

Abstract
The characterization of the interacting behaviors of complex biological systems is a primary objective in protein–protein network analysis and computational biology. In this paper we present FunMod, an innovative Cytoscape version 2.8 plugin that is able to mine undirected protein–protein networks and to infer sub-networks of interacting proteins intimately correlated with relevant biological pathways. This plugin may enable the discovery of new pathways involved in diseases. In order to describe the role of each protein within the relevant biological pathways, FunMod computes and scores three topological features of the identified sub-networks. By integrating the results from biological pathway clustering and topological network analysis, FunMod proved to be useful for the data interpretation and the generation of new hypotheses in two case studies.
Keywords: Systems biology, Network analysis, Pathway analysis, Network modules, Cytoscape, Budd–Chiari syndrome
Introduction
Systems biology broadly uses networks to model and discover emerging properties among genes, proteins and other relevant biomolecules. Theoretical studies have indicated that biological networks share many features with other types of networks, as computer or social networks [1]. Therefore, biological network analyses allow the application of mathematical and computational methods of the graph theory to biological studies [2]. The computational analysis of biological networks has therefore become increasingly useful to mine the complex cellular processes and signaling pathways [3]. Many types of biological networks exist, depending on the information associated with their nodes and edges. In general, biological networks can be classified as directed and undirected networks [4]. In directed networks, the nodes are molecules and edges represent causal biological interactions, such as the transcription and translation regulations [5]. In contrast, in undirected networks, an edge indicates a shared property, such as the sequence similarity [6], gene co-expression [7], protein–protein interaction [8], or the term co-occurrence in the scientific literature [9–11]. In order to extract relevant biological implication from undirected networks, which are also called informative networks [12], it is useful to complement the topological information with the independent biological information retrieved from Gene Ontology (GO) and pathway databases. Often the goal is to identify densely-interconnected areas and correlate them with a specific biological function [13,14]. Several algorithms and bioinformatic tools have been proposed for partitioning the network into structural modules, or for clustering sub-network modules within an informative network [15,16]. Researchers in Bioinformatics developed Cytoscape plugins [17] to mine functional modules in a varieties of network types, such as Clust&See [18], clusterMaker [19], CyClus3D [20], GLay [21] and Enrichment Map [22]. These plugins mainly function on the basis of topological properties. The groups of highly-interconnected nodes may form clusters based on a “first topological clustering” strategy, which aimed at partitioning complex networks into modules. The biological functions are then assigned assuming that members within each sub-network shared a similar biological function [23]. However, clusters are identified solely on the basis of the topology. Therefore, the possibility of co-occurrence events cannot be ruled out using these methods. Moreover, these strategies are heavily influenced by the topological structure of the network itself, and the way that the network is constructed [24].
In practice, the connectivity of the informative networks is established by experimental methods, which can lead to sampling of a subnet of the real biological network [25,26]. Often, biases in the sampling strategies lead to apparent scale-free topologies, which do not reflect the actual complete network topology. As an alternate to the “first topological clustering” methods, some authors used a “first pathway enrichment” strategy [27], which enables analyzing gene networks and extracting functional modules starting from the biological enrichment analysis. Several Cytoscape plugins thus have been implemented owning to this strategy, such as BiNGO [28], ClueGO [29], ClusterViz [30], JEPETTO [31] and Reactome [32].
BiNGO and ClueGO are widely used tools to determine which biological functions are statistically over-represented in a list of genes or a network. These plugins offer the possibility to calculate enrichment by using different statistical algorithms. However, they do not evaluate the connectivity between genes, and do not take into account the possibility that nodes spread in the network could still represent a significant biological function.
A recent focus of bioinformatics has been to develop the computational tools that are able to mine the connectivity of gene networks and uncover the sets of molecules that participate in a common biological function [33]. Reactome is established based on an un-weighted human protein functional interaction network and the functional interaction score was calculated with Pearson correlation coefficients among all gene pairs in the biological database. The weighted network was clustered into a series of gene interaction modules using the Markov clustering algorithm. Each module of the Reactome consists of a set of genes that are both connected in the protein functional interaction network and highly-correlated in biological databases [34]. This approach, however, does not consider the topology or the connectivity of gene interaction modules.
JEPETTO identifies functional associations between genes and pathways using protein interaction networks and topological analyses. Although JEPPETTO combines network analysis with functional enrichment, this tool requires a list of genes in input and the selection of a database of interest from which the reference gene sets will be extracted.
Reactome and JEPETTO are based on an internal gene interaction database, in which the users are not able to filter this pre-defined database or to analyze their own protein informative networks. Nowadays there are a great number of tools able to produce undirected protein networks from a user-defined query, such as protein–protein interaction network (STRING, BioGRID, etc.), co-expression network (COXPRESdb) [35] and co-occurrence network (ProteinQuest) [36].
To the best of our knowledge, there is no software available to identify sub-networks of genes that are highly connected and belong to the same biological pathways. Moreover, the aforementioned plugins do not allow the analysis of user-defined undirected networks, such as protein–protein interaction, functional association, gene co-expression and literature co-occurrence.
In this work we developed a new approach using a “first biological assignment” strategy and we implemented our method as a Cytoscape plugin, called FunMod. According to the principle that interacting proteins drive common biological processes, FunMod analyzes informative networks combing topological and biological information to identify and extract sub-network modules in proteins that are involved in the same biological pathway. Moreover, in order to describe the shape of the modules and discriminate the proteins’ topological proprieties within the single sub-network, FunMod analyzes sub-network features by using three topological scores. The sub-networks that are statistically overrepresented can act as building blocks of complex informative networks and carry out a specific biological function [24]. Assessment of the sub-network topological proprieties and shapes can consequently be used for the gene ranking in the context of a specific research domain, such as a disease [35].
In the present study, FunMod proves to be a useful method for identifying functional sub-networks in an informative protein network, exploring biomedical information and inferring automated functional hypotheses between an user defined protein–protein network [36]. FunMod is unique at its capability of analyzing user-defined undirected networks, in order to provide more realistic models that incorporate information from certain cellular types, developmental states and/or disease conditions.
Methods
FunMod analyzes the informative protein network displayed in the Cytoscape Main Network View window. The plugin supports many standard protein dictionaries, the protein nodes can be identified (node ID) by six different dictionaries: Entrez Gene ID, Ensembl, Official Symbol (HGNC symbol), NCBI Reference Sequence, UniGene and Uniprot Entry Name.
FunMod iteratively selects all edges of the network and assigns a functional annotation to an edge when two linked nodes are annotated in the same biological group or pathway in the ConsensusPathDB (DB) database [37]. In other words, FunMod considers an order pair network G = (V, E), where vertices (V, nodes) join with edges (E), and collects, for each ConsensusPathDB pathway, pairs of linked nodes to model a functional sub-network Gp = (Vp, Ep), where Vp ⊆ ends on the pathways and Ep ∈ E. FunMod performs a global enrichment analysis screening the pathways whose proteins are co-annotated with their neighbors. Accordingly, all the pathways identified by FunMod are also significant in a global enrichment analysis, since the connected nodes are a fraction of nodes in the network. So pathways enriched in a sub-network are also enriched in the global network, but only a few pathways enriched in the global network are enriched in a cluster. Our algorithm thus could find the pathways enriched in the global network and whose proteins are densely connected within a sub-network.
FunMod extracts all pairs of nodes annotated for the same pathway in a new sub-network. Subsequently, FunMod tests the statistical significance and calculates the topological properties of the sub-network to identify the sub-networks that are statistically enriched in biological functions and that exhibiting interesting topological features. The statistical significance of the sub-network is determined by performing a hypergeometric test, a well-established method used in gene enrichment analyses [27]. The hypergeometric probability (h) is based on the following formula:
| (1) |
where x is the number of nodes of the sub-network (the items in the sample that are classified as success), n is the number of genes in the network (items in the sample); X is the number of genes annotated in the DB with that pathway (items in the population that are classified as success) and N is the number of all genes annotated in DB (items in the population). FunMod preserves the sub-networks with a P value <0.05.
For a better understanding of the systemic functions and the cooperative interactions between genes within the functional modules, FunMod checks whether the sub-network topology fits into a specific module. Network modules represent patterns occurring significantly more often than random in the complex networks. They consist of sub-graphs of local interconnections between network elements. FunMod calculates a fitting score of each sub-network for three types of modules: clique, star and path [38].
A clique is a sub-network in which all nodes are connected to each other. Cliques are the most widely-used modules for assigning a biological function to a topological sub-network. FunMod calculates the tendency to be a clique by graph density (GD), a score that can also be defined as the local clustering coefficient, using the formula:
| (2) |
where E is the number of edges in the sub-network and n is the number of genes in the sub-network.
The star module is particularly interesting for identifying drug targets. It is characterized by a central gene with a high degree connection to a set of first-degree neighbors, which are loosely connected between each other. In a star sub-network, the central gene (the hub gene) has influence on its neighbor genes and possibly on the whole network. To identify a star module, FunMod calculates the sub-network centralization (CE) using the formula:
| (3) |
where max(k) is the highest degree of central gene connection in the sub-network and n is the number of genes in the sub-network.
The path module corresponds to a real pathway where the genes contribute to a signal transduction. The path score is evaluated by the sub-network diameter (D), which uses the maximum length of all shortest paths between any two connected nodes, using the formula:
| (4) |
where is the minimum path between two nodes i and j of the network.
Pathways identified using FunMod were displayed in the Cytoscape Results Panel and ranked based on their P values. For each pathway, FunMod displays its clique, star and path coefficients. By clicking the “Pathway” button, FunMod selects the corresponding nodes in the network. And by using the “View subnet” function, it creates a new network containing only those genes and edges annotated within that pathway. Moreover, FunMod enables saving the results in a tab-delimited file. FunMod plugin, user’s guide, screenshot and demo networks can be freely downloaded from the SourceForge project page at: http://sourceforge.net/projects/funmodnetwork/. Developed in Java, FunMod is a platform independent plugin for Cytoscape 2.8.4, which is freely available without charge for non-commercial purposes.
Results and discussion
Gene Ontology (GO) provides information on the location of genes and gene products in a cell or the extracellular environment and also on the molecular function they carry out. However, GO does not provide information about the interaction of proteins in the same biological context. For example, GO does not allow us to describe genes in terms of which cells or tissues they are expressed in, which developmental stages they are expressed at, or their involvement in disease (http://www.geneontology.org/GO.doc.shtml). We thus chose ConsensusPathDB [39] to identify proteins that are strictly involved in the same pathways. We also assess the topological shape of each sub-network in order to reveal evidence of its biological function and the function of its components. Three topological scores are calculated to describe the global features of the sub-network: graph density, network centralization and shortest path. Other topological scores, such as centrality and degree, describe the relative importance of a single node within the sub-network and thus left out in FunMod.
To demonstrate the usage and performance of FunMod, we showed here case studies to analyze two different informative networks: a bibliometric network of proteins related to the Budd–Chiari syndrome and a co-expression network of proteins related to spinal muscular atrophy. Identification of the sub-network functional modules in the undirected protein networks allows the reduction in network complexity, clustering of proteins on the basis of common biological functions and discovery of the mechanisms underlying a disease [40].
Case study 1: Analysis of a bibliometric network
Budd–Chiari syndrome is an uncommon condition characterized by obstruction of the hepatic venous outflow tract [41]. Budd–Chiari syndrome is difficult for diagnosis and management and often fatal if not treated optimally, since none of the current medical therapies are based on the clinical evidence due to the difficulty in performing appropriate clinical studies on a sufficient number of patients with this rare disease [42]. In this circumstance, a meta-analysis can be useful to provide valuable information for researchers to understand the mechanism of this disease [43].
In this case study, we used ProteinQuest (PQ), an advanced text-mining tool that resorts to the web services offered by PubMed to identify all proteins co-cited in the same abstract or figure legend [44] and to export the literature co-occurrence information as a network file that can be managed in Cytoscape. We submitted a query to PQ using the starting search terms “Budd–Chiari syndrome” and its NCBI medical subject heading (MeSH) term alias. PQ retrieved more than 2200 papers. The network obtained by PQ was an undirected protein–protein network consisting of 76 nodes that represent 76 proteins cited in at least one document and 174 edges indicating co-occurrence of two proteins in at least one document. A screenshot of the plugin is shown in Figure 1, whereas a quick guide of the Budd–Chiari syndrome network can be downloaded at the SourceForge project page (http://sourceforge.net/projects/funmodnetwork/).
Figure 1.
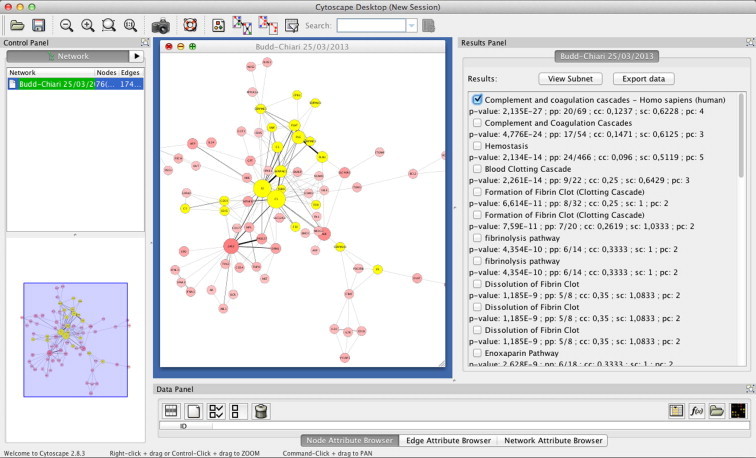
Screenshot of FunMod plugin in action
Screenshot shows the bibliometric network of proteins related to Budd–Chiari syndrome. Proteins are displayed as pink nodes and the size and color density are proportional to the occurrence of the protein term in the literature of Budd–Chiari syndrome. The line between nodes represents the co-occurrence, and its thickness is proportional to the number of times that the proteins are co-cited in the same abstract or figure caption. Cytoscape “Results Panel” displays the results of FunMod analysis. When a particular pathway is selected, all the proteins on this pathway are selected in the network as yellow nodes.
Using FunMod we identified 33 different biological pathways that are significantly enriched in the Budd–Chiari syndrome network. Table 1 shows 13 most relevant pathways. The most significant one is related to the coagulation cascades, which is in agreement with the previous reports that Budd–Chiari is a disorder frequently characterized by the thrombotic obstruction of hepatic venous outflow [45,46]. This result indicated that FunMod, relying on the ABC principle (A and C have no direct connection but are connected via shared B intermediates) [47,48] to establish new relationships between proteins from literature co-occurrence, can be effectively used to combine information from the relevant scientific literature and from pathway databases, supporting the discovery of new knowledge. Moreover, we calculated the three types of topological coefficients of each pathway sub-network with FunMod and selected the most relevant pathways using three types of coefficients. As shown in Figure 2, fibrinolysis pathway was exemplified as a clique module (Figure 2A), the JAK-STAT pathway as an example of a star module (Figure 2B), and the platelet activation, signaling and aggregation as an example of path module (Figure 2C).
Table 1.
The most relevant biological pathways identified in the Budd−Chiari syndrome network
| Pathway | Pathway coverage | P value | Clique coefficient | Star coefficient | Path coefficient |
|---|---|---|---|---|---|
| Complement and coagulation cascades | 20/69 | 2.13E−27 | 0.1237 | 0.6228 | 4 |
| Homeostasis | 24/466 | 2.13E−14 | 0.096 | 0.5119 | 5 |
| Fibrinolysis pathway | 6/14 | 4.35E−10 | 0.3333 | 1 | 2 |
| Heparin pathway | 6/18 | 2.63E−9 | 0.3333 | 1 | 2 |
| Vitamin B12 metabolism | 8/51 | 3.57E−9 | 0.1786 | 1.0952 | 2 |
| Platelet degranulation | 9/83 | 1.03E−8 | 0.0972 | 0.3571 | 3 |
| Response to elevated platelet cytosolic Ca2+ | 9/88 | 1.75E−8 | 0.0972 | 0.3571 | 3 |
| Folate metabolism | 8/66 | 2.94E−8 | 0.1786 | 1.0952 | 2 |
| JAK/STAT molecular variation 1 | 9/99 | 4.95E−8 | 0.125 | 0.8036 | 2 |
| Selenium pathway | 8/77 | 1.01E−7 | 0.1786 | 1.0952 | 2 |
| Lepirudin pathway | 5/17 | 1.24E−7 | 0.3 | 1.1667 | 2 |
| Dicumarol pathway | 5/18 | 1.71E−7 | 0.3 | 1.1667 | 2 |
| PI3K−Akt signaling pathway | 7/334 | 1.29E−2 | 0.1429 | 0.7333 | 2 |
Figure 2.
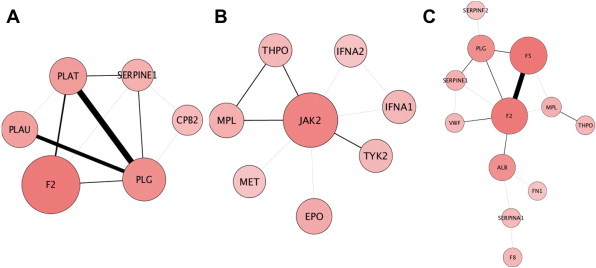
Three sub-networks extracted from Budd–Chiari network
The three sub-network functional modules selected according to the highest coefficient for each type of topological module—cluster module (A), star module (B) and pathway module (C). The three sub-networks were extracted from the Budd–Chiari network by clicking on the “View Subnet” button.
By examining the topological feature of a sub-network functional module, we can extract information of pathways functioning in Budd–Chiari syndrome. The cliques represent a highly connected sub-network, where the proteins of the pathway are highly co-studied and co-cited. The fibrinolysis pathway is the central pathway in Budd–Chiari syndrome, consistent with the fact that prothrombotic tendency is caused by abnormalities in the coagulation of fibrinolysis pathways [49]. These data may explain why the fibrinolysis pathway functional sub-network is represented by a clique. A star module represents a functional sub-network where a protein plays a central role in the pathway. Indeed JAK2 is the hub (the central node) in the JAK-STAT pathway sub-network. Moreover, a mutation in JAK2 tyrosine kinase (JAK2 V617F) is frequently found in patients with Budd–Chiari syndrome [50,51]. Finally, the path module represents a significant pathway in the context of the disease, which is not studied comprehensively. The platelet activation, signaling and aggregation pathway is a therapeutic target for the treatment of Budd–Chiari syndrome [52]. von Willebrand factor (vWF) [53] and SerpinA1 [54], two peripheral sub-network proteins that were recently described as markers of abnormalities of coagulation play roles in provoking thrombosis [55]. The information extracted by FunMod will provide guidance for the future study on Budd–Chiari syndrome.
Case study 2: Analysis of a co-expression network
Spinal muscular atrophy (SMA) is an inheritable neuromuscular disorder, leading to degeneration of α-motor neurons, due to recessive mutations of the SMN1 gene [56]. No treatment is currently available for curing SMA patients, and the development of a computational approach could facilitate a better understanding of this disease and identification of potential therapeutic targets. In the second case study, we applied FunMod to analyze a co-expression network obtained using PQ and COXPRESdb. The protein–protein interactions within this co-expression network reveal proteins examined in the context of SMA and that are simultaneously documented to be co-regulated at mRNA level. The analysis of gene co-expression network provides powerful searching ability that allowed us to identify more than 400 functional clusters related to SMA.
In this case study, we examined all proteins involved in SMA using PQ and found that 1482 proteins were cited in PubMed articles describing SMA. This list of proteins was submitted to COXPRESdb [35], a publicly-available co-expression database, which provides a tool to export the networks in Cytoscape. We used a co-expression network to enhance the predictive power of the informative network. We submitted the list of SMA proteins to COXPRESdb and obtained a co-expression protein network with 2022 nodes and 9166 edges (Figure 3). Further analyzing the SMA co-expression network using FunMod, we identified 429 functional modules and the 13 most relevant modules were shown in Table 2.
Figure 3.
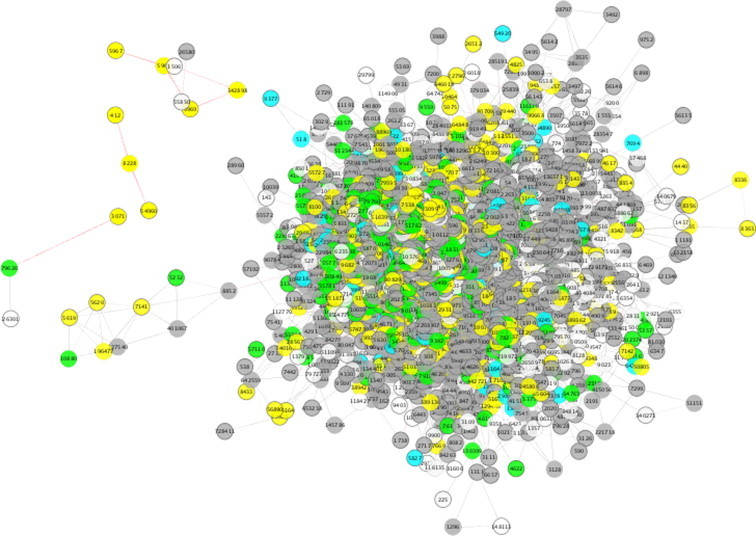
The SMA co-expression network
COXPRESdb provides NetworkDrawer, a tool for drawing and visualizing the protein co-expression network in Cytoscape Web. The network can be downloaded in the XGMML format, the standard format for saving and importing network into Cytoscape, and imported into a stand-alone Cytoscape distribution for advanced analyses. NetworkDrawer performs cluster detection and Gene Ontology enrichment analysis, providing a node color scale to indicate protein localization: cytosol (green); cytoskeleton, endoplasmic reticulum, Golgi body, lysosome, peroxisome and plasma membrane (gray); mitochondrion (cyan); nucleus (yellow); and extracellular matrix (white). The edge represents the type of interaction between two nodes: gene co-expression (solid in black) and indicates known protein–protein interaction (dotted in red). SMA, spinal muscular atrophy; XGMML, extensible graph markup and modeling language.
Table 2.
The most relevant biological pathways identified in spinal muscular atrophy network
| Pathway | Pathway coverage | P value | Clique coefficient | Star coefficient | Path coefficient |
|---|---|---|---|---|---|
| AGE−RAGE pathway | 39/66 | 3.75E−12 | 0.0789 | 0.3051 | 6 |
| BDNF signaling pathway | 61/140 | 1.93E−10 | 0.0484 | 0.2602 | 6 |
| Prolactin signaling pathway | 40/76 | 2.74E−10 | 0.0814 | 0.2922 | 5 |
| Inhibition of cellular proliferation by Gleevec | 17/22 | 1.25E−08 | 0.136 | 0.3417 | 4 |
| Leptin signaling pathway | 32/61 | 1.86E−08 | 0.1008 | 0.3742 | 5 |
| Androgen receptor | 59/148 | 2.22E−08 | 0.0649 | 0.6289 | 4 |
| ATF-2 transcription factor network | 31/61 | 8.13E−08 | 0.0753 | 0.3471 | 5 |
| Glucocorticoid receptor regulatory network | 37/80 | 1.11E−07 | 0.0691 | 0.3675 | 4 |
| MAPK signaling pathway | 61/161 | 1.16E−07 | 0.0522 | 0.2562 | 5 |
| Chronic myeloid leukemia | 34/73 | 2.94E−07 | 0.066 | 0.3163 | 5 |
| mRNA processing | 50/126 | 3.07E−07 | 0.0861 | 0.3142 | 5 |
| Striated muscle contraction | 22/38 | 3.26E−07 | 0.0736 | 0.3905 | 6 |
| ErbB2/ErbB3 signaling events | 23/42 | 6.89E−07 | 0.1206 | 0.3658 | 4 |
FunMod identified a few pathways known to be related to systemic aspects of neurodegenerative conditions, such as the AGE-RAGE pathway [57], striated muscle contraction [58] and leptin signaling pathway [59]. FunMod identified the brain-derived neurotropic factor (BDNF) signaling among the most relevant pathway (Figure 4A). The BDNF promotes neurogenesis, neuronal survival and synaptic plasticity [60]. Therefore, this finding validates the ability of our software to reveal new relevant functional modules. Interestingly, FunMod identified the androgen receptor signaling pathway in SMA co-expression network as well (Figure 4B). Androgens interact with BDNF during development to regulate non-pathological death of motor neurons. Androgens regulate BDNF levels in the target musculature and moreover, androgenic actions at the muscle regulate BDNF levels in motoneurons [61]. Therefore, these interactions have important implications for the maintenance of motoneuron morphology. These findings provide further insights into the pathogenic role of BDNF and androgens in SMA, which might be a preferential route for targeting motor neurons [62].
Figure 4.
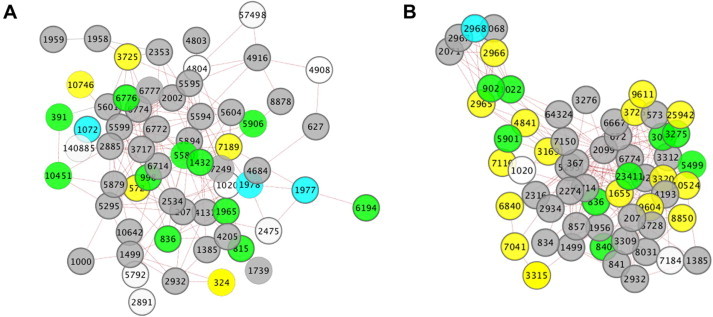
Two sub-networks identified within the SMA co-expression network
A. The BDNF signaling pathway. B. The androgen receptor pathway. The Entrez Gene IDs were used for the node label as required by COXPRESdb for input. The node color scale is inherited from COXPRESdb. Larger nodes are the query genes and smaller gray nodes are additional nodes with one or more edges to at least one query node. Solid lines and red dotted lines indicate gene co-expression and protein–protein interactions from the Human Protein Reference Database (HPRD) and IntAct databases, respectively.
Conclusion
Using a “first topological assignment” strategy to identify sub-network functional modules, such as stars and cliques, can be tricky because informative networks are known to have a huge number of edges that are not always pertinent to biological functions. In this work we presented FunMod, a new Cytoscape 2.8 plugin, which can analyze undirected protein networks, such as co-occurrence and co-expression networks, and guide the discovery of sub-network functional modules. A functional module can be considered as a distinct group of interacting proteins [32] within a pathway relevant to a condition of interest.
FunMod identifies within an informative network, pairs of nodes belonging to the same biological pathways and assesses their statistical significance. It then analyzes the topology of the identified sub-network to infer the topological relations (motifs) of its nodes. In this work, the network topology is influenced by the biomedical knowledge since the link between two proteins was established when two gene symbols appear in the same MEDLINE record. The study of the connection between biomedical concepts by using co-occurrence network extracted from MEDLINE proved capable of guiding the discovery of novel knowledge from scientific literature [63]. This sub-network profiling combined with information from the biological database will help us to better understand the biological significance of the protein–protein network.
FunMod was tested using the co-occurrence network of proteins cited in Budd–Chiari syndrome papers, identifying 33 different biological pathways that are significantly enriched; and using the co-expression network of proteins discussed in publications on SMA. FunMod proves to be a useful tool for a better understanding of the cooperative interactions between proteins and discriminating the biological role played by each protein within a functional module.
Authors’ contributions
AB and SDC conceived the idea and supervised the study. MN designed and implemented the network analysis method, and performed data analysis. AB and SDC analyzed the results and drafted the manuscript. EF edited the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors have declared that no competing interests exist.
Acknowledgements
This work was conducted as a part of the project Open Source Drug Discovery (CUP: B15G13000010006) and partially funded by Valle d’Aosta Region Government under the Grant for Creation and Development of Research Units (dgr. 1988/2011; dgr. 538/2012) regional funding program of Italy.
Footnotes
Peer review under responsibility of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China.
References
- 1.Barabasi A.L., Oltvai Z.N. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 2.Huber W., Carey V.J., Long L., Falcon S., Gentleman R. Graphs in molecular biology. BMC Bioinformatics. 2007;8:S8. doi: 10.1186/1471-2105-8-S6-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spirin V., Mirny L.A. Protein complexes and functional modules in molecular networks. Proc Natl Acad Sci U S A. 2003;100:12123–12128. doi: 10.1073/pnas.2032324100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pieroni E., de la Fuente van Bentem S., Mancosu G., Capobianco E., Hirt H., de la Fuente A. Protein networking: insights into global functional organization of proteomes. Proteomics. 2008;8:799–816. doi: 10.1002/pmic.200700767. [DOI] [PubMed] [Google Scholar]
- 5.Li J., Hua X., Haubrock M., Wang J., Wingender E. The architecture of the gene regulatory networks of different tissues. Bioinformatics. 2012;28:i509–i514. doi: 10.1093/bioinformatics/bts387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuchaiev O., Przulj N. Integrative network alignment reveals large regions of global network similarity in yeast and human. Bioinformatics. 2011;27:1390–1396. doi: 10.1093/bioinformatics/btr127. [DOI] [PubMed] [Google Scholar]
- 7.Prifti E., Zucker J.D., Clément K., Henegar C. Interactional and functional centrality in transcriptional coexpression networks. Bioinformatics. 2010;26:3083–3089. doi: 10.1093/bioinformatics/btq591. [DOI] [PubMed] [Google Scholar]
- 8.Chen L., Xuan J., Riggins R.B., Wang Y., Clarke R. Identifying protein interaction sub-networks by a bagging Markov random field-based method. Nucleic Acids Res. 2013;41:e42. doi: 10.1093/nar/gks951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gatti S., Leo C., Gallo S., Sala V., Bucci E., Natale M. Gene expression profiling of HGF/Met activation in neonatal mouse heart. Transgenic Res. 2012;3:579–593. doi: 10.1007/s11248-012-9667-2. [DOI] [PubMed] [Google Scholar]
- 10.Lee I., Ambaru B., Thakkar P., Marcotte E.M., Rhee S.Y. Rational association of genes with traits using a genome-scale gene network for Arabidopsis thaliana. Nat Biotechnol. 2010;28:149–156. doi: 10.1038/nbt.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gabow A.P., Leach S.M., Baumgartner W.A., Hunter L.E., Goldberg D.S. Improving protein function prediction methods with integrated literature data. BMC Bioinformatics. 2008;9:198. doi: 10.1186/1471-2105-9-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lysenko A., Defoin-Platel M., Hassani-Pak K., Taubert J., Hodgman C., Rawlings C.J. Assessing the functional coherence of modules found in multiple-evidence networks from Arabidopsis. BMC Bioinformatics. 2011;12:203. doi: 10.1186/1471-2105-12-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bu D., Zhao Y., Cai L., Xue H., Zhu X., Lu H. Topological structure analysis of the protein–protein interaction network in budding yeast. Nucleic Acids Res. 2003;31:2443–2450. doi: 10.1093/nar/gkg340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weatheritt R.J., Jehl P., Dinkel H., Gibson T.J. IELM-a web server to explore short linear motif-mediated interactions. Nucleic Acids Res. 2012;40:W364–W369. doi: 10.1093/nar/gks444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas S., Bonchev D. A survey of current software for network analysis in molecular biology. Hum Genomics. 2010;4:353–360. doi: 10.1186/1479-7364-4-5-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen R., Goonesekere N.C., Guda C. Mining functional subgraphs from cancer protein–protein interaction networks. BMC Syst Biol. 2012;6:S2. doi: 10.1186/1752-0509-6-S3-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smoot M.E., Ono K., Ruscheinski J., Wang P.L., Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spinelli L., Gambette P., Chapple C.E., Robisson B., Baudot A., Garreta H. Clust&See: a Cytoscape plugin for the identification, visualization and manipulation of network clusters. Biosystems. 2013;113:91–95. doi: 10.1016/j.biosystems.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 19.Morris J.H., Apeltsin L., Newman A.M., Baumbach J., Wittkop T., Su G. ClusterMaker: a multi-algorithm clustering plugin for Cytoscape. BMC Bioinformatics. 2011;12:436. doi: 10.1186/1471-2105-12-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Audenaert P., Van Parys T., Brondel F., Pickavet M., Demeester P., Van de Peer Y. CyClus3D: a Cytoscape plugin for clustering network motifs in integrated networks. Bioinformatics. 2011;27:1587–1588. doi: 10.1093/bioinformatics/btr182. [DOI] [PubMed] [Google Scholar]
- 21.Su G., Kuchinsky A., Morris J.H., States D.J., Meng F. GLay: community structure analysis of biological networks. Bioinformatics. 2010;26:3135–3137. doi: 10.1093/bioinformatics/btq596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Merico D., Isserlin R., Stueker O., Emili A., Bader G.D. Enrichment Map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One. 2010;5:e13984. doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milo R., Shen-Orr S., Itzkovitz S., Kashtan N., Chklovskii D., Alon U. Network motifs: simple building blocks of complex networks. Science. 2002;298:824–827. doi: 10.1126/science.298.5594.824. [DOI] [PubMed] [Google Scholar]
- 24.Dong J., Horvath S. Understanding network concepts in modules. BMC Syst Biol. 2007;4:1–24. doi: 10.1186/1752-0509-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aittokallio T., Schwikowski B. Graph-based methods for analysing networks in cell biology. Brief Bioinform. 2006;7:243–255. doi: 10.1093/bib/bbl022. [DOI] [PubMed] [Google Scholar]
- 26.Heatha A.P., Kavrakia L.E. Computational challenges in systems biology. Comput Sci Rev. 2009;3:117. [Google Scholar]
- 27.Griswold A.J., Ma D., Cukier H.N., Nations L.D., Schmidt M.A., Chung R.H. Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum Mol Genet. 2012;21:3513–3523. doi: 10.1093/hmg/dds164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maere S., Heymans K., Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 29.Bindea G., Mlecnik B., Hackl H., Charoentong P., Tosolini M., Kirilovsky A. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cai J, Chen G, Wang J. ClusterViz: a Cytoscape plugin for graph clustering and visualization. Changsha, Hunan: School of Information Science and Engineering, <http://chianti.ucsd.edu/cyto_web/plugins/displayplugininfo.php?name=ClusterViz>; 2010 [accessed 22.03.10].
- 31.Glaab E., Baudot A., Krasnogor N., Schneider R., Valencia A. EnrichNet: network-based gene set enrichment analysis. Bioinformatics. 2012;28:i451–i457. doi: 10.1093/bioinformatics/bts389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jupe S., Akkerman J.W., Soranzo N., Ouwehand W.H. Reactome – A curated knowledgebase of biological pathways: megakaryocytes and platelets. J Thromb Haemost. 2012;10:2399–2402. doi: 10.1111/j.1538-7836.2012.04930.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitra K., Carvunis A.R., Ramesh S.K., Ideker T. Integrative approaches for finding modular structure in biological networks. Nat Rev Genet. 2013;14:719–732. doi: 10.1038/nrg3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu G., Stein L. A network module-based method for identifying cancer prognostic signatures. Genome Biol. 2012;13:R112. doi: 10.1186/gb-2012-13-12-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Obayashi T., Okamura Y., Ito S., Tadaka S., Motoike I.N., Kinoshita K. COXPRESdb: a database of comparative gene coexpression networks of eleven species for mammals. Nucleic Acids Res. 2013;41:D1014–D1020. doi: 10.1093/nar/gks1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benso A., Cornale P., Di Carlo S., Politano G., Savino A. Reducing the complexity of complex gene coexpression networks by coupling multiweighted labeling with topological analysis. Biomed Res Int. 2013;2013:676328. doi: 10.1155/2013/676328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liekens A.M., De Knijf J., Daelemans W., Goethals B., De Rijk P., Del-Favero J. BioGraph: unsupervised biomedical knowledge discovery via automated hypothesis generation. Genome Biol. 2011;12:R57. doi: 10.1186/gb-2011-12-6-r57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dolinski K., Chatr-Aryamontri A., Tyers M. Systematic curation of protein and genetic interaction data for computable biology. BMC Biol. 2013;11:43. doi: 10.1186/1741-7007-11-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamburov A., Stelzl U., Lehrach H., Herwig R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2013;41:D793–D800. doi: 10.1093/nar/gks1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Royer L., Reimann M., Andreopoulos B., Schroeder M. Unraveling protein networks with power graph analysis. PLoS Comput Biol. 2008;4:e1000108. doi: 10.1371/journal.pcbi.1000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.MacNicholas R., Olliff S., Elias E., Tripathi D. An update on the diagnosis and management of Budd–Chiari syndrome. Expert Rev Gastroenterol Hepatol. 2012;6:731–744. doi: 10.1586/egh.12.56. [DOI] [PubMed] [Google Scholar]
- 42.Walker E., Hernandez A.V., Kattan M.W. Meta-analysis: its strengths and limitations. Cleve Clin J Med. 2008;75:431–439. doi: 10.3949/ccjm.75.6.431. [DOI] [PubMed] [Google Scholar]
- 43.Jaqua N.T., Stratton A., Yaccobe L., Tahir U., Kenny P., Kerns T. A review of the literature on three extraintestinal complications of ulcerative colitis: an ulcerative colitis flare complicated by Budd–Chiari syndrome, cerebral venous thrombosis and idiopathic thrombocytopenia. Acta Gastroenterol. 2013;76:311–316. [PubMed] [Google Scholar]
- 44.Giordano M., Natale M., Cornaz M., Ruffino A., Bonino D., Bucci E.M. IMole, a web based image retrieval system from biomedical literature. Electrophoresis. 2013;34:1965–1968. doi: 10.1002/elps.201300085. [DOI] [PubMed] [Google Scholar]
- 45.Wang R., Meng Q., Qu L., Wu X., Sun N., Jin X. Treatment of Budd–Chiari syndrome with inferior vena cava thrombosis. Exp Ther Med. 2013;5:1254–1258. doi: 10.3892/etm.2013.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Akbulut S., Yilmaz M., Kahraman A., Yilmaz S. Budd–Chiari syndrome due to giant hydatid cyst: a case report and brief literature review. J Infect Dev Ctries. 2013;7:489–493. doi: 10.3855/jidc.2712. [DOI] [PubMed] [Google Scholar]
- 47.Frijters R., van Vugt M., Smeets R., van Schaik R., de Vlieg J., Alkema W. Literature mining for the discovery of hidden connections between drugs, genes and diseases. PLoS Comput Biol. 2010;6:e1000943. doi: 10.1371/journal.pcbi.1000943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li C., Liakata M., Rebholz-Schuhmann D. Biological network extraction from scientific literature: state of the art and challenges. Brief Bioinform. 2013;23:e237–e244. doi: 10.1093/bib/bbt006. [DOI] [PubMed] [Google Scholar]
- 49.Smalberg J.H., Kruip M.J., Janssen H.L., Rijken D.C., Leebeek F.W., de Maat M.P. Hypercoagulability and hypofibrinolysis and risk of deep vein thrombosis and splanchnic vein thrombosis: similarities and differences. Arterioscler Thromb Vasc Biol. 2011;31:485–493. doi: 10.1161/ATVBAHA.110.213371. [DOI] [PubMed] [Google Scholar]
- 50.Qi X., Yang Z., Bai M., Shi X., Han G., Fan D. Meta-analysis: the significance of screening for JAK2V617F mutation in Budd–Chiari syndrome and portal venous system thrombosis. Aliment Pharmacol Ther. 2011;33:1087–1103. doi: 10.1111/j.1365-2036.2011.04627.x. [DOI] [PubMed] [Google Scholar]
- 51.Patel R.K., Lea N.C., Heneghan M.A., Westwood N.B., Milojkovic D., Thanigaikumar M. Prevalence of the activating JAK2 tyrosine kinase mutation V617F in the Budd–Chiari syndrome. Gastroenterology. 2006;130:2031–2038. doi: 10.1053/j.gastro.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 52.Chaudhuri M., Jayaranganath M., Chandra V.S. Percutaneous recanalization of an occluded hepatic vein in a difficult subset of pediatric Budd–Chiari syndrome. Pediatr Cardiol. 2012;33:806–810. doi: 10.1007/s00246-012-0188-9. [DOI] [PubMed] [Google Scholar]
- 53.Alkim H., Ayaz S., Sasmaz N., Oguz P., Sahin B. Hemostatic abnormalities in cirrhosis and tumor-related portal vein thrombosis. Clin Appl Thromb Hemost. 2012;18:409–415. doi: 10.1177/1076029611427900. [DOI] [PubMed] [Google Scholar]
- 54.Hourigan S.K., Anders R.A., Mitchell S.E., Schwarz K.B., Lau H., Karnsakul W. Chronic diarrhea, ascites, and protein-losing enteropathy in an infant with hepatic venous outflow obstruction after liver transplantation. Pediatr Transplant. 2012;16:E328–E331. doi: 10.1111/j.1399-3046.2012.01686.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ding P.X., Li Z., Han X.W., Zhang W.G., Zhou P.L., Wang Z.G. Spontaneous intrahepatic portosystemic shunt in Budd–Chiari syndrome. Ann Vasc Surg. 2014;28:742.e1–742.e4. doi: 10.1016/j.avsg.2013.06.031. [DOI] [PubMed] [Google Scholar]
- 56.Van Meerbeke J.P., Sumner C.J. Progress and promise: the current status of spinal muscular atrophy therapeutics. Discov Med. 2011;12:291–305. [PubMed] [Google Scholar]
- 57.Li J., Liu D., Sun L., Lu Y., Zhang Z. Advanced glycation end products and neurodegenerative diseases: mechanisms and perspective. J Neurol Sci. 2012;317:1–5. doi: 10.1016/j.jns.2012.02.018. [DOI] [PubMed] [Google Scholar]
- 58.Katsuno M., Adachi H., Tanaka F., Sobue G. Spinal and bulbar muscular atrophy: ligand-dependent pathogenesis and therapeutic perspectives. J Mol Med. 2004;82:298–307. doi: 10.1007/s00109-004-0530-7. [DOI] [PubMed] [Google Scholar]
- 59.Paz-Filho G., Wong M.L., Licinio J. The procognitive effects of leptin in the brain and their clinical implications. Int J Clin Pract. 2010;64:1808–1812. doi: 10.1111/j.1742-1241.2010.02536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Teixeira A.L., Barbosa I.G., Diniz B.S., Kummer A. Circulating levels of brain-derived neurotrophic factor: correlation with mood, cognition and motor function. Biomark Med. 2010;4:871–887. doi: 10.2217/bmm.10.111. [DOI] [PubMed] [Google Scholar]
- 61.Verhovshek T., Rudolph L.M., Sengelaub D.R. Brain-derived neurotrophic factor and androgen interactions in spinal neuromuscular systems. Neuroscience. 2013;239:103–114. doi: 10.1016/j.neuroscience.2012.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dupuis L., Echaniz-Laguna A. Skeletal muscle in motor neuron diseases: therapeutic target and delivery route for potential treatments. Curr Drug Targets. 2010;11:1250–1261. doi: 10.2174/1389450111007011250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Narayanasamy V., Mukhopadhyay S., Palakal M., Potter D.A. TransMiner: mining transitive associations among biological objects from text. J Biomed Sci. 2004;11:864–873. doi: 10.1007/BF02254372. [DOI] [PubMed] [Google Scholar]