

Abstract
The vast majority of multi-exon genes in higher eukaryotes are alternatively spliced and changes in alternative splicing (AS) can impact gene function or cause disease. High-throughput RNA sequencing (RNA-seq) has become a powerful technology for transcriptome-wide analysis of AS, but RT-PCR still remains the gold-standard approach for quantifying and validating exon splicing levels. We have developed PrimerSeq, a user-friendly software for systematic design and visualization of RT-PCR primers using RNA-seq data. PrimerSeq incorporates user-provided transcriptome profiles (i.e., RNA-seq data) in the design process, and is particularly useful for large-scale quantitative analysis of AS events discovered from RNA-seq experiments. PrimerSeq features a graphical user interface (GUI) that displays the RNA-seq data juxtaposed with the expected RT-PCR results. To enable primer design and visualization on user-provided RNA-seq data and transcript annotations, we have developed PrimerSeq as a stand-alone software that runs on local computers. PrimerSeq is freely available for Windows and Mac OS X along with source code at http://primerseq.sourceforge.net/. With the growing popularity of RNA-seq for transcriptome studies, we expect PrimerSeq to help bridge the gap between high-throughput RNA-seq discovery of AS events and molecular analysis of candidate events by RT-PCR.
Keywords: Alternative splicing, RNA-seq, Primer design, Transcriptome, Visualization
Introduction
Alternative splicing (AS) is a prevalent mechanism of gene regulation in higher eukaryotes [1]. AS plays an important role in both normal biological processes [2] and disease [3]. In recent years, high-throughput RNA sequencing (RNA-seq) has become a powerful and popular technology for global analysis of AS [4]. From the massive amount of RNA-seq reads, one can discover novel splicing events, quantify the usage level of alternatively spliced exons in any RNA sample of interest, and identify genome-wide changes in AS between different biological states. However, RT-PCR is still regarded as the most reliable and standard approach to quantify and validate exon splicing levels [5]. In fact, researchers customarily perform RT-PCR validation of AS events discovered from RNA-seq data prior to downstream functional studies.
A widely used measure of AS is the percent-spliced-in (PSI, or ψ) metric, which measures the percent inclusion level of an alternatively spliced exon (or splice site) in the final mRNA products [4]. In an RNA-seq project, the PSI value of an AS event can be first estimated from RNA-seq data using software like mixture-of-isoforms (MISO) [4] or multivariate analysis of transcript splicing (MATS) [6] and then validated independently by RT-PCR. However, the design of RT-PCR primers for AS analysis is typically a time-consuming step that requires tedious manual operations. Software that allow input of a template sequence for primer design like Primer3 [7] and associated interfaces Primer3Web, Primer3Plus and BatchPrimer3 [8] can theoretically design primers for any AS event of interest. However, it is left to the user to manually extract sequences for primer design, which is time-consuming and error-prone. This is particularly challenging for RNA-seq projects, where researchers may need to validate tens or even hundreds of AS events identified from the transcriptome-wide AS analysis (for example, see [9,10]). Consequently validation of AS from big RNA-seq data has become a major bottleneck between high-throughput discovery of AS events and in-depth analysis of molecular function and regulation.
It should also be noted that the repertoire of expressed genes and mRNA isoforms is dynamically regulated, while current tools and databases for RT-PCR primer design use static (pre-defined) gene and transcript annotations and do not incorporate transcriptome profiles for the specific sample(s) of interest. Primer databases like GETPrime [11], RTPrimerDB [12], PrimerBank [13] and qPrimerDepot [14] only contain primers for a restricted set of species and are built on pre-defined gene annotations. Most primer design tools (e.g., PerlPrimer [15] and QuantPrime [16]) or primer databases (mentioned above) focus on gene expression and occasionally transcript expression rather than AS events. In general, there is a lack of primer design tools or databases for AS analysis, with a few exceptions being GETPrime (gene and transcript specific) [11] and RASE (alternative splicing) [17]. RASE is the method most specifically designed for AS analysis, but its web interface only works with human genes and requires time-consuming manual input of sequences. It should also be noted that RNA-seq is a flexible technology, which can be applied to any organism of interest. In fact, researchers have used RNA-seq to study AS in a wide variety of organisms such as honey bee [18], silkworm [19], Plasmodium falciparum [20] and frog [21]. Additionally, computational tools such as Cufflinks [22] and Scripture [23] can be used to construct transcript annotations de novo from RNA-seq data aligned to the genome. Therefore, an ideal primer design tool for AS analysis should have the flexibility to work with user-provided RNA-seq data on a diverse range of organisms, instead of being restricted to a small set of species and pre-defined transcript annotations.
Here we present PrimerSeq, a user-friendly stand-alone software for systematic design and visualization of RT-PCR primers for AS analysis. PrimerSeq has a graphical user interface (GUI) and “one-click” type installation for convenient access by a wide range of users. It utilizes user-provided RNA-seq data to define splicing patterns, estimate exon inclusion levels (PSI, or ψ), select suitable regions for placement of RT-PCR primers and visualize RNA-seq data along with highlighting expected RT-PCR results. Users can conveniently compare the graphical output of PrimerSeq to their RT-PCR experimental result.
Methods
PrimerSeq workflow and algorithm
PrimerSeq designs RT-PCR primers for AS analysis. The design process can incorporate the transcriptome profiles of the samples of interest through user-provided RNA-seq data files, or only utilize pre-defined gene and transcript annotations. As shown in the flow diagram (Figure 1), the input to PrimerSeq includes a genome sequence file (FASTA), a gene and transcript annotation file (GTF), mapped RNA-seq reads (BAM, recommended but optional) and a list of exon coordinates representing the events of interest. Visualizing read density also requires a BigWig file, although this visualization step is optional. For each AS event, PrimerSeq attempts to place a pair of forward and reverse PCR primers on suitable flanking exons. Such flanking exons can be specified by users in the input. Alternatively, PrimerSeq can automatically choose appropriate flanking exons by finding the nearest suitable flanking exons whose inclusion levels (PSI) are above a user-defined threshold (95% by default), a procedure that typically finds constitutive exons. PrimerSeq then runs Primer3 [7] to perform primer design on the selected flanking exons. Through configuration options, users can fully customize the parameters for primer design, such as the size range of the PCR products.
Figure 1.
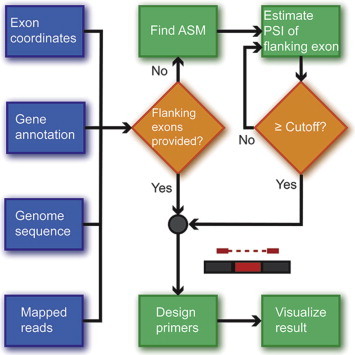
The flow diagram of PrimerSeq PrimerSeq flow diagram designates inputs as blue, computations as green and decisions as orange. If flanking exons are specified by the user, PrimerSeq will immediately design primers. If not specified, PrimerSeq will first identify the alternative splicing module (ASM) containing the target exon and then iteratively search for the closest flanking exons above a user-defined PSI (ψ) cutoff. Results are subsequently visualized through plotting the RNA-seq data juxtaposed with the expected RT-PCR results, which include estimated ψ values for the target exon. Visualizing read density, an optional feature, requires a BigWig file. PSI stands for percent-spliced-in.
As part of the primer design procedure, PrimerSeq utilizes the biconnected components algorithm [24] as a generalized definition of AS events called alternative splicing modules (ASMs). Conceptually, if we consider the transcript structure of an alternatively spliced gene as a directed acyclic graph (i.e., “splice graph” [25,26]), each ASM represents a subset of the splice graph, where the transcripts diverge from a single point and then converge back to a single point. In the case of a simple exon skipping (ES) event, the ASM includes two splice forms corresponding to the exon inclusion and skipping isoforms. To understand this, it is clear for an ES event that an upstream constitutive exon is used by both isoforms before the skipped exon of interest. Similarly, a downstream constitutive exon is also utilized for both the skipping and inclusion isoforms. The two isoforms differ by whether the middle exon is utilized (included) or skipped. Therefore a simple ES event fits our definition of an ASM, because an ES event has two paths that start from a common origin of the upstream constitutive exon and reconvene at the downstream constitutive exon. However, an ASM could contain more than two splice forms if multiple alternative splicing events are coupled. Using this generalized definition of AS, PrimerSeq can design primers for all types of AS events, such as exon skipping, alternative splice sites and mutually exclusive exons. For a more technically detailed description, please see the technical manual at http://primerseq.sourceforge.net/technical_manual.pdf. Ideally, primers should be placed on constitutive exons flanking an ASM, so the RT-PCR analysis can obtain the PSI measurements for all splice forms within the ASM. If users do not specify flanking exons, PrimerSeq uses RNA-seq read counts to estimate relative isoform abundance using an expectation maximization (EM) algorithm [27] (also see the technical manual of PrimerSeq at http://primerseq.sourceforge.net/technical_manual.pdf), then places primers on flanking exons with constitutive splicing, or at minimum exons with high inclusion levels (PSI > 95% by default). Regardless of whether flanking exons are specified by the user or selected by PrimerSeq, the abundance estimates from the EM algorithm are used to calculate PSI estimates for the target AS exon. Such estimates will be used for predicting the relative ratios of the RT-PCR products in the subsequent visualization step.
Novel isoforms supported by RNA-seq reads can optionally be considered in the design process. If this option is enabled (disabled by default), our current algorithm adds novel exon-exon junctions detected from the RNA-seq data to the supplied transcript annotations. For a more refined control of novel isoforms, or for organisms with poor transcript annotations, users are suggested to perform novel transcript assembly using tools like Cufflinks [22] and then load the resulting gene and transcript annotations (GTF) into PrimerSeq.
Following primer design, PrimerSeq visualizes RNA-seq data along the expected RT-PCR results. Specifically, PrimerSeq can automatically generate figures that display the expected sizes and relative ratios of the RT-PCR products, together with the RNA-seq read density profile along the transcripts. Displaying the read density plot requires a BigWig file. The entire AS module will be displayed, if flanking exons are selected by PrimerSeq, i.e., not specified by the user. PrimerSeq can display figures within the GUI and save the results as a static web page (HTML). Additionally, PrimerSeq provides links to UCSC In-Silico PCR [28] for users to inspect potential off-target amplifications.
Implementation and availability
PrimerSeq is mainly written in Python using the wxPython library (http://wxpython.org/) to create a GUI. The identification of AS events using the biconnected components algorithm was performed using the NetworkX library [29] in Python. The Java libraries SAM-JDK v1.77 (http://picard.sourceforge.net/) and BigWig API r39 (revision 39, https://code.google.com/p/bigwig/) were used to enhance the performance of handling RNA-seq data and read density files, respectively. PrimerSeq uses standard file formats for gene and transcript annotations (GTF), RNA-seq data (SAM/BAM), genome sequence (FASTA) and read density (BigWig). BAM, FASTA and BigWig files are indexed, which provides significant speed improvements for handling large datasets. Primer3 v2.3.4 [7] is used to perform primer design after the appropriate exonic sequences are retrieved from the FASTA file.
The stand-alone PrimerSeq software is free and open to all users and there is no login requirement to download the software. PrimerSeq is available as a Windows installer and a Mac OS X binary on SourceForge at http://primerseq.sourceforge.net/. Source code for PrimerSeq is hosted on GitHub at https://github.com/ctokheim/PrimerSeq. The technical manual of PrimerSeq which includes a detailed description of nomenclature and algorithms can be found at http://primerseq.sourceforge.net/technical_manual.pdf. User tutorials can also be found on the PrimerSeq website at http://primerseq.sf.net/getting_started.html and http://primerseq.sf.net/user_tutorial.html.
RT-PCR validation of PrimerSeq design
Total RNA samples from human heart and testes were purchased from Applied Biosystems (Foster City, CA, USA) and Clontech (Mountain View, CA, USA), respectively. RT-PCR was carried out and 5% TBE-PAGE gel was used for resolving PCR products as described before [30].
Results
As an example, we compared the splicing profiles of human heart and testes using RNA-seq data from the Illumina Human Body Map 2.0 Project (NCBI GEO Accession No. GSE30611). From the top 100 differential AS events detected by MATS (version 3.0.6.beta) [6], five were chosen at random for primer design by PrimerSeq and the efficacy of the primers was evaluated by RT-PCR experiments (see Table S1 for details regarding the RT-PCR primers). All five AS events had successful primer design as evidenced by target amplification during RT-PCR. Figure 2 illustrates the AS event in the gene TJP1 encoding tight junction protein 1. The PSI estimates matched well between RNA-seq (Figure 2A) and RT-PCR (Figure 2B). In another gene, HNRPLL encoding heterogeneous nuclear ribonucleoprotein L-like, the RNA-seq data suggested a novel isoform and this was confirmed by RT-PCR (Figure S1). In all five events, the RT-PCR primers successfully amplified the target regions and the sizes of PCR products were consistent with the design results from PrimerSeq (Figure 1 and Figures S1–4).
Figure 2.
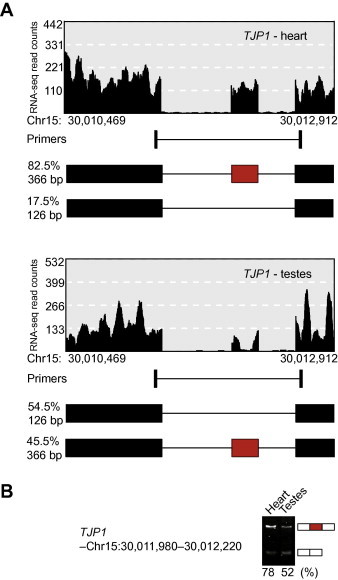
Example result from PrimerSeq Example automatically generated figures (A) for a selected AS event in the TJP1 gene and corresponding RT-PCR results (B) with the ψ values displayed. The RNA-seq data are from heart and testes in the Illumina Human Body Map 2.0 project [Gene Expression Omnibus (GEO) Accession No. GSE30611]. TJP1, tight junction protein 1; Chr, chromosome.
Discussion
To the best of our knowledge, PrimerSeq is the only software that incorporates RNA-seq data in the design and visualization of RT-PCR primers for AS analysis. This has several advantages. By incorporating RNA-seq data, we ensure that the primers will be placed on flanking exons with constitutive splicing (or at minimum, high inclusion levels of close to 100%) in the sample(s) of interest. Second, we produce figures based on the RNA-seq data to illustrate the expected results of the RT-PCR experiments (Figure 2), so that researchers can easily compare the RT-PCR results to RNA-seq predictions. In addition, novel isoforms not present in the transcript annotations can be identified and visualized before performing RT-PCR (Figure S1).
We have chosen to develop PrimerSeq as a stand-alone software that runs on local computers, as opposed to a web-based tool. This is important given the typical size of RNA-seq data files and the goal of working with a diverse range of organisms. For example, the RNA-seq BAM files of human heart and testes in the Illumina Body Map 2.0 dataset [Gene Expression Omnibus (GEO) Accession No. GSE30611] are 3.5 GB and 2.7 GB, respectively. Due to computational costs and network speeds, it is impractical to process and manipulate such big RNA-seq data files through typical web-based tools. By implementing PrimerSeq as a stand-alone software, we have the flexibility to utilize any user-provided RNA-seq data and transcript annotations. For organisms with poor transcript annotations, we suggest researchers to use de novo RNA-seq transcript assembly tools, such as Cufflinks [22] and Scripture [23], to generate transcript annotations from their RNA-seq data, which can then be loaded into PrimerSeq for primer design. With the growing popularity of RNA-seq for transcriptome studies, we expect PrimerSeq to help bridge the gap between high-throughput RNA-seq discovery of AS events and molecular validation of candidate events by RT-PCR.
Authors’ contributions
YX and CT conceived the idea of PrimerSeq. CT implemented PrimerSeq as a software package. JWP helped with implementation of RNA-seq data handling and alternative splicing analysis. CT and YX wrote the manuscript. All authors had final approval of the manuscript.
Competing interests
The authors declare no competing financial interests.
Acknowledgements
This work was supported by the National Institutes of Health of USA (Grant No. R01GM088342) awarded to YX. We thank Zhixiang Lu for testing PrimerSeq and performing RT-PCR experiments.
Footnotes
Peer review under responsibility of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China.

Supplementary material
PrimerSeq design result and RT-PCR analysis of an AS event in HNRPLL Automatically generated figures by PrimerSeq based on RNA-seq data for human heart (A) and testes (B) along with the RT-PCR result (C) for HNRPLL. The RT-PCR result of testes supports a novel isoform (the 114 bp PCR product) not present in the UCSC Known Genes annotation. HNRPLL, heterogeneous nuclear ribonucleoprotein L-like.
PrimerSeq design result and RT-PCR analysis of an AS event in VCL Automatically generated figures by PrimerSeq based on RNA-seq data for human heart (A) and testes (B) along with the RT-PCR result (C) for VCL. An alternative 3’ splice site event for the upstream flanking exon results in displaying two 113 bp isoforms in (A) and (B). VCL, vinculin.
PrimerSeq design result and RT-PCR analysis of an AS event in TRIP10 Automatically generated figures by PrimerSeq based on RNA-seq data for human heart (A) and testes (B) along with the RT-PCR result (C) for TRIP10. The RT-PCR primers successfully amplified the target regions and the results were mostly consistent with the RNA-seq data. However, a 127 bp product in the heart according to the RNA-seq data was not supported by RT-PCR. Asterisk denotes an unspecific PCR band. TRIP10, thyroid hormone receptor interactor 10.
PrimerSeq design result and RT-PCR analysis of an AS event in NF2 Automatically generated figures by PrimerSeq based on RNA-seq data for human heart (A) and testes (B) along with the RT-PCR result (C) for NF2. The intended target exon (in the 290 bp PCR band) had alternative splice sites which resulted in two additional predicted PCR bands of 275 bp and 316 bp. RT-PCR of the heart sample showed two prominent bands at 275 bp and 290 bp which matched the two most abundant isoforms according to RNA-seq. Testes had a prominent PCR band at 230 bp corresponding to the exon skipping isoform, consistent with the RNA-seq data. NF2, neurofibromin 2.
Five randomly-chosen AS events for RT-PCR primer design by PrimerSeq.
References
- 1.Nilsen T.W., Graveley B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalsotra A., Cooper T.A. Functional consequences of developmentally regulated alternative splicing. Nat Rev Genet. 2011;12:715–729. doi: 10.1038/nrg3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang G.S., Cooper T.A. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007;8:749–761. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- 4.Katz Y., Wang E.T., Airoldi E.M., Burge C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods. 2010;7:1009–1015. doi: 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Venables J.P., Klinck R., Koh C., Gervais-Bird J., Bramard A., Inkel L. Cancer-associated regulation of alternative splicing. Nat Struct Mol Biol. 2009;16:670–676. doi: 10.1038/nsmb.1608. [DOI] [PubMed] [Google Scholar]
- 6.Shen S., Park J.W., Huang J., Dittmar K.A., Lu Z.X., Zhou Q. MATS: a Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic Acids Res. 2012;40:e61. doi: 10.1093/nar/gkr1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Untergasser A., Cutcutache I., Koressaar T., Ye J., Faircloth B.C., Remm M. Primer3–new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.You F.M., Huo N., Gu Y.Q., Luo M.C., Ma Y., Hane D. BatchPrimer3: a high throughput web application for PCR and sequencing primer design. BMC Bioinformatics. 2008;9:253. doi: 10.1186/1471-2105-9-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dittmar K.A., Jiang P., Park J.W., Amirikian K., Wan J., Shen S. Genome-wide determination of a broad ESRP-regulated posttranscriptional network by high-throughput sequencing. Mol Cell Biol. 2012;32:1468–1482. doi: 10.1128/MCB.06536-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen S., Lin L., Cai J.J., Jiang P., Kenkel E.J., Stroik M.R. Widespread establishment and regulatory impact of Alu exons in human genes. Proc Natl Acad Sci U S A. 2011;108:2837–2842. doi: 10.1073/pnas.1012834108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gubelmann C., Gattiker A., Massouras A., Hens K., David F., Decouttere F. GETPrime: a gene- or transcript-specific primer database for quantitative real-time PCR. Database. 2011;2011:bar040. doi: 10.1093/database/bar040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lefever S., Vandesompele J., Speleman F., Pattyn F. RTPrimerDB: the portal for real-time PCR primers and probes. Nucleic Acids Res. 2009;37:D942–D945. doi: 10.1093/nar/gkn777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X., Spandidos A., Wang H., Seed B. PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res. 2012;40:D1144–D1149. doi: 10.1093/nar/gkr1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cui W., Taub D.D., Gardner K. QPrimerDepot: a primer database for quantitative real time PCR. Nucleic Acids Res. 2007;35:D805–D809. doi: 10.1093/nar/gkl767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marshall O.J. PerlPrimer: cross-platform, graphical primer design for standard, bisulphite and real-time PCR. Bioinformatics. 2004;20:2471–2472. doi: 10.1093/bioinformatics/bth254. [DOI] [PubMed] [Google Scholar]
- 16.Arvidsson S., Kwasniewski M., Riano-Pachon D.M., Mueller-Roeber B. QuantPrime – a flexible tool for reliable high-throughput primer design for quantitative PCR. BMC Bioinformatics. 2008;9:465. doi: 10.1186/1471-2105-9-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brosseau J.P., Lucier J.F., Lapointe E., Durand M., Gendron D., Gervais-Bird J. High-throughput quantification of splicing isoforms. RNA. 2010;16:442–449. doi: 10.1261/rna.1877010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li-Byarlay H., Li Y., Stroud H., Feng S., Newman T.C., Kaneda M. RNA interference knockdown of DNA methyl-transferase 3 affects gene alternative splicing in the honey bee. Proc Natl Acad Sci U S A. 2013;110:12750–12755. doi: 10.1073/pnas.1310735110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shao W., Zhao Q.Y., Wang X.Y., Xu X.Y., Tang Q., Li M. Alternative splicing and trans-splicing events revealed by analysis of the Bombyx mori transcriptome. RNA. 2012;18:1395–1407. doi: 10.1261/rna.029751.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sorber K., Dimon M.T., DeRisi J.L. RNA-Seq analysis of splicing in Plasmodium falciparum uncovers new splice junctions, alternative splicing and splicing of antisense transcripts. Nucleic Acids Res. 2011;39:3820–3835. doi: 10.1093/nar/gkq1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amin N.M., Tandon P., Osborne Nishimura E., Conlon F.L. RNA-seq in the tetraploid Xenopus laevis enables genome-wide insight in a classic developmental biology model organism. Methods. 2014;66:398–409. doi: 10.1016/j.ymeth.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guttman M., Garber M., Levin J.Z., Donaghey J., Robinson J., Adiconis X. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol. 2010;28:503–510. doi: 10.1038/nbt.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sacomoto G.A., Kielbassa J., Chikhi R., Uricaru R., Antoniou P., Sagot M.F. KISSPLICE: de-novo calling alternative splicing events from RNA-seq data. BMC Bioinformatics. 2012;13:S5. doi: 10.1186/1471-2105-13-S6-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xing Y., Resch A., Lee C. The multiassembly problem: reconstructing multiple transcript isoforms from EST fragment mixtures. Genome Res. 2004;14:426–441. doi: 10.1101/gr.1304504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heber S., Alekseyev M., Sze S.H., Tang H., Pevzner P.A. Splicing graphs and EST assembly problem. Bioinformatics. 2002;18:S181–S188. doi: 10.1093/bioinformatics/18.suppl_1.s181. [DOI] [PubMed] [Google Scholar]
- 27.Xing Y., Yu T., Wu Y.N., Roy M., Kim J., Lee C. An expectation-maximization algorithm for probabilistic reconstructions of full-length isoforms from splice graphs. Nucleic Acids Res. 2006;34:3150–3160. doi: 10.1093/nar/gkl396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuhn R.M., Haussler D., Kent W.J. The UCSC genome browser and associated tools. Brief Bioinform. 2013;14:144–161. doi: 10.1093/bib/bbs038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hagberg AA, Schult DA, Swart PJ. Exploring network structure, dynamics, and function using networkX. In: Proceedings of the 7th Python in science conference; 2008. p. 11–5.
- 30.Lin L., Shen S., Tye A., Cai J.J., Jiang P., Davidson B.L. Diverse splicing patterns of exonized Alu elements in human tissues. PLoS Genet. 2008;4:e1000225. doi: 10.1371/journal.pgen.1000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PrimerSeq design result and RT-PCR analysis of an AS event in HNRPLL Automatically generated figures by PrimerSeq based on RNA-seq data for human heart (A) and testes (B) along with the RT-PCR result (C) for HNRPLL. The RT-PCR result of testes supports a novel isoform (the 114 bp PCR product) not present in the UCSC Known Genes annotation. HNRPLL, heterogeneous nuclear ribonucleoprotein L-like.
PrimerSeq design result and RT-PCR analysis of an AS event in VCL Automatically generated figures by PrimerSeq based on RNA-seq data for human heart (A) and testes (B) along with the RT-PCR result (C) for VCL. An alternative 3’ splice site event for the upstream flanking exon results in displaying two 113 bp isoforms in (A) and (B). VCL, vinculin.
PrimerSeq design result and RT-PCR analysis of an AS event in TRIP10 Automatically generated figures by PrimerSeq based on RNA-seq data for human heart (A) and testes (B) along with the RT-PCR result (C) for TRIP10. The RT-PCR primers successfully amplified the target regions and the results were mostly consistent with the RNA-seq data. However, a 127 bp product in the heart according to the RNA-seq data was not supported by RT-PCR. Asterisk denotes an unspecific PCR band. TRIP10, thyroid hormone receptor interactor 10.
PrimerSeq design result and RT-PCR analysis of an AS event in NF2 Automatically generated figures by PrimerSeq based on RNA-seq data for human heart (A) and testes (B) along with the RT-PCR result (C) for NF2. The intended target exon (in the 290 bp PCR band) had alternative splice sites which resulted in two additional predicted PCR bands of 275 bp and 316 bp. RT-PCR of the heart sample showed two prominent bands at 275 bp and 290 bp which matched the two most abundant isoforms according to RNA-seq. Testes had a prominent PCR band at 230 bp corresponding to the exon skipping isoform, consistent with the RNA-seq data. NF2, neurofibromin 2.
Five randomly-chosen AS events for RT-PCR primer design by PrimerSeq.