

Abstract
The thymine DNA glycosylase (TDG) is a multifunctional enzyme, which is essential for embryonic development. It mediates the base excision repair (BER) of G:T and G:U DNA mismatches arising from the deamination of 5-methyl cytosine (5-MeC) and cytosine, respectively. Recent studies have pointed at a role of TDG during the active demethylation of 5-MeC within CpG islands. TDG interacts with the histone acetylase CREB-binding protein (CBP) to activate CBP-dependent transcription. In addition, TDG also interacts with the retinoic acid receptor α (RARα), resulting in the activation of RARα target genes. Here we provide evidence for the existence of a functional ternary complex containing TDG, CBP and activated RARα. Using global transcriptome profiling, we uncover a coupling of de novo methylation-sensitive and RA-dependent transcription, which coincides with a significant subset of CBP target genes. The introduction of a point mutation in TDG, which neither affects overall protein structure nor BER activity, leads to a significant loss in ternary complex stability, resulting in the deregulation of RA targets involved in cellular networks associated with DNA replication, recombination and repair. We thus demonstrate for the first time a direct coupling of TDG’s epigenomic and transcription regulatory function through ternary complexes with CBP and RARα.
Keywords: CREB-binding protein, Thymine DNA glycosylase, Retinoic acid receptor α, Transcription regulation, Cytosine DNA methylation, Epigenomics
Introduction
The base excision repair (BER) enzyme thymine DNA glycosylase (TDG) plays an important role in the maintenance of genetic stability by correcting guanine:thymine (G:T) and guanine:uracil (G:U) DNA mismatches [1,2]. These mispaired basepairs arise from the spontaneous or catalyzed deamination of 5-methyl cytosine (5-MeC) at CpG sites and cytosine, respectively and would, if uncorrected, lead to G:C to A:T transitions [3,4]. Depending on their location within the genome, such mutations can result in the loss of CpG dinucleotides with potential impact on gene regulation or in codon changes within coding regions, both of which can have serious consequences such as cancer formation [5,6].
Apart from its function during BER, TDG interacts with the retinoic acid receptor α (RARα) and the retinoid X receptor (RXR) in a ligand-independent manner to enhance receptor affinity for their DNA target sites [7]. RAR and RXR belong to a family of ligand-activated nuclear transcription factors, which form homo- or heterodimers in order to activate gene expression via binding to RA response elements (RAREs) in target promoters [8]. In the absence of ligand, the RXR/RARα heterodimer represses transcription by recruiting corepressors such as NCOR1, SMRT (NCOR2) and histone deacetylases [9,10], whereas ligand binding mediates a conformational change, allowing the recruitment of coactivators such as histone acetyltransferases (HATs) and the basic transcription machinery [11,12]. RARα can be activated by all-trans RA (atRA) and 9-cis RA (9cRA), while RXR activation is restricted to 9cRA only.
Moreover, TDG interacts with the HATs CREB-binding protein (CBP) and p300, thereby enhancing their gene expression-activating capacities [13]. CBP and p300 are highly related coactivators for a variety of transcription factors, including CREB, the AP-1 proteins Jun and Fos, nuclear receptors and the tumor suppressor p53 [14]. Via their HAT activity, CBP/p300 are involved in chromatin remodeling at target promoters in order to activate gene expression [15,16]. As reported for TDG, CBP has also been shown to interact with RAR, leading to the increased expression of RARE-driven reporters; but unlike TDG, the binding of CBP is limited to ligand-activated RAR [17]. In line with these findings, a CBP knockdown results in decreased expression of RARE-driven reporters [18].
Recent studies are pointing at an involvement of the BER pathway in DNA demethylation [19]. In this context, TDG may play a dual role in active demethylation as well as in the inhibition of de novo DNA methylation, since it has been shown to inhibit the activity of the DNA methyltransferases Dnmt3a and Dnmt3b [20,21]. The homozygous knockout of TDG leads to strong developmental defects and prenatal death in mice [22,23]. Notably, some of the specific lethal phenotypes of TDG null mice are comparable to those previously described for CBP-knockout mouse embryos and for defects in RA signaling [24,25]. The promoters of down-regulated genes in TDG-deficient mouse embryonic fibroblasts (MEFs) show a decreased CBP-binding [22] and elevated levels of CpG methylation, supporting a role of TDG in DNA demethylation [22,23].
Here, we investigate the interplay of RA-dependent and DNA methylation-sensitive transcription with CBP target genes in HEK293 cells. We provide evidence for a functional ternary complex composed of TDG, CBP and activated RARα, which consistently controls the expression of RA-dependent target genes that are involved in important cellular processes such as DNA replication, cell survival or cell cycle regulation.
Results and discussion
RA-dependent transcription coincides with de novo methylation-sensitive gene expression
Given that TDG influences RARα- and CBP-dependent gene expression and in view of a direct involvement of TDG in the active demethylation of 5-MeC within CpG contexts, we investigated whether there is coherence in gene expression regulation between these different pathways, which would point at TDG as a connector of epigenetic DNA modification, RA and CBP gene regulatory functions.
We compared gene expression profiles of HEK293 cells overexpressing CBP with those of RA and 5-aza-2′-deoxycytidine (5-aza-dC)-treated cells (Figure 1). Incorporation of 5-aza-dC has been shown to efficiently decrease de novo DNA methylation by an irreversible inhibition of Dnmt activity [26]. As expected by the transcription activating roles of CBP, RA and DNA demethylation, all three conditions resulted in gene activation in the majority of cases (Figure 1A). Thereby, overexpression of CBP results in the statistically significant up-regulation of 1344 genes and down-regulation of 534 genes (P < 0.01) (Figure 1A, left panel). RA treatment leads to the up-regulation of 418 genes and down-regulation of 20 genes (Figure 1A, middle panel) and the inhibition of de novo methylation by 5-aza-dC treatment affects the expression of 174 genes, of which expression of 145 and 29 genes is up-regulated and down-regulated, respectively (Figure 1A, right panel). When comparing the regulated gene sets of all three conditions, we observe statistically significant common subsets of 256 (RA- vs. CBP-regulated genes), 102 (5-aza-dC- vs. CBP-regulated genes) and 94 (RA- vs. 5-aza-dC-regulated) genes, as indicated by the corresponding hypergeometric distribution P values (Figure 1B). Moreover, there are 86 genes of each common subset, which are statistically significantly regulated in all three conditions (Figure 1B, red). Comparing the log2 fold changes in gene expression upon RA treatment with those upon 5-aza-dC treatment reveals a highly significant (P < 0.001) positive correlation (R = 0.76, red), which is even retained when comparing those genes significantly regulated in only one of the two conditions (R = 0.72, 5-aza-dC, brown; R = 0.57, RA, green; Figure 1C, left panel). In similar comparisons, the CBP target genes split up into two groups. While the common subsets with both 5-aza-dC target genes (R = 0.74; Figure 1C, middle panel) and RA target genes (R = 0.80; Figure 1C, right panel) show statistically significant (P < 0.001) positive correlations with CBP targets, genes with expression only significantly regulated by CBP do not show any responsiveness to de novo methylation inhibition and RA treatment. Notably, the whole sets of 5-aza-dC targets and RA targets retained responsiveness to CBP, as indicated by the corresponding Pearson correlation coefficients of 0.66 (middle panel) and 0.78 (right panel), respectively (Figure 1C). We validated our findings in human HEK293 cells using COS-7 cells from the African green monkey and revealed the same strong positive correlation between CBP, RA and 5-aza-dC target genes in a different species (Figure S1). Taken together, these results support a direct linkage between CBP-mediated gene activation and CpG demethylation at promoter sites of RA target genes. The classical RA pathway includes ligand-mediated activation of homo- or heterodimers of RAR and RXR bound to RAREs in the promoter region of target genes, which subsequently affects gene expression directly or via the recruitment of other transcription factors [27]. Our findings suggest that gene activation by RA may include active or passive DNA demethylation of CpG and potentially also of non-CpG sites in gene promoters. Indeed, several recent studies have linked RA treatment with a global loss of CpG methylation in HL-60 and U937 cell lines [28]. RA has been shown to up-regulate the expression of the tumor suppressors p16 and p21 as well as E-cadherin expression by inducing promoter hypomethylation [29,30]. In the case of p16 and p21, a RA-induced decrease of Dnmt expression was held responsible for promoter hypomethylation [29].
Figure 1.
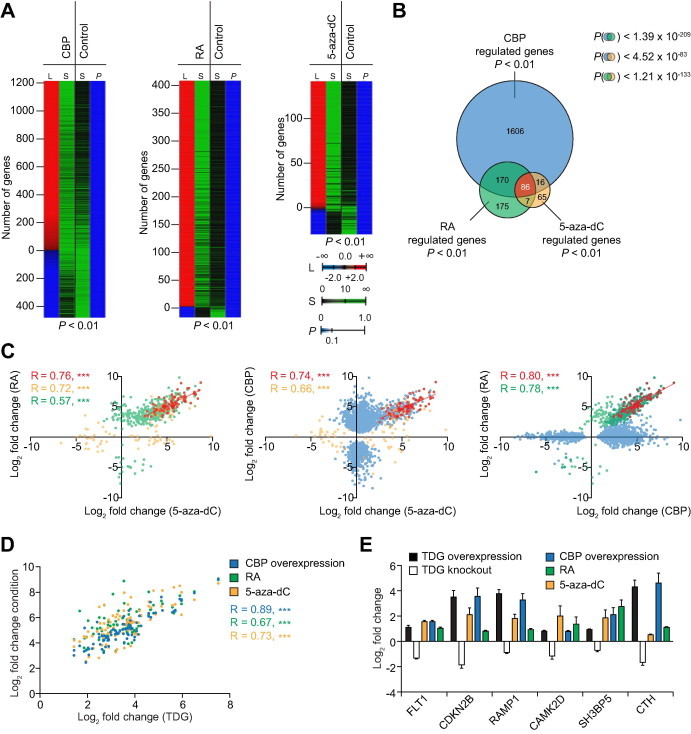
Comparison of CBP, 5-aza-dC and RA target genes by transcriptome profiling A. Heatmaps of a substraction profile of significantly (post hoc pFDR P < 0.01) regulated genes after CBP overexpression (left), RA treatment (middle) and 5-aza-dC treatment (right) of HEK293 cells. Mock transfected, ethanol- or DMSO-treated cells were used as controls, respectively. The number of genes (L, column 1) that are positively (red shading) and negatively (blue shading) regulated is shown together with the average over three independent biological replicate signals for each condition (S, column 2) and control (S, column 3). The final column (column 4) represents the post hoc P values (P). B. Venn diagram for the overlap of genes regulated in a statistically significant manner in all three conditions studied in A. The P values (hypergeometric distribution) for chance occurrence of each overlap are indicated. Note that the overall overlap of 86 genes is almost entirely composed of genes which are derepressed by 5-aza-dC treatment and concomittantly activated by atRA treatment and CBP overexpression. C. Scatter plots of the overlaps of methylation and RA-regulated genes (left), methylation and CBP-regulated genes (middle), and RA- and CBP- regulated genes (right) reveals clear Pearson’s correlations (R). Genes with expression significantly affected under both conditions compared in each panel are indicated in red. Genes with expression significantly affected upon CBP overexpression only (blue), by RA treatment only (green) and by 5-aza-dC treatment only (brown) are also indicated in different colors. ∗∗∗P < 0.001. D. Scatter-plot of the common subset from panel B in each condition (Y axis) compared to the overexpression of human TDG in HEK293 cells (X axis). E. Gene expression changes of an assigned set of the common subset from panel B in HEK293 cells overexpressing human TDG and in TDG knockout mouse embryonic fibroblasts [23]. TDG, thymine DNA glycosylase; RA, retinoic acid; 5-aza-dC, 5-azacytidine; FLT1, fls-related tyrosine kinase 1; CDKN2B, cyclin-dependent kinase inhibitor 2B; RAMP1, receptor activity modifying protein 1; CAMK2D, calcium/calmodulin-dependent protein kinase II delta; SH3BP5, SH3-domain binding protein 5; CTH, cystathionase.
Since TDG has been shown to interact with CBP [13] and RARα [7] in a functional manner and has been associated with the active demethylation of CpG sites in gene promoters [20,21], we further investigated whether there is a TDG-dependent functional connection of these three pathways. By using transcriptome profiling, we assessed the gene expression changes of the common subset of CBP, RA and 5-aza-dC targets upon overexpression of human TDG in HEK293 cells. Our results unveiled that expression of all genes are concomitantly up-regulated by TDG, as judged by the highly statistically significant Pearson correlation coefficients (0.67 for RA, 0.73 for 5-aza-dC and 0.89 for CBP) (Figure 1D). Indeed, the expression of a randomly picked, assigned set of these genes is down-regulated in mice upon knockout of murine TDG, as assessed by gene expression profiling of previously published data from TDG knockout MEFs [23], excluding the possibility that artifacts induced by the overexpression of TDG are responsible for the TDG responsiveness of these genes (Figure 1E).
A point mutation in TDG affects CBP-binding and ternary complex stability
The catalytic domain of TDG, which is responsible for BER activity, is important for RARα binding (Figure 2A) [7]. The CBP binding interface of TDG is split into two distinct regions, one located in the N-terminus and including the regulatory domain, which is known to be essential for G:T but not G:U DNA repair activity [31], and the other located C-terminally (Figure 2A) [13]. The former domain is important for interacting with the HAT domain as well as the CH3 domain of CBP, while the latter one is restricted to CH3 interaction only. Furthermore, the TDG regulatory domain contains four lysine (K) residues, which are acetylated by CBP and thus represent sites of very tight TDG/CBP interaction [13]. Hence, lysine residues are not only crucial for CBP recruitment and activity, but are also hotspots for DNA binding by establishing electrostatic interactions [32]. Notably, there is an enrichment of proline (P) residues in the regulatory domain of TDG of which several are located between two sites for CBP acetylation (Figure 2A, K59 on one hand and K83/K84/K87 on the other hand). Proline residues are known to increase the rigidity of polypeptide chains, making these residues possibly responsible for the previously observed lack of flexibility of the regulatory domain, as compared to the extreme N-terminus of TDG [32]. Given the importance of the regulatory domain for the interaction with CBP, we hypothesize that a proline mutation in this region has the potential to modify the structural dynamics in this region, which might lead to an altered CBP affinity.
Figure 2.
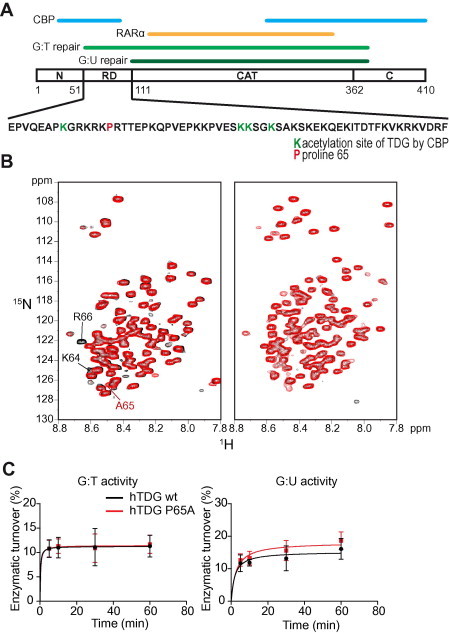
Structure and activity of wild type TDG and the mutant P65A A. Schematic structure of hTDG. The regions required for G:T and G:U mismatch recognition and processing (light and dark green, respectively), the N- and C-termini, the regulatory domain (RD), the catalytic (CAT) domain as well as the interfaces with CBP (blue) and RARα (brown) are indicated. The amino acid sequence of the RD is provided and the lysines that can be acetylated by CBP and the proline 65 that was mutated in this study are highlighted in green and red, respectively. B.1H–15N HSQC spectra of the N-terminus (amino acid residues 1–111) (left panel) and full length hTDG (right panel). Spectra for wild-type and the P65A mutant are shown in black and red, respectively. The resonances of the neighboring amino acids of P65 as well as the resonance of the introduced alanine are indicated. C. Glycosylase kinetics of human TDG wild-type and P65A mutant on G:T and G:U repair. DNA nicking assays were performed on a 25-mer dsDNA containing either a central G:T (left panel) or a G:U (right panel) mismatch. A 25-mer dsRNA containing a central canonical G:C pair was used as a control.
We thus generated a mutant form of TDG, TDG P65A, by introducing a proline to alanine mutation using directed mutagenesis at amino acid residue 65, which is located between the aforementioned acetylation sites. We performed NMR studies in order to assess the structure of TDG P65A and compared it to that of wild-type TDG (Figure 2B). Importantly the 1H–15N HSQC spectrum of the mutated N-terminal domain of TDG (amino acid residues 1–111, Figure 2B, left panel, red) differs from that of the wild-type TDG N-terminal counterpart (Figure 2B, left panel, black) solely by the resonances of the amino acids directly flanking the site of mutation, unveiling no global structural consequences of the mutation on the TDG N-terminus. Similarly, in the context of the full length protein, P65A did not result in significant structural changes (Figure 2B, right panel).
The site of mutation, aside from serving as the interface with CBP, is located in the region responsible for G:T but not for G:U activity. We therefore performed glycosylase activity assays with G:T and G:U mismatch-containing DNA substrates to examine whether glycosylase activity would be altered as a consequence of the mutation. Our results indicated that compared to wild-type TDG (black curve), neither G:T (left panel) nor G:U (right panel) repair kinetics were significantly altered when using TDG P65A (red curve) (Figure 2C).
In order to analyze the capability of TDG P65A to interact with CBP, we performed microscale thermophoresis (MST) experiments with YFP-tagged CBP and HA-fused versions of TDG P65A or wild-type TDG (Figure 3A). The MST technique relies on the fact that molecules move in a temperature gradient from hot to cold (thermophoresis) [33,34]. Such movement is impacted by complex formation, since complexes exhibit an altered Brownian velocity when compared to their compounds in a free state. MST techniques allow to follow the thermophoresis of a fluorescently labeled molecule (here YFP-CBP) as a function of increasing concentrations of a binding partner (here HA-TDG or HA-TDG P65A). While these analyses with wild-type HA-TDG show saturating binding kinetics (Figure 3A, black curve, and Figure S2), HA-TDG P65A affinity for YFP-CBP is significantly decreased by a factor >3 (Figure 3A, red curve).
Figure 3.
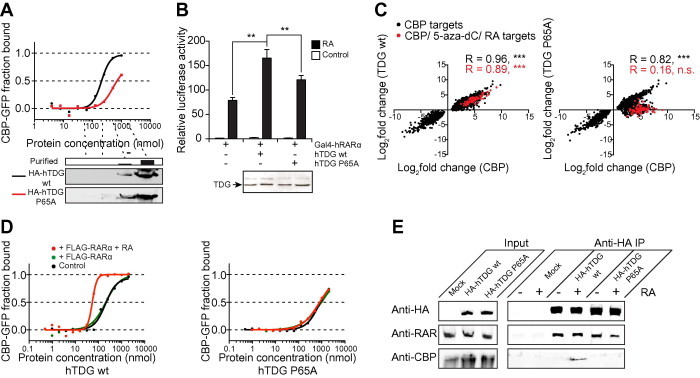
The TDG P65A mutation results in reduced TDG/CBP/RARα ternary complex stability A. A representative microscale thermophoresis (MST) experiment comparing HA-tagged hTDG wt to HA-tagged hTDG P65A. Ten successive 1:1 dilutions of immuno-purified HA-hTDG (wt or P65A mutant) in binding buffer were used for MST analyses with YFP-CBP. The amounts of HA-fusion protein in every second dilution were monitored by Western blot using anti-HA antibody. The protein concentration of the non-diluted HA-hTDG samples was measured by Bradford assay. B. Promoter activation assay using a minimal Gal4 promoter driving the expression of firefly luciferase. The reporter construct was co-transfected with a Gal4-RARα expression vector and, where indicated, with either wild-type or P65A mutant HA-hTDG. Luciferase activity was normalized by protein amounts. Comparable TDG protein expression in the different conditions was verified by Western blotting. ∗∗P < 0.01 in a students t-test. C. Scatter plot of the genes with expression significantly (P < 0.01) regulated by CBP overexpression (X axis) upon overexpression of TDG (Y axis) wild-type (left panel) or TDG P65A (right panel). Pearson correlation coefficients for all CBP target genes were shown in black and those for the common subset with the 5-aza-dC and RA treatments from Figure 1D are shown in red. ∗∗∗P < 0.001. D. MST experiments using increasing concentrations of purified HA-hTDG wild-type (left panel) or P65A mutant (right panel) in the presence of non-activated (green) or RA-activated (red) purified FLAG-tagged hRARα compared to HA-hTDG wt or P65A mutant alone (control). E. Immunoprecipitation of HA-TDG wt or P65A mutant from nuclear extracts of transiently transfected HEK293 cells in the presence or absence of RA using anti-HA agarose. Co-immnoprecipitated proteins were monitored for RAR and CBP using the appropriate antibodies. Mock transfected cells were used as control. Only input without RA treatment was included since addition of RA does not affect expression of the relevant proteins (data not shown).
To investigate the effects of TDG P65A on the RA-mediated transactivation of RARα-dependent genes, we performed RA-dependent luciferase reporter assays using a GAL4 DNA binding domain (DBD) fusion of RARα and a GAL reporter gene in cells overexpressing wild-type TDG or TDG P65A (Figure 3B). In the absence of TDG overexpression, RA treatment results in an approximate 75-fold increase of reporter expression, which is statistically significantly (P < 0.01) boosted by a factor greater than two in the presence of wild-type TDG (Figure 3B). The overexpression of TDG P65A results in an approximate 1.5-fold significant (P < 0.05) boost in RA-dependent reporter expression, when compared to mock transfected cells, but the effect is significantly compromised in comparison to wild-type TDG overexpression as determined by paired students t-test (P < 0.01). In order to investigate the effect of the P65A mutation on CBP-dependent transcription, we monitored the expression of CBP target genes — defined as being responsive to CBP — upon overexpression of wild-type TDG in comparison to the overexpression of TDG P65A. The expression of CBP targets (black spots), including those among the common subset with 5-aza-dC and RA targets (red spots), is concomitantly regulated by the overexpression of wild-type TDG (Figure 3C, left panel). In the case of TDG P65A overexpression (Figure 3C, right panel), expression of 5-aza-dC and RA-dependent CBP targets (red spots) are not affected, while expression of the majority of the 5-aza-dC and RA-independent CBP targets (black spots) are concomitantly regulated.
Our data indicate that the P65A mutation influences the TDG/CBP interaction as well as RA-dependent transcription activation via RARα and the transcription of CBP target genes responsive to 5-aza-dC and RA. This could be potentially explained by a ternary complex forming between all three compounds, as it has been previously shown for TDG, RAR and the CBP-related HAT p300 [23]. In order to answer this question, we performed another set of MST experiments with YFP-CBP and dilution series of immuno-purified wild-type HA-TDG or HA-TDG P65A in the presence or absence of non-activated (green curve) / RA-activated (red curve) purified FLAG-RARα (Figures 3D and S2). The presence of non-activated RARα does not lead to significant alterations in CBP affinity for either wild-type or mutant TDG protein (Figure 3D, green lines versus black lines), whereas the presence of RA-activated RARα significantly increases the wild-type TDG affinity for CBP by a factor >4 (Figure 3D, left panel, red curve), but leaves the TDG P65A/CBP binding unchanged (Figure 3D, right panel, red line). We performed co-immunoprecipitation experiments of wild-type HA-TDG and HA-TDG P65A from nuclear extracts of HEK293 cells in the presence or absence of RA to verify these findings (Figure 3E). RAR co-immunoprecipitates with both wild-type and mutant TDG proteins regardless of the presence of RA, confirming previous reports [7]. However, detectable amounts of CBP only co-precipitate with wild-type TDG in the presence of RA, strongly arguing for a ternary complex formation of TDG, CBP and activated RARα in vivo, which is destabilized by the P65A mutation of TDG. Remarkably, such a ternary complex of TDG, RAR and p300 has been discovered recently, in which TDG is required for the p300/RAR interaction and for the recruitment of p300 to RA-regulated gene promoters [23]. Given the significantly different roles of p300 and CBP, as well as the fact that CBP directly interacts with RARs in a ligand-dependent manner in the absence of TDG, it is not only more challenging to show such a ternary complex, but also of significant interest to uncover a mutual stabilization within the complex. The fact that the P65A mutation prevents ternary complex formation with CBP and activated RARα in vitro makes TDG P65A a useful tool to functionally characterize the ternary TDG/CBP/RARα complex.
Disruption of TDG/CBP/RARα ternary complex deregulates RA-dependent gene expression
In order to assess the role of the complex of TDG and CBP with activated RARα in gene expression regulation, we co-transfected HEK293 cells with wild-type TDG and CBP and compared the RA-responsive transcriptome profile with cells co-expressing TDG P65A and CBP. Thereby, we focused on genes consistently co-regulated upon RA treatment in the wild-type TDG/CBP condition and in non-treated cells, in order to avoid side-effects caused by TDG/CBP overexpression. In both conditions, a total of 152 genes were significantly (P < 0.05) differentially expressed upon RA treatment, among which, expression of 144 and 8 genes was up-regulated and down-regulated, respectively (Figure 4A, Conditions 1 and 4). Notably, these genes showed similar expression changes upon 5-aza-dC treatment and CBP overexpression (Figure 4A, Conditions 2 and 3). Assessing their differential expression upon RA treatment in the TDG P65A/CBP condition reveals a global deregulation of gene expression (Figure 4A, Condition 6). It is worth noting that the vast majority of these genes do not respond to RA in a significant manner when either wild-type TDG or TDG P65A is overexpressed alone (Figure 4A, Conditions 5 and 7), arguing for CBP and TDG being in conjunction important for the RA responsiveness of those genes. A comparison of the log2-fold changes of the RA-dependent genes in the wild-type TDG/CBP condition (X axis) with those in the TDG P65A/CBP condition (Y axis) shows that expression of the majority of genes is not significantly regulated upon RA treatment in TDG P65A/CBP expressing cells (Figure 4B, black spots). Expression of only 30 genes (20%) is significantly (P < 0.05) regulated in the TDG P65A/CBP condition as well, of which notably expression of 19 genes is regulated in the opposite manner, when compared to the wild-type TDG/CBP condition (Figure 4B, red spots). We calculated the change in RA responsiveness caused by the introduction of TDG P65A as the difference of the RA-induced change of gene expression between TDG P65A/CBP and wild-type TDG/CBP expressing cells, which is normalized to the wild-type TDG/CBP condition (Figure 4C). While expression of only 6 genes (4%) shows a stronger RA responsiveness in the presence of TDG P65A/CBP (Figure 4C, red), the RA responsiveness of the remaining 146 genes is severely reduced when compared to wild-type TDG/CBP (Figure 4C, blue). Notably, we observe a set of genes, whose responsiveness to RA is inversed by the introduction of TDG P65A (Figure 4C, change in RA responsiveness lower than −1).
Figure 4.
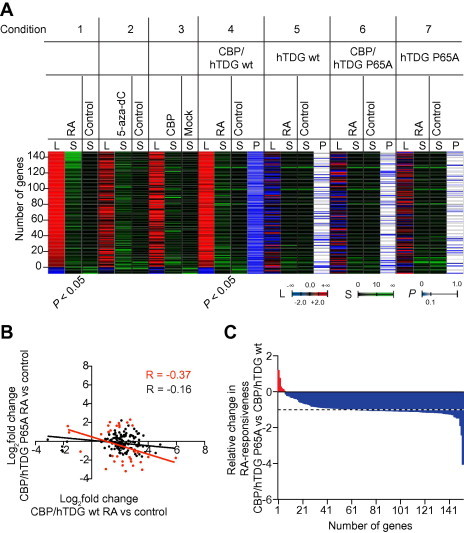
TDG P65A influences the expression of genes sensitive to de novo DNA demethylation, RA and CBP A. Heatmap of significant (P < 0.05) RA-responsive genes in non-transfected (Condition 1) and in CBP and wild-type hTDG overexpressing HEK293 cells (Condition 4). The number of genes (L) that are positively (red shading) or negatively (blue shading) regulated is shown together with the average over three independent biological replicate signals of each experimental condition and the corresponding control condition (S). Expression changes of these genes upon 5-aza-dC treatment (Condition 2), CBP overexpression (Condition 3) as well as RA treatment during simultaneous overexpression CBP and hTDG P65A (Condition 6) are displayed as heat maps. Cells overexpressing TDG wt (Condition 5) or P65A mutant (Condition 7) alone and subjected to RA treatment were used as control. Post-hoc P values are indicated as color code (P). B. Scatter plot of the genes shown in panel A. RA-mediated differential expression was compared between cells overexpressing CBP and hTDG wt (X axis) and cells overexpressing CBP and hTDG P65A mutant (Y axis). Genes with expression significantly (P < 0.05) impacted in the hTDG P65A condition are highlighted in red. Pearson correlation coefficients for both non-significant (black) and significant subsets are indicated. C. Relative difference in RA-responsiveness of cells overexpressing CBP and hTDG P65A mutant in relative to cells overexpressing CBP and hTDG wt. The RA-responsiveness was normalized to the CBP/hTDG wt condition. Genes whose expression is significantly (P < 0.05) affected by RA treatment during CBP and hTDG P65A expression are highlighted in red, whereas genes with RA-dependent expression significantly (P < 0.05) affected during CBP and wild-type hTDG expression are highlighted in blue.
Conclusion
Our findings point at the existence of a transcriptionally active ternary complex composed of TDG, CBP and activated RARα. The single interactions between each two of the three compounds add up to a high affinity complex in vitro, which may activate RA-dependent gene expression in vivo in a similar way as it has been suggested for RARα, TDG and p300 [23] (Figure 5). However, functional studies of ternary complexes composed of independent catalytically active components constitute a major challenge in vivo. Here, we have functionally characterized a TDG point mutation, P65A, which is capable of substantially affecting ternary complex stability and transcription regulatory activity, and thus may serve as a powerful tool for future in vivo studies in order to address the biological role of the TDG/CBP/RARα complex. The recent findings of a point mutation, R66G, in the very same region of TDG in patients with colorectal cancer further substantiates the functional importance of this region and thus may provide a link between the ternary TDG/CBP/RARα complex and the development of this pathology [32,35].
Figure 5.
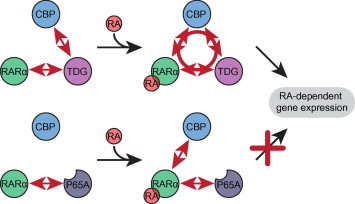
Model of ternary CBP:TDG:RAR complex action The interaction of CBP with RA-activated RARα cooperates with the interactions of TDG with CBP and RARα in order to stabilize a ternary complex composed of all three molecules, which affects the expression of RA-dependent genes (top). The P65A mutant of TDG is still able to interact with RARα, but fails to bind CBP, resulting in the inability to form a stable functional ternary complex (bottom).
Materials and methods
Plasmids
The TDG P65A mutant was produced by site-directed mutagenesis, exchanging the codon for proline 65 (CCC) into an alanine codon (GCC). TDG wt and the mutant TDG P65A were cloned into the BglII/SacI cloning sites of pSG5 plasmid (Agilent) to obtain HA-fusion proteins. CBP was cloned into the BglII cloning site of pEYFP plasmid (Clontech). Flag-RARα fusion proteins have been described previously [7]. For bacterial expression, full-length TDG (residues 1–410), its isolated N-terminal domain (residues 1–111) and the corresponding P65A mutants were cloned into the BamHI/EcoRI cloning sites of pGEX-6P-1 plasmid (GE Healthcare).
Cell culture
COS-7 and HEK293 cells were cultured at 37 °C in 5% CO2 in DMEM medium supplemented with 10% fetal calf serum (FCS) and with 1% penicillin/streptomycin. Transfections were performed using FugeneHD (Roche) according to the manufacturer’s instructions. Cells were treated with a final concentration of 5 × 10−7 M atRA dissolved in ethanol or 5-aza-2′-deoxycytidine (5-aza-dC) dissolved in DMSO for 24 h. Cells were grown to 4–6 × 105 cells/ml prior to harvesting for extract preparation.
Extract preparations
Cells were pelleted by centrifugation at 5000 g for 5 min at room temperature. The cell pellet was resuspended in cold phosphate buffered saline (PBS) and collected by centrifugation at 5000 g for 5 min at room temperature. All subsequent steps were performed at 4 °C. The cells were suspended in three volumes of lysis buffer (50 mM Tris/HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100) and homogenized for 30 min at 4 °C, mixing the sample head-over-tail. Cellular debris was removed by high speed centrifugation (16,000 g for 10 min at 4 °C). The supernatant was subsequently dialyzed against buffer D (0.1 M KCl, 20 mM HEPES pH 7.9, 0.2 mM EDTA, 20% glycerol) and directly used for immunoprecipitation experiments. Nuclear extracts were prepared as described previously [36].
Immunoprecipitations
20 μl of anti-FLAG agarose or anti-HA agarose (Sigma Aldrich) was washed 3 times with 500 μl of IP buffer (20 mM HEPES/KOH pH 7.9, 100 mM KCl, 0.2 mM EDTA and 20% glycerol, Sigma Aldrich) supplemented with 0.1% Tween 20. A 50% mixture of whole cell extracts or nuclear extracts and IP buffer was added and incubated for 1 h to overnight mixing head-over-tail at 4 °C. Empty FLAG and HA expression vectors were transfected to generate the appropriate control extracts. The bead slurry was washed 3 times using IP buffer, before the purified protein was eluted by the addition of IP elution buffer (Sigma Aldrich) and subsequently dialyzed against buffer D. In the case of co-immunoprecipitation analyses, the proteins were eluted by incubating with Laemmli sample buffer at 95 °C prior to Western blot analyses. Protein concentrations were determined using the Bradford method (Bio-Rad).
Western blot analyses
A total of 20–50 μg of soluble protein or eluates from immunopurifications were separated using SDS–polyacrylamide gels, transferred to a nitrocellulose membrane (Bio-Rad) and incubated with blocking buffer (1× PBS with 0.1% Tween 20 and 5% skimmed milk) for 1 h at room temperature. Blocked membranes were washed twice with PBS-T (1× PBS with 0.1% Tween 20) for 5 min before incubation with a 1:2500 dilution of the primary antibody for 45 min at room temperature in PBS-T. Antibodies used were mouse monoclonal anti-HA (clone12CA5) antibody (Roche), mouse monoclonal anti-FLAG antibody (Sigma Aldrich), rabbit polyclonal anti-CBP antibody (Santa Cruz) and mouse polyclonal anti-RAR antibody (Millipore). Membranes were washed three times with PBS-T at room temperature for 5 min and subsequently incubated with a horseradish peroxidase–conjugated anti-mouse or anti-rabbit secondary antibody (Sigma Aldrich) at a dilution of 1:5000 in blocking buffer for 45 min at room temperature. After three washing steps of 5 min each with PBS-T at room temperature, detection of the signals was carried out using the ECL Plus Western Blotting Substrate (Pierce).
Glycosylase activity assays
Oligonucleotide primers used to generate 25-mer dsDNA containing either a central G:T or G:U mismatch basepair were as follows: 5′-GAT AGG TTC CAC G(G)G TAC TCG AAG C-3′ as the forward primer and 5′-GCT TCG AGT AC(T/U) CGT GGA ACC TAT G-3′ as the reverse primer (the nucleotides involved in the G:T(U) mismatches are written in brackets). DNA nicking assays were performed as described previously [37] on this 25-mer dsDNA and a 25-mer dsRNA containing a central canonical G:C pair as a control. Briefly, oligonucleotides corresponding to the complementary strand were labeled on the primary amine modified 3′-end with the AlexaFluor® 488 dye (Invitrogen) and oligonucleotide annealing was performed by heating 1 mM solutions for 5 min at 100 °C and cooling down the mixtures slowly to room temperature. TDG proteins were incubated at 0.5 μM final concentrations with dsDNA at 5 μM in 80 μl nicking buffer (25 mM HEPES/KOH pH 7.8, 1 mM EDTA, 1 mM DTT) at 37 °C. 20 μl aliquots were withdrawn at different incubation time points. DNA was precipitated in 70% ethanol solution containing 300 mM NaCl and then incubated with 0.01 N NaOH for 30 min at 50 °C. Oligonucleotides were separated by denaturing polyacrylamide gel electrophoresis and quantified using a GeneGenius bioimaging system (SynGene, Ozyme). Three independent replicates of glycosylase reactions were performed for each time point of the kinetic studies. The enzymatic turnover was normalized by the amount of protein used, as detected by the Bradford assay.
Microscale thermophoresis
Microscale thermophoresis (MST) technique capitalizes from the fact that molecules move in a temperature gradient from hot to cold (thermophoresis). This movement is impacted by complex formation, since molecules in a complex show an altered velocity. MST technique is used to measure the thermophoresis of a fluoresently labeled molecule (here YFP-CBP) subject to increasing concentrations of a binding partner (here immuno-purified TDG or TDG P65A). To obtain a series of successively decreasing TDG concentrations, immuno-purified HA-TDG samples (or HA-TDG P65A) were diluted up to 14 times with buffer D (1:1, 1:4, 1:8, etc.). The same amount of YFP-CBP containing cellular extract was added to each dilution in the presence or absence of immuno-purified FLAG-tagged RARα either in the presence or in the absence of RA. The reaction conditions were set to 20 mM MgCl2. MST experiments were conducted using a Monolith NT.115 (Nanotemper Technologies) as described previously [33,34] (laser-power 20%, laser-on time 60 s, LED-power 30%).
Luciferase reporter assays
Cells were transfected with a mixture of plasmids containing 1 μg 17-mer β-globin-Luc, 50 ng pRL SV40, 50 ng Gal4-hRARα 1.5 μg pSG5-hTDGwt or pSG5-hTDG P65A and 1.5 μg pSG5 DNA in 50 μl total volume to which 12 μl of Exgen 500 (Fermentas) was added according to the manufacturer’s instructions. After 6 h, the transfection reaction was stopped by adding 180 μl of DMEM to each well. Then cells were treated with atRA at 5 × 10−7 M for 24 h. After incubation, cells were washed with PBS and lysed with 20 μl of Passive Lysis Buffer (Promega) at room temperature for 15 min before reading. Luciferase activity was subsequently measured using GloMax®-96 Microplate Luminometer (Promega) according to the manufacturer’s instructions and normalized by protein amounts.
NMR studies
Full-length TDG wild-type and the mutant TDG P65A and their isolated N terminal domains for NMR studies were overexpressed as GST fusion proteins in Escherichia coli BL21 (DE3) strain. Protein expression and purification were performed as described previously [37,38]. NMR experiments were performed at 293 K on a Bruker DMX 600 MHz spectrometer (Bruker, Karlsruhe, Germany) equipped with a cryogenic triple resonance probe head. All 1H spectra were calibrated with 1 mM sodium 3-trimethylsilyl-d(3,3′,2,2′)-propionate as a reference. All 1H–15N HSQC spectra were recorded in an aqueous buffer composed of 100 mM NaiPO4 pH 6.6, 1 mM EDTA, 1 mM DTT and 5% D2O. 1H–15N HSQC spectra were recorded on 100 μM samples of 15N-labeled proteins with 128 scans per increment and 128 dummy scans, 128 points in the nitrogen dimension and 1024 points in the proton dimension.
Total RNA preparation
Total RNA from transfected or non-transfected cells was prepared using the RNeasy Midi Kit (Qiagen) as recommended by the manufacturer.
Transcriptome analyses
Microarray analyses, RNA amplification, labeling, hybridization and detection were performed following the protocols supplied by Applied Biosystems using the corresponding kits (Applied Biosystems). The microarray data were extracted using the Bioconductor limma package [39] and median normalized. Data quality was determined using a QC procedure [40]. Data were normalized using NeONORM with k = 0.02 [41–43]. Subtraction profiling was performed as described previously [44,45] using the CDS test [46]. Differentially expressed genes were classified using Ingenuity Pathway Analysis software to detect network- and pathway-enrichments. Transcriptome data were deposited in the public database MACE (http://mace.ihes.fr) using Accession Nos: 3167467256 (5-aza-dC treatment HEK293 cells), 2426124024 (RA treatment HEK293 cells), 2147989240 (CBP overexpression HEK293 cells), 2267526904 (RA treatment of TDG(TDGP65A)/CBP expressing HEK293 cells), 2586598264 (5-aza-dC treatment COS-7 cells), 2283559800 (RA treatment COS-7 cells), 2740738936 (CBP overexpression COS-7 cells), 2763807608 (RA treatment of TDG/CBP expressing HEK293 cells), 2549898104 (RA treatment of TDGP65A/CBP expressing HEK293 cells), 2901761912 (wild-type TDG overexpression in HEK293 cells) and 2890751864 (TDG P65A overexpression in HEK293 cells).
Authors’ contributions
HL has performed the majority of experiments; CS has performed the NMR studies; AA has performed the luciferase reporter assays. SM has supervised the study; AGB has designed and supervised the study; and SE has supervised the study, analyzed the data and written the manuscript. All authors read and approved the final manuscript.
Completing interests
The authors have no competing interests to declare.
Acknowledgements
SE is recipient of a post-doctoral fellowship of the Agence Nationale de recherches sur le SIDA et les hépatites virales (ANRS). This work was funded by the Centre National de la Recherche Scientifique (CNRS) and the Genopole Evry. The 600 MHz-NMR facility used in this study was funded by CNRS, the Région Nord-Pas de Calais (France), the University of Lille 1 and the Institut Pasteur de Lille.
Footnotes
Peer review under responsibility of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China.

Supplementary material
Comparison of CBP target genes with de novo methylation inhibition targets and RA-regulated genes A. Heatmap of a substraction profile of significantly (post hoc pFDR P < 0.01) regulated genes from cells overexpressing CBP, compared to mock-transfected controls. The number of genes (L, column 1) that are positively (red shading) or negatively (blue shading) regulated is shown together with the average over three independent biologic replicate signals for CBP overexpression (S, column 2) and mock control (S, column 3). The final column (column 4) represents the post hoc P values (P). B. Heatmap of a substraction profile for atRA-mediated regulation in biological replicates. Organization and coloring as in panel A. C. Heatmap of a substraction profile of significantly regulated genes from cells after 24 h of 5-aza-dC treatment compared to DMSO-treated controls. Organization and coloring as in panel A. D. Venn diagram for the overlap of genes regulated in a significant manner in all three conditions examined in A–C. The P values (hypergeometric distribution) for chance occurence of each overlap are indicated. E. Scatter plot of the overlap of methylation and atRA regulated genes, but also of the target genes, whose expression is affected in a significant manner in each single condition only reveals clear Pearson’s correlations (R). Genes with expression significantly affected under both conditions compared in each panel are indicated in red. Genes with expression significantly affected upon CBP overexpression only (blue), by RA treatment only (green) and by 5-aza-dC treatment only (brown) are also indicated in different colors. ∗∗∗P < 0.001. F. Scatter plot of the methylation and CBP regulated genes as in E. G. Scatter plot of the RA and CBP regulated genes as in E.
Kds for MST measurements The Kd values for MST experiments in Figure 3 were plotted as a graph. Compared to experiments with hTDG wt, Kd values were significantly increased when purified hTDG P65A were used for MST. Significant differences were indicated with # (compared to hTDG wt), $ (compared to hTDG wt in presence of RARα) and & (comapred to hTDG wt in presence of RARα and RA), respectively. #, $ and &, all P < 0.001.
References
- 1.Schär P., Fritsch O. DNA repair and the control of DNA methylation. Prog Drug Res. 2011;67:51–68. doi: 10.1007/978-3-7643-8989-5_3. [DOI] [PubMed] [Google Scholar]
- 2.Deem A.K., Li X., Tyler J.K. Epigenetic regulation of genomic integrity. Chromosoma. 2012;121:131–151. doi: 10.1007/s00412-011-0358-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ehrlich M., Zhang X.Y., Inamdar N.M. Spontaneous deamination of cytosine and 5-methylcytosine residues in DNA and replacement of 5-methylcytosine residues with cytosine residues. Mutat Res. 1990;238:277–286. doi: 10.1016/0165-1110(90)90019-8. [DOI] [PubMed] [Google Scholar]
- 4.Krokan H.E., Drabløs F., Slupphaug G. Uracil in DNA – occurrence, consequences and repair. Oncogene. 2001;21:8935–8948. doi: 10.1038/sj.onc.1205996. [DOI] [PubMed] [Google Scholar]
- 5.Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009;1:239–259. doi: 10.2217/epi.09.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peltomäki P. DNA mismatch repair and cancer. Mutat Res. 2001;488:77–85. doi: 10.1016/s1383-5742(00)00058-2. [DOI] [PubMed] [Google Scholar]
- 7.Um S., Harbers M., Benecke A., Pierrat B., Losson R., Chambon P. Retinoic acid receptors interact physically and functionally with the T:G mismatch-specific thymine-DNA glycosylase. J Biol Chem. 1998;273:20728–20736. doi: 10.1074/jbc.273.33.20728. [DOI] [PubMed] [Google Scholar]
- 8.Chambon P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996;10:940–954. [PubMed] [Google Scholar]
- 9.Hörlein A.J., Näär A.M., Heinzel T., Torchia J., Gloss B., Kurokawa R. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature. 1995;377:397–404. doi: 10.1038/377397a0. [DOI] [PubMed] [Google Scholar]
- 10.Chen J.D., Evans R.M. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. 1995;377:454–457. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- 11.Benecke A. Genomic plasticity and information processing by transcription coregulators. ComPlexUs. 2003;1:65–76. [Google Scholar]
- 12.Benecke A. Chromatin code, local non-equilibrium dynamics, and the emergence of transcription regulatory programs. Eur Phys J E Soft Matter. 2006;19:353–366. doi: 10.1140/epje/i2005-10068-8. [DOI] [PubMed] [Google Scholar]
- 13.Tini M., Benecke A., Um S.J., Torchia J., Evans R.M., Chambon P. Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol Cell. 2002;9:265–277. doi: 10.1016/s1097-2765(02)00453-7. [DOI] [PubMed] [Google Scholar]
- 14.Goodman R.H., Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- 15.Bannister A.J., Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- 16.Ogryzko V.V., Schiltz R.L., Russanova V., Howard B.H., Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- 17.Kamei Y., Xu L., Heinzel T., Torchia J., Kurokawa R., Gloss B. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- 18.Dietze E.C., Troch M.M., Bowie M.L., Yee L., Bean G.R., Seewaldt V.L. CBP/p300 induction is required for retinoic acid sensitivity in human mammary cells. Biochem Biophys Res Commun. 2003;302:841–848. doi: 10.1016/s0006-291x(03)00266-3. [DOI] [PubMed] [Google Scholar]
- 19.Hajkova P., Jeffries S.J., Lee C., Miller N., Jackson S.P., Surani M.A. Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science. 2010;329:78–82. doi: 10.1126/science.1187945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y.Q., Zhou P.Z., Zheng X.D., Walsh C.P., Xu G.L. Association of Dnmt3a and thymine DNA glycosylase links DNA methylation with base-excision repair. Nucleic Acids Res. 2007;35:390–400. doi: 10.1093/nar/gkl1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boland M.J., Christman J.K. Characterization of Dnmt3b:thymine-DNA glycosylase interaction and stimulation of thymine glycosylase-mediated repair by DNA methyltransferase(s) and RNA. J Mol Biol. 2008;379:492–504. doi: 10.1016/j.jmb.2008.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cortázar D., Kunz C., Selfridge J., Lettieri T., Saito Y., MacDougall E. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature. 2011;470:419–423. doi: 10.1038/nature09672. [DOI] [PubMed] [Google Scholar]
- 23.Cortellino S., Xu J., Sannai M., Moore R., Caretti E., Cigliano A. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146:67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tanaka Y., Naruse I., Hongo T., Xu M., Nakahata T., Maekawa T. Extensive brain hemorrhage and embryonic lethality in a mouse null mutant of CREB-binding protein. Mech Dev. 2000;95:133–145. doi: 10.1016/s0925-4773(00)00360-9. [DOI] [PubMed] [Google Scholar]
- 25.Vermot J., Niederreither K., Garnier J.M., Chambon P., Dollé P. Decreased embryonic retinoic acid synthesis results in a DiGeorge syndrome phenotype in newborn mice. Proc Natl Acad Sci U S A. 2003;100:1763–1768. doi: 10.1073/pnas.0437920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Christman J.K. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21:5483–5495. doi: 10.1038/sj.onc.1205699. [DOI] [PubMed] [Google Scholar]
- 27.Balmer J.E., Blomhoff R. Gene expression regulation by retinoic acid. J Lipid Res. 2002;43:1773–1808. doi: 10.1194/jlr.r100015-jlr200. [DOI] [PubMed] [Google Scholar]
- 28.Miftakhova R., Sandberg T., Hedblom A., Nevzorova T., Persson J.L., Bredberg A. DNA methylation in ATRA-treated leukemia cell lines lacking a PML-RAR chromosome translocation. Anticancer Res. 2012;32:4715–4722. [PubMed] [Google Scholar]
- 29.Lim J.S., Park S.H., Jang K.L. All-trans retinoic acid induces cellular senescence by up-regulating levels of p16 and p21 via promoter hypomethylation. Biochem Biophys Res Commun. 2011;412:500–505. doi: 10.1016/j.bbrc.2011.07.130. [DOI] [PubMed] [Google Scholar]
- 30.Woo Y.J., Jang K.L. All-trans retinoic acid activates E-cadherin expression via promoter hypomethylation in the human colon carcinoma HCT116 cells. Biochem Biophys Res Commun. 2012;425:944–949. doi: 10.1016/j.bbrc.2012.08.038. [DOI] [PubMed] [Google Scholar]
- 31.Gallinari P., Jiricny J. A new class of uracil-DNA glycosylases related to human thymine-DNA glycosylase. Nature. 1996;383:735–738. doi: 10.1038/383735a0. [DOI] [PubMed] [Google Scholar]
- 32.Smet-Nocca C., Wieruszeski J.M., Chaar V., Leroy A., Benecke A. The thymine-DNA glycosylase regulatory domain: residual structure and DNA binding. Biochemistry. 2008;47:6519–6530. doi: 10.1021/bi7022283. [DOI] [PubMed] [Google Scholar]
- 33.Wienken C.J., Baaske P., Rothbauer U., Braun D., Duhr S. Protein-binding assays in biological liquids using microscale thermophoresis. Nat Commun. 2010;1:100. doi: 10.1038/ncomms1093. [DOI] [PubMed] [Google Scholar]
- 34.Eilebrecht S., Wilhelm E., Benecke B.J., Bell B., Benecke A.G. HMGA1 directly interacts with TAR to modulate basal and Tat-dependent HIV transcription. RNA Biol. 2013;10:436–444. doi: 10.4161/rna.23686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Broderick P., Bagratuni T., Vijayakrishnan J., Lubbe S., Chandler I., Houlston R.S. Evaluation of NTHL1, NEIL1, NEIL2, MPG, TDG, UNG and SMUG1 genes in familial colorectal cancer predisposition. BMC Cancer. 2006;6:243. doi: 10.1186/1471-2407-6-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eilebrecht S., Brysbaert G., Wegert T., Urlaub H., Benecke B.J., Benecke A. 7SK small nuclear RNA directly affects HMGA1 function in transcription regulation. Nucleic Acids Res. 2011;39:2057–2072. doi: 10.1093/nar/gkq1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smet-Nocca C., Wieruszeski J.M., Léger H., Eilebrecht S., Benecke A. SUMO-1 regulates the conformational dynamics of thymine-DNA glycosylase regulatory domain and competes with its DNA binding activity. BMC Biochem. 2011;12:4. doi: 10.1186/1471-2091-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eilebrecht S., Smet-Nocca C., Wieruszeski J.M., Benecke A. SUMO-1 possesses DNA binding activity. BMC Res Notes. 2010;3:146. doi: 10.1186/1756-0500-3-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smyth G.K., Michaud J., Scott H.S. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics. 2005;21:2067–2075. doi: 10.1093/bioinformatics/bti270. [DOI] [PubMed] [Google Scholar]
- 40.Brysbaert G., Pellay F.X., Noth S., Benecke A. Quality assessment of transcriptome data using intrinsic statistical properties. Genomics Proteomics Bioinformatics. 2010;8:57–71. doi: 10.1016/S1672-0229(10)60006-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Noth S., Brysbaert G., Benecke A. Normalization using weighted negative second order exponential error functions (NeONORM) provides robustness against asymmetries in comparative transcriptome profiles and avoids false calls. Genomics Proteomics Bioinformatics. 2006;4:90–109. doi: 10.1016/S1672-0229(06)60021-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Noth S., Brysbaert G., Pellay F.X., Benecke A. High-sensitivity transcriptome data structure and implications for analysis and biologic interpretation. Genomics Proteomics Bioinformatics. 2006;4:212–229. doi: 10.1016/S1672-0229(07)60002-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Noth S., Benecke A. Avoiding inconsistencies over time and tracking difficulties in Applied Biosystems AB1700/Panther probe-to-gene annotations. BMC Bioinformatics. 2005;6:307. doi: 10.1186/1471-2105-6-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eilebrecht S., Benecke B.J., Benecke A. 7SK snRNA-mediated, gene-specific cooperativity of HMGA1 and P-TEFb. RNA Biol. 2011;8:1084–1093. doi: 10.4161/rna.8.6.17015. [DOI] [PubMed] [Google Scholar]
- 45.Eilebrecht S., Bécavin C., Léger H., Benecke B.J., Benecke A. HMGA1-dependent and independent 7SK RNA gene regulatory activity. RNA Biol. 2011;8:143–157. doi: 10.4161/rna.8.1.14261. [DOI] [PubMed] [Google Scholar]
- 46.Tchitchek N., Dzib J.F., Targat B., Noth S., Benecke A., Lesne A. CDS: a fold-change based statistical test for concomitant identification of distinctness and similarity in gene expression analysis. Genomics Proteomics Bioinformatics. 2012;10:127–135. doi: 10.1016/j.gpb.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparison of CBP target genes with de novo methylation inhibition targets and RA-regulated genes A. Heatmap of a substraction profile of significantly (post hoc pFDR P < 0.01) regulated genes from cells overexpressing CBP, compared to mock-transfected controls. The number of genes (L, column 1) that are positively (red shading) or negatively (blue shading) regulated is shown together with the average over three independent biologic replicate signals for CBP overexpression (S, column 2) and mock control (S, column 3). The final column (column 4) represents the post hoc P values (P). B. Heatmap of a substraction profile for atRA-mediated regulation in biological replicates. Organization and coloring as in panel A. C. Heatmap of a substraction profile of significantly regulated genes from cells after 24 h of 5-aza-dC treatment compared to DMSO-treated controls. Organization and coloring as in panel A. D. Venn diagram for the overlap of genes regulated in a significant manner in all three conditions examined in A–C. The P values (hypergeometric distribution) for chance occurence of each overlap are indicated. E. Scatter plot of the overlap of methylation and atRA regulated genes, but also of the target genes, whose expression is affected in a significant manner in each single condition only reveals clear Pearson’s correlations (R). Genes with expression significantly affected under both conditions compared in each panel are indicated in red. Genes with expression significantly affected upon CBP overexpression only (blue), by RA treatment only (green) and by 5-aza-dC treatment only (brown) are also indicated in different colors. ∗∗∗P < 0.001. F. Scatter plot of the methylation and CBP regulated genes as in E. G. Scatter plot of the RA and CBP regulated genes as in E.
Kds for MST measurements The Kd values for MST experiments in Figure 3 were plotted as a graph. Compared to experiments with hTDG wt, Kd values were significantly increased when purified hTDG P65A were used for MST. Significant differences were indicated with # (compared to hTDG wt), $ (compared to hTDG wt in presence of RARα) and & (comapred to hTDG wt in presence of RARα and RA), respectively. #, $ and &, all P < 0.001.