Seshadri et al. also used a three-stage design [19] and found two loci with significant association, including rs744373 near BIN1 and rs597668 near EXOC3L2/BLOC1S3/MARK4. These two loci, together with the previously identified loci in CLU and PICALM, were further confirmed in the Spanish sample.
In the most recent large-scale GWAS study of AD, 74,046 individuals were included in the meta-analysis where a two-stage design was adopted [20]. In addition to APOE, 19 loci were found to be significantly associated with AD, among which 11 were newly discovered in this study. The new risk loci included INPP5D (rs35349669), MEF2C (rs190982), NME8 (rs2718058), ZCWPW1 (rs1476679), CELF1 (rs10838725), FERMT2 (rs17125944), CASS4 (rs7274581), SORL1 (rs11218343), PTK2B (rs28834970), HLA-DRB5/DRB1 (rs9271192) and SLC24A4/RIN3 (rs10498633). This work clearly demonstrated the power of large sample size in GWAS studies.
Due to the high heritability of ASD, GWAS studies of ASD usually involve families rather than case-control design adopted in most GWAS studies of LOAD. In an earlier study, a SNP between CDH10 and CDH9 (rs4307059) was found to be significantly associated with ASD in both discovery and replication stages [21]. In another study, a significant association of SEMA5A with ASD was found, which was further supported by the reduced expression of SEMA5A in autistic brains [22]. In a later study, MACROD2 (rs4141463) was found to be significant in the discovery stage and was then reasonably replicated [23]. In a study of rare copy number variants (CNVs), it was found that the ASD group carried a higher burden of rare CNVs compared to the control group [24]. The implicated genes included SHANK2, SYNGAP1, DLGAP2 and DDX53/PTCHD1.
Whole exome/genome sequencing studies
The heritability of LOAD has been estimated to be 58%–79% [25]. The classical SNP association studies have identified dozens of genetic variants associated with AD [20]. But the genetic alterations of AD found by GWAS studies cannot fully explain the estimated heritability [26]. The potential sources of the “missing heritability” include large number of “small effect” variants, rare genetic variants, structural genetic variations and possible gene–gene interactions. To explore the missing heritability, rare variants associated with AD have been extensively examined with whole genome/exome sequencing technology.
TREM2 is a gene located on chromosome 6, which encodes a membrane protein that plays a role in immune response. TREM2 on microglial is also important for the clearance of neuronal debris produced by the impaired CNS [27,28]. Jonsson et al. reported that a missense mutation (rs75932628, R47H) within TREM2 is strongly associated with AD [29]. Similar results were obtained in another study with a smaller discovery dataset [30]. In this study, the functional relevance of TREM2 was demonstrated by the higher mRNA expression level in an AD mouse model compared to the wild type control. Later, the prevalence of R47H mutation was examined in a Spanish population comprising 180 EOAD, 324 LOAD patients and 550 controls [31] and R47H mutation was found in 1.4% of the AD patients but not in the controls. Significant risk conferred by rs75932628 was also revealed in another EOAD related study examining 726 patients and 783 controls [32].
Besides association studies based on large sample size of case-control design, family-based studies have also been carried out for AD. Guerreiro et al. reported a mutation of NOTCH3 (R1231C) in a Turkish family [33], whereas mutations in SORL1 were identified as associated with FEOAD [34]. In this study, 7 samples in a total of 29 FEOAD cases carried SORL1 mutations, while none of 1500 controls carried those mutations. SORL1 gene is located on chromosome 11, and the protein product is involved in the Aβ production. It is of note that common SNPs of SORL1 were also reported to be associated with AD [35]. PLD3, which encodes a membrane protein catalyzing the hydrolysis of membrane phospholipids, was also reported to be associated with AD [36]. It was found that PLD3 mutation V232M was segregated with disease status in two independent families. Furthermore, overexpression of PLD3 led to a significant decrease in intra/extra-cellular APP related species, whereas knockdown of PLD3 gene led to an increase in extracellular Aβ level.
Besides novel genes reported to be associated with AD, novel mutations in classical AD genes, such as APP and MAPT have also been reported. A missense mutation in the APP gene, which leads to an alanine to threonine substitution at position 673 (A673T), was significantly associated with AD (0.62% in control vs. 0.13% in AD, odds ratio (OR) = 5.29, P = 4.78 × 10−7) in a study with 1795 samples [37]. The association was further confirmed with 3661 additional samples in the same study. This is one of the very few rare mutations found to be protective against AD. In another work, a rare mutation residing in MAPT (A152T) was found to increase the risk of AD (OR = 2.3, P = 0.004) [38]. However, further replication would be needed to validate this association due to the less significant P value.
In the first trio-based (parents plus child) whole exome sequencing (WES) study for ASD, 21 de novo mutations were found [39]. Among them, four mutations were predicted to be causative, including mutations in FOXP1, GRIN2B, SCN1A and LAMC3. In a large-scale WES study of ASD with 928 individuals [40], 279 de novo coding mutations were discovered, with the strongest evidence showing SCN2A to be causative. A WES study of 16 families [41] identified candidate recessive mutations in four genes including UBE3B, CLTCL1, NCKAP5L and ZNF18. Genes disrupting de novo mutations were twice as frequent in probands compared to unaffected siblings and many of these genes were targets of FMRP gene, as revealed by a WES study of 343 families [42]. Moreover, rare complete gene knockout was found to contribute to a small portion of ASD in a large WES case-control study [43], whereas partial loss-of-function in several genes was found to contribute to ASD in another WES study that compared consanguineous to non-consanguineous families [44].
Compared to WES, whole genome sequencing (WGS) can provide variations in the non-coding regions of the genome, which may also have major contribution to the disease development. In the first large-scale WGS study of ASD, it was found that risk genes tended to reside in hypermutability regions [45]. Later on, seven candidate genes were found with the recessive model and 59 candidate variants were found with the model free approach in a WGS study of a large pedigree with two probands [46]. In another WGS study with 32 families [47], deleterious de novo mutations were found in six families and X-linked autosomal inherited alterations were found in ten families. With the richer information from WGS and fast dropping sequencing cost, WGS may soon replace WES as the main deep-sequencing technology for genetic studies.
Epigenomic studies
Neurological conditions are not only reflected on genomic mutations and transcriptomic dysregulations, but also on the change of epigenome. Among the various types of epigenomic modifications, DNA methylation and histone modifications have attracted high attention and been the most widely studied. Additionally, the expression level of microRNA (miRNA) adds another layer of complexity in the epigenomic landscape. During human brain development, widespread methylome reconfiguration occurs and the highly conserved non-CG methylation accumulates in the neuronal genome [48]. Dynamic alterations in the epigenome play a critical role in regulating cellular phenotype during differentiation, and distinct tissue-specific patterns of DNA methylation have been identified across multiple human brain regions [49].
Several studies have been conducted on the change of DNA methylation and other types of modifications in AD. In a recent investigation of four types of cytosine modifications [50], it was found that the abundance of 5hmC is lower in the entorhinal cortex and cerebellum of AD patients compared to healthy elderly controls. In an earlier study on hippocampus [51], both 5mC and 5hmC were found to be lower in AD patients. However, in another study focusing on 5mC and 5hmC, a global hypermethylation was observed in the middle frontal gyrus and the middle temporal gyrus [52]. Nevertheless, due to the small sample size in most of the epigenomic studies, some of the results should be interpreted with caution.
The dysregulation of miRNA has been investigated in AD brains, blood and CSF. A pioneering miRNA study on AD was done by Cogswell and his colleagues [53]. They examined miRNA expression in both brain and CSF and showed that expression of let-7i was significantly altered in both comparing AD patients with controls. Further pathway enrichment analysis suggested that deregulated miRNAs might be related to amyloid processing, neurogenesis, insulin resistance and immunity. miRNA profiling has also been performed in the hippocampus and prefrontal cortex and miR-132a-3p was down regulated in both brain regions [54]. As for the function of critical miRNAs in the AD brains, it has been reported that miR-124 may have a role in regulating the APP alternative splicing [55], and miR-15a was reported to be associated with neuritic plaque score [56].
Other works on miRNAs are focused on the discovery of biomarkers. In Villa’s work, an inverse relationship between SP1 mRNA and miR-29b levels in PBMCs was observed [57]. This was of particular interest because SP1 is a dysregulated transcription factor in AD brains [58]. In a recent study on serum, six miRNAs were identified as peripheral biomarkers for AD [59], whereas a 7-miRNA signature displayed >95% accuracy in discriminating AD from controls in another peripheral biomarker study [60]. In yet another study, a panel of 12 miRNAs was identified from blood samples, which showed 93% accuracy in differentiating AD from controls and 74%−78% in discriminating AD from other neurological diseases, such as Parkinson’s disease [61]. In a study on CSF, the level of hsa-miR-27a-3p was found to be lower in AD patients [62].
Compared to AD, few epigenomic studies have been conducted on ASD. In a study of DNA methylation in autistic brains, four differentially methylated regions were discovered, with three of them independently validated [63]. In addition, several differentially methylated regions were identified in a recent study on monozygotic twins discordant for ASD [64], among which some were strongly correlated with quantitative behavioral traits. Different DNA methylation patterns were also observed in monozygotic twins with Rett syndrome (a subtype of ASD) when no distinct difference could be found in SNP mutations and number of indels and CNVs [65].
iPSC technology in neurological disorders
The iPSC technology is an attractive approach to model a live neuron in neurological disorders. iPSCs provide researchers with a source of patient-specific stem cells and potential applications including cellular modeling, drug discovery and cell-based therapy [66]. Direct reprogramming of fibroblasts to neurons could be a promising approach in neurological disease modeling [67].
iPSC as an experimental research tool is now widely used in the neurological disorders including AD [68–72]. Fibroblasts reprogrammed to neuron cells could offer clues to the mechanisms related to Aβ production and processing [68]. By increasing gene regulation of GSK-3β, phosphorylated tau protein was found in the neurons derived from familial and sporadic AD (fAD and sAD) patients. Levels of early endosome and synapse were also found to be different in patient derived neurons. The development of AD pathology could also be studied in iPSC using the Down syndrome induced neurons with much shorter duration of pathology development compared to in vivo [70]. Despite all the promising progress, we shall note that several pitfalls exist in the application of iPSC technology to neurological disease research. For example, neurons might not fully mature and different phenotypes may exist between in vivo and cultured neurons [73].
Database resources for AD and ASD
To facilitate the discovery of novel risk genes, the research communities of AD and ASD have formulated policies for genetic data sharing. As a pioneer, the National Institute on Aging (NIA) has set up the NIA Genetics of Alzheimer’s Disease Data Storage site (NIAGADS), a national genetic data repository which provides access to genotypic data for the study of the genetics of LOAD upon approval by the data access committee (Table 1). Currently, NIAGADS has collected about 20 AD GWAS datasets.
Table 1.
Genomic data resources for Alzheimer’s disease and autism spectrum disorder
| Abbreviation | Full name | Description | Web link |
|---|---|---|---|
| NIAGADS | National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site | A national genetics data repository (USA) that facilitates access of genotypic data to qualified investigators for the study of the genetics of late-onset Alzheimer’s disease | https://www.niagads.org/ |
| ADSP | Alzheimer’s Disease Sequencing Project | Aiming to develop and execute a large-scale sequencing project to analyze the genomes of a large number of well-characterized individuals in order to identify a broad range of Alzheimer’s disease risk and protective gene variants, with the ultimate goal of facilitating the identification of new pathways for therapeutic approaches and prevention | https://www.niagads.org/adsp/ |
| ADNI | Alzheimer’s Disease Neuroimaging Initiative | Working to define the progression of Alzheimer’s disease and to collect, validate and utilize data such as MRI and PET images, genetics, cognitive tests, CSF and blood biomarkers as predictors for the disease | http://adni.loni.usc.edu/ |
| AGD | Autism Genetic Database | A comprehensive database for autism susceptibility genes and CNVs, integrated with known noncoding RNAs and fragile sites | http://wren.bcf.ku.edu/ |
| AutDB | Autism Database | An integrated resource for the autism research community, which is built on information extracted from the studies on molecular genetics and biology of autism spectrum disorder | http://autism.mindspec.org/autdb/ |
| NDAR | National Database for Autism Research | An NIH (USA)-funded research data repository that aims to accelerate progress in autism spectrum disorder research through data sharing, data harmonization and reporting of research results | http://ndar.nih.gov/ |
Note: MRI, magnetic resonance imaging; PET, positron emission tomography; CSF, cerebrospinal fluid; CNV, copy number variation.
Compared to the SNP-array technology, next-generation sequencing technology can provide a deeper insight into the missing heritability of AD. Currently, there are two ongoing sequencing projects for AD: the Alzheimer’s Disease Neuroimaging Initiative (ADNI) and Alzheimer’s Disease Sequencing Project (ADSP) [74]. The ADNI project has made publicly available the whole genome sequencing data of about 818 participants including AD, mild cognitive impairment (MCI) and control. The ADSP project plans to complete whole genome sequencing of about 111 AD families and whole exome sequencing of about additional 11,000 subjects, where the first batch of data has been released to the public.
Autism Genetic Database (AGD) is a comprehensive database for autism susceptibility [75]. It currently contains the full list of autism susceptibility genes as well as CNVs related to autism. Additionally, all non-coding RNA molecules, such as small nucleolar RNA (snoRNA), miRNA and Piwi-interacting RNA (piRNA), and chemically induced fragile sites are archived as well. The information in this database can be accessed through a human genome browser focusing specifically on the chromosomal features associated with autism or a tabular format broken down by chromosome.
Autism Database (AutDB) supports an integrated resource for the autism research community [76]. The main focus of this database is to provide an up to date, annotated list of ASD candidate genes in the form of reference datasets for interrogating molecular mechanisms underlying the disorder. The information in AutDB is extracted from the published scientific literature on molecular genetics and biology of ASD and organized to optimize its use through experts. The database provides users four modules for free, including information on human genes, animal models, protein interactions and copy number variants.
The National Database for Autism Research (NDAR) is an NIH-funded research data repository that aims to accelerate progress in ASD research through data sharing, and reporting of research results. NDAR also serves as a scientific community platform and portal to multiple other research repositories, allowing for integration and secondary analysis of data. Comparing with the two above-described databases, this database is mainly aimed at supporting an open access platform that allows researchers to obtain and integrate the public data, so as to facilitate novel discoveries for the prevention and cure of the disease.
Concluding remarks
The human brain remains to be one of the biggest mysteries in biological sciences. Preventing and curing neurological disorders require better understanding of the brain. The ongoing big brain initiatives in Europe, USA and China will take this grand challenge, where the main focus is future computing, brain wiring and brain disorders, respectively. The genomic approaches described in this review together with the state-of-the-art brain imaging technologies will be the main workhorses in the foreseeable future for the investigation of neurological disorders. Mapping out the 100 billion neurons and 100 trillion connections in the human brain is certainly not a trivial task. Efficient integration with genomic technologies will undoubtedly lead to breakthroughs. The exciting new progress in this field will be closely followed. In addition, stem cell technology may be a viable option in the future for treating neurodegenerative disorders especially for patients at the middle or late stage of the disease. Nevertheless, much of the focus in this field is to detect the disease early and intervene before massive neurodegeneration occurs. As for the childhood disorders such as autism, non-invasive diagnosis of mutations and genome correction are the technologies to watch.
Competing interests
The authors declare that there are no conflicts of interest.
Acknowledgements
This work was supported by the grant from the National Basic Research Program of China (973 Program, Grant No. 2014CB964901) awarded to HL from the Ministry of Science and Technology of China.
Footnotes
Peer review under responsibility of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China.
References
- 1.Krystal J.H., State M.W. Psychiatric disorders: diagnosis to therapy. Cell. 2014;157:201–214. doi: 10.1016/j.cell.2014.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCarroll S.A., Feng G., Hyman S.E. Genome-scale neurogenetics: methodology and meaning. Nat Neurosci. 2014;17:756–763. doi: 10.1038/nn.3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lei H. Amyloid and Alzheimer’s disease. Protein Cell. 2010;1:312–314. doi: 10.1007/s13238-010-0046-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guerreiro R., Brás J., Hardy J. SnapShot: genetics of Alzheimer’s disease. Cell. 2013;155:968–968.e1. doi: 10.1016/j.cell.2013.10.037. [DOI] [PubMed] [Google Scholar]
- 5.Callaway E. Alzheimer’s drugs take a new tack. Nature. 2012;489:13–14. doi: 10.1038/489013a. [DOI] [PubMed] [Google Scholar]
- 6.Schaafsma S.M., Pfaff D.W. Etiologies underlying sex differences in autism spectrum disorders. Front Neuroendocrinol. 2014;35:255–271. doi: 10.1016/j.yfrne.2014.03.006. [DOI] [PubMed] [Google Scholar]
- 7.Ronemus M., Iossifov I., Levy D., Wigler M. The role of de novo mutations in the genetics of autism spectrum disorders. Nat Rev Genet. 2014;15:133–141. doi: 10.1038/nrg3585. [DOI] [PubMed] [Google Scholar]
- 8.Sun J., Feng X., Liang D., Duan Y., Lei H. Down-regulation of energy metabolism in Alzheimer’s disease is a protective response of neurons to the microenvironment. J Alzheimers Dis. 2012;28:389–402. doi: 10.3233/JAD-2011-111313. [DOI] [PubMed] [Google Scholar]
- 9.Feng X., Bai Z., Wang J., Xie B., Sun J., Han G. Robust gene dysregulation in Alzheimer’s disease brains. J Alzheimers Dis. 2014;41:587–597. doi: 10.3233/JAD-140147. [DOI] [PubMed] [Google Scholar]
- 10.Twine N.A., Janitz K., Wilkins M.R., Janitz M. Whole transcriptome sequencing reveals gene expression and splicing differences in brain regions affected by Alzheimer’s disease. PLoS One. 2011;6:e16266. doi: 10.1371/journal.pone.0016266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang B., Gaiteri C., Bodea L.G., Wang Z., McElwee J., Podtelezhnikov A.A. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013;153:707–720. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simpson J.E., Ince P.G., Shaw P.J., Heath P.R., Raman R., Garwood C.J. Microarray analysis of the astrocyte transcriptome in the aging brain: relationship to Alzheimer’s pathology and APOE genotype. Neurobiol Aging. 2011;32:1795–1807. doi: 10.1016/j.neurobiolaging.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 13.Hallock P., Thomas M.A. Integrating the Alzheimer’s disease proteome and transcriptome: a comprehensive network model of a complex disease. OMICS. 2012;16:37–49. doi: 10.1089/omi.2011.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Voineagu I., Wang X., Johnston P., Lowe J.K., Tian Y., Horvath S. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Antunez C., Boada M., Gonzalez-Perez A., Gayan J., Ramirez-Lorca R., Marin J. The membrane-spanning 4-domains, subfamily A (MS4A) gene cluster contains a common variant associated with Alzheimer’s disease. Genome Med. 2011;3:33. doi: 10.1186/gm249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lambert J.C., Heath S., Even G., Campion D., Sleegers K., Hiltunen M. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 17.Harold D., Abraham R., Hollingworth P., Sims R., Gerrish A., Hamshere M.L. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Naj A.C., Jun G., Beecham G.W., Wang L.S., Vardarajan B.N., Buros J. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seshadri S., Fitzpatrick A.L., Ikram M.A., DeStefano A.L., Gudnason V., Boada M. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303:1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lambert J.C., Ibrahim-Verbaas C.A., Harold D., Naj A.C., Sims R., Bellenguez C. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang K., Zhang H., Ma D., Bucan M., Glessner J.T., Abrahams B.S. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459:528–533. doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weiss L.A., Arking D.E., Daly M.J., Chakravarti A. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009;461:802–808. doi: 10.1038/nature08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anney R., Klei L., Pinto D., Regan R., Conroy J., Magalhaes T.R. A genome-wide scan for common alleles affecting risk for autism. Hum Mol Genet. 2010;19:4072–4082. doi: 10.1093/hmg/ddq307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pinto D., Pagnamenta A.T., Klei L., Anney R., Merico D., Regan R. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gatz M., Reynolds C.A., Fratiglioni L., Johansson B., Mortimer J.A., Berg S. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63:168–174. doi: 10.1001/archpsyc.63.2.168. [DOI] [PubMed] [Google Scholar]
- 26.Manolio T.A., Collins F.S., Cox N.J., Goldstein D.B., Hindorff L.A., Hunter D.J. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neumann H., Daly M.J. Variant TREM2 as risk factor for Alzheimer’s disease. N Engl J Med. 2013;368:182–184. doi: 10.1056/NEJMe1213157. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi K., Rochford C.D., Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005;201:647–657. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jonsson T., Stefansson H., Steinberg S., Jonsdottir I., Jonsson P.V., Snaedal J. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368:107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guerreiro R., Wojtas A., Bras J., Carrasquillo M., Rogaeva E., Majounie E. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benitez B.A., Cooper B., Pastor P., Jin S.C., Lorenzo E., Cervantes S. TREM2 is associated with the risk of Alzheimer’s disease in Spanish population. Neurobiol Aging. 2013;34:1711. doi: 10.1016/j.neurobiolaging.2012.12.018. e15–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pottier C., Wallon D., Rousseau S., Rovelet-Lecrux A., Richard A.C., Rollin-Sillaire A. TREM2 R47H variant as a risk factor for early-onset Alzheimer’s disease. J Alzheimers Dis. 2013;35:45–49. doi: 10.3233/JAD-122311. [DOI] [PubMed] [Google Scholar]
- 33.Guerreiro R.J., Lohmann E., Kinsella E., Brás J.M., Luu N., Gurunlian N. Exome sequencing reveals an unexpected genetic cause of disease: NOTCH3 mutation in a Turkish family with Alzheimer’s disease. Neurobiol Aging. 2012;33:1008. doi: 10.1016/j.neurobiolaging.2011.10.009. e17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pottier C., Hannequin D., Coutant S., Rovelet-Lecrux A., Wallon D., Rousseau S. High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Mol Psychiatry. 2012;17:875–879. doi: 10.1038/mp.2012.15. [DOI] [PubMed] [Google Scholar]
- 35.Reitz C., Cheng R., Rogaeva E., Lee J.H., Tokuhiro S., Zou F. Meta-analysis of the association between variants in SORL1 and Alzheimer disease. Arch Neurol. 2011;68:99–106. doi: 10.1001/archneurol.2010.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cruchaga C., Karch C.M., Jin S.C., Benitez B.A., Cai Y., Guerreiro R. Rare coding variants in Phospholipase D3 (PLD3) confer risk for Alzheimer’s disease. Nature. 2014;505:550–554. doi: 10.1038/nature12825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jonsson T., Atwal J.K., Steinberg S., Snaedal J., Jonsson P.V., Bjornsson S. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 38.Coppola G., Chinnathambi S., Lee J.J., Dombroski B.A., Baker M.C., Soto-Ortolaza A.I. Evidence for a role of the rare p. A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer’s diseases. Hum Mol Genet. 2012;21:3500–3512. doi: 10.1093/hmg/dds161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Roak B.J., Deriziotis P., Lee C., Vives L., Schwartz J.J., Girirajan S. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanders S.J., Murtha M.T., Gupta A.R., Murdoch J.D., Raubeson M.J., Willsey A.J. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chahrour M.H., Yu T.W., Lim E.T., Ataman B., Coulter M.E., Hill R.S. Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet. 2012;8:e1002635. doi: 10.1371/journal.pgen.1002635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iossifov I., Ronemus M., Levy D., Wang Z., Hakker I., Rosenbaum J. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lim E.T., Raychaudhuri S., Sanders S.J., Stevens C., Sabo A., MacArthur D.G. Rare complete knockouts in humans: population distribution and significant role in autism spectrum disorders. Neuron. 2013;77:235–242. doi: 10.1016/j.neuron.2012.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu T.W., Chahrour M.H., Coulter M.E., Jiralerspong S., Okamura-Ikeda K., Ataman B. Using whole-exome sequencing to identify inherited causes of autism. Neuron. 2013;77:259–273. doi: 10.1016/j.neuron.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Michaelson J.J., Shi Y., Gujral M., Zheng H., Malhotra D., Jin X. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell. 2012;151:1431–1442. doi: 10.1016/j.cell.2012.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shi L., Zhang X., Golhar R., Otieno F.G., He M., Hou C. Whole-genome sequencing in an autism multiplex family. Mol Autism. 2013;4:8. doi: 10.1186/2040-2392-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang Y.H., Yuen R.K., Jin X., Wang M., Chen N., Wu X. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet. 2013;93:249–263. doi: 10.1016/j.ajhg.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lister R., Mukamel E.A., Nery J.R., Urich M., Puddifoot C.A., Johnson N.D. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davies M.N., Volta M., Pidsley R., Lunnon K., Dixit A., Lovestone S. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012;13:R43. doi: 10.1186/gb-2012-13-6-r43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Condliffe D., Wong A., Troakes C., Proitsi P., Patel Y., Chouliaras L. Cross-region reduction in 5-hydroxymethylcytosine in Alzheimer’s disease brain. Neurobiol Aging. 2014;35:1850–1854. doi: 10.1016/j.neurobiolaging.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chouliaras L., Mastroeni D., Delvaux E., Grover A., Kenis G., Hof P.R. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol Aging. 2013;34:2091–2099. doi: 10.1016/j.neurobiolaging.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coppieters N., Dieriks B.V., Lill C., Faull R.L., Curtis M.A., Dragunow M. Global changes in DNA methylation and hydroxymethylation in Alzheimer’s disease human brain. Neurobiol Aging. 2014;35:1334–1344. doi: 10.1016/j.neurobiolaging.2013.11.031. [DOI] [PubMed] [Google Scholar]
- 53.Cogswell J.P., Ward J., Taylor I.A., Waters M., Shi Y., Cannon B. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J Alzheimers Dis. 2008;14:27–41. doi: 10.3233/jad-2008-14103. [DOI] [PubMed] [Google Scholar]
- 54.Lau P., Bossers K., Janky R., Salta E., Frigerio C.S., Barbash S. Alteration of the microRNA network during the progression of Alzheimer’s disease. EMBO Mol Med. 2013;5:1613–1634. doi: 10.1002/emmm.201201974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith P., Al Hashimi A., Girard J., Delay C., Hebert S.S. In vivo regulation of amyloid precursor protein neuronal splicing by microRNAs. J Neurochem. 2011;116:240–247. doi: 10.1111/j.1471-4159.2010.07097.x. [DOI] [PubMed] [Google Scholar]
- 56.Bekris L.M., Lutz F., Montine T.J., Yu C.E., Tsuang D., Peskind E.R. MicroRNA in Alzheimer’s disease: an exploratory study in brain, cerebrospinal fluid and plasma. Biomarkers. 2013;18:455–466. doi: 10.3109/1354750X.2013.814073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Villa C., Ridolfi E., Fenoglio C., Ghezzi L., Vimercati R., Clerici F. Expression of the transcription factor Sp1 and its regulatory hsa-miR-29b in peripheral blood mononuclear cells from patients with Alzheimer’s disease. J Alzheimers Dis. 2013;35:487–494. doi: 10.3233/JAD-122263. [DOI] [PubMed] [Google Scholar]
- 58.Citron B.A., Dennis J.S., Zeitlin R.S., Echeverria V. Transcription factor Sp1 dysregulation in Alzheimer’s disease. J Neurosci Res. 2008;86:2499–2504. doi: 10.1002/jnr.21695. [DOI] [PubMed] [Google Scholar]
- 59.Tan L., Yu J.T., Tan M.S., Liu Q.Y., Wang H.F., Zhang W. Genome-wide serum microrna expression profiling identifies serum biomarkers for Alzheimer’s disease. J Alzheimers Dis. 2014;40:1017–1027. doi: 10.3233/JAD-132144. [DOI] [PubMed] [Google Scholar]
- 60.Kumar P., Dezso Z., MacKenzie C., Oestreicher J., Agoulnik S., Byrne M. Circulating miRNA biomarkers for Alzheimer’s disease. PLoS One. 2013;8:e69807. doi: 10.1371/journal.pone.0069807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leidinger P., Backes C., Deutscher S., Schmitt K., Mueller S.C., Frese K. A blood based 12-miRNA signature of Alzheimer disease patients. Genome Biol. 2013;14:R78. doi: 10.1186/gb-2013-14-7-r78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sala Frigerio C, Lau P, Salta E, Tournoy J, Bossers K, Vandenberghe R, et al. Reduced expression of hsa-miR-27a-3p in CSF of patients with Alzheimer disease. Neurology 2013;81:2103–6. [DOI] [PubMed]
- 63.Ladd-Acosta C., Hansen K.D., Briem E., Fallin M.D., Kaufmann W.E., Feinberg A.P. Common DNA methylation alterations in multiple brain regions in autism. Mol Psychiatry. 2014;19:862–871. doi: 10.1038/mp.2013.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wong C.C., Meaburn E.L., Ronald A., Price T.S., Jeffries A.R., Schalkwyk L.C. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol Psychiatry. 2014;19:495–503. doi: 10.1038/mp.2013.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miyake K., Yang C., Minakuchi Y., Ohori K., Soutome M., Hirasawa T. Comparison of genomic and epigenomic expression in monozygotic twins discordant for Rett syndrome. PLoS One. 2013;8:e66729. doi: 10.1371/journal.pone.0066729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cundiff P.E., Anderson S.A. Impact of induced pluripotent stem cells on the study of central nervous system disease. Curr Opin Genet Dev. 2011;21:354–361. doi: 10.1016/j.gde.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wapinski O.L., Vierbuchen T., Qu K., Lee Q.Y., Chanda S., Fuentes D.R. Hierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell. 2013;155:621–635. doi: 10.1016/j.cell.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Israel M.A., Yuan S.H., Bardy C., Reyna S.M., Mu Y., Herrera C. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482:216–220. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kondo T., Asai M., Tsukita K., Kutoku Y., Ohsawa Y., Sunada Y. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Aβ and differential drug responsiveness. Cell Stem Cell. 2013;12:487–496. doi: 10.1016/j.stem.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 70.Shi Y, Kirwan P, Smith J, MacLean G, Orkin SH, Livesey FJ. A human stem cell model of early Alzheimer’s disease pathology in Down syndrome. Sci Transl Med 2012;4:124ra29. [DOI] [PMC free article] [PubMed]
- 71.Yahata N., Asai M., Kitaoka S., Takahashi K., Asaka I., Hioki H. Anti-Aβ drug screening platform using human iPS cell-derived neurons for the treatment of Alzheimer’s disease. PLoS One. 2011;6:e25788. doi: 10.1371/journal.pone.0025788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang D., Pekkanen-Mattila M., Shahsavani M., Falk A., Teixeira A.I., Herland A. A 3D Alzheimer’s disease culture model and the induction of P21-activated kinase mediated sensing in iPSC derived neurons. Biomaterials. 2014;35:1420–1428. doi: 10.1016/j.biomaterials.2013.11.028. [DOI] [PubMed] [Google Scholar]
- 73.Soldner F., Jaenisch R. Medicine. iPSC disease modeling. Science. 2012;338:1155–1156. doi: 10.1126/science.1227682. [DOI] [PubMed] [Google Scholar]
- 74.Bryant C., Giovanello K.S., Ibrahim J.G., Chang J., Shen D., Peterson B.S. Mapping the genetic variation of regional brain volumes as explained by all common SNPs from the ADNI study. PLoS One. 2014;8:e71723. doi: 10.1371/journal.pone.0071723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Matuszek G., Talebizadeh Z. Autism Genetic Database (AGD): a comprehensive database including autism susceptibility gene-CNVs integrated with known noncoding RNAs and fragile sites. BMC Med Genet. 2009;10:102. doi: 10.1186/1471-2350-10-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Basu S.N., Kollu R., Banerjee-Basu S. AutDB: a gene reference resource for autism research. Nucleic Acids Res. 2009;37:D832–D836. doi: 10.1093/nar/gkn835. [DOI] [PMC free article] [PubMed] [Google Scholar]