

Abstract
Next-generation sequencing (NGS) technology has revolutionized and significantly impacted metagenomic research. However, the NGS data usually contains sequencing artifacts such as low-quality reads and contaminating reads, which will significantly compromise downstream analysis. Many quality control (QC) tools have been proposed, however, few of them have been verified to be suitable or efficient for metagenomic data, which are composed of multiple genomes and are more complex than other kinds of NGS data. Here we present a metagenomic data QC method named Meta-QC-Chain. Meta-QC-Chain combines multiple QC functions: technical tests describe input data status and identify potential errors, quality trimming filters poor sequencing-quality bases and reads, and contamination screening identifies higher eukaryotic species, which are considered as contamination for metagenomic data. Most computing processes are optimized based on parallel programming. Testing on an 8-GB real dataset showed that Meta-QC-Chain trimmed low sequencing-quality reads and contaminating reads, and the whole quality control procedure was completed within 20 min. Therefore, Meta-QC-Chain provides a comprehensive, useful and high-performance QC tool for metagenomic data. Meta-QC-Chain is publicly available for free at: http://computationalbioenergy.org/meta-qc-chain.html.
Keywords: Quality control, Metagenomic data, Parallel computing, Next-generation sequencing
Introduction
Next-generation sequencing (NGS) technologies have become a common practice in life science [1]. Quality control (QC) is the very first step of NGS data processing. Although many QC tools are available, there are still limitations in various aspects, such as speed and difficulties in contamination screening. Metagenomic data, which are composed of NGS reads from multiple genomes (usually unknown in advance) present in microbial communities, face a more serious problem if data QC cannot be performed accurately and efficiently.
Among the QC problems for raw metagenomic NGS data, low sequencing-quality reads and contaminating reads pose two major challenges in data QC. Existence of both types of reads can significantly compromise downstream analyses. It is reported that sequencing-quality filtering can vastly improve the accuracy of microbial diversity from metagenomic sequencing [2]. For metagenomic data, higher eukaryotic species are usually considered as contaminations that have to be identified and filtered before further analyses to prevent erroneous results and conclusions. For both sequencing-quality trimming and contamination screening processes, current QC tools, such as Prinseq [3], NGS QC Toolkit [4] and Fastx-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/), are time-consuming and highly dependent on pre-defined information such as the source of contaminations. Moreover, the processing speed has become another bottleneck in handling large amounts of NGS data. We previously reported QC-Chain, a fast and holistic NGS data QC package which can achieve fast and de novo contamination screening on NGS data [5]. However, it is a general QC pipeline, which is not specifically optimized or well suited to metagenomic data.
Here we report Meta-QC-Chain, an open-source and parallel-computation based NGS data QC method specific for metagenomic data. Meta-QC-Chain can perform raw data status checking, sequencing-quality trimming and de novo identification of contamination in raw NGS data. Using Meta-QC-Chain, low sequencing-quality reads can be trimmed and all the unexpected contaminating species in metagenomic data, especially higher eukaryotic genomes, can be identified and filtered. Additionally, Meta-QC-Chain is highly efficient since it is based on parallel computing developed by Linux C++ and multi-thread technology on X86-64 multi-core CPU platform.
Methods
The workflow of Meta-QC-Chain
Meta-QC-Chain has enabled multiple QC procedures, including data technical tests, read quality trimming and contamination screening. The workflow of Meta-QC-Chain consists of four steps, which are described below and illustrated in Figure 1.
Figure 1.
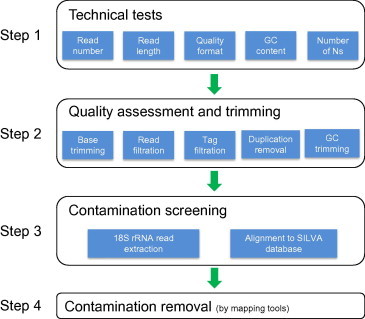
The workflow and functions of Meta-QC-Chain for metagenomic data quality control
Step 1 Technical tests on the input metagenomic dataset
This step checks the data status itself, including the total read number, average read length, quality format, average GC content, GC distribution and number of ambiguous (N) bases. The statistics will be exported in a report, and the GC distribution will be exported as a graph. Technical tests can alert users to basic technical errors and help users to overview the input data before further QC processing.
Step 2 Sequencing-quality trimming
Sequencing-quality trimming function is executed by a tool named Meta-Parallel-QC and includes: (1) base trimming to cut reads into a specified length from both 5′ and 3′ ends; (2) quality trimming to filter low-quality reads with user-defined base quality value and percentage; (3) GC trimming to filter reads by GC content; (4) duplication trimming to identify and remove duplicated reads and (5) tag trimming to remove reads that can be mapped to multiple tag sequences at both 5′ and 3′ ends with user-defined mismatches in a single run.
Step 3 Contamination screening and identification
Since higher eukaryotic species are commonly regarded as the possible contaminating sources for metagenomic data, Meta-QC-Chain identifies contaminating reads using 18S rRNA as the organismal biomarker. First, 18S rRNA reads are predicted and extracted from the input data, which are then aligned to the ribosomal RNA database SILVA [6]. Subsequently, taxonomy information is produced to parse out the possible contaminating higher eukaryotic species. Benefitting from this de novo contamination screening approach, Meta-QC-Chain can identify and filter contaminations without any prior information of the input data. Additionally, Meta-QC-Chain outputs the species information in the dynamic and interactive graph [7], which can help users to obtain the contamination status of the sample quickly and in a visualized manner.
Step 4 Contaminating read removal
Once the contaminating species are identified by Step 3, and if the candidate contaminating species has a reference genome, read alignment tools can be used to remove the contaminating reads. Presently, more than 60 mapping tools are available, with different advantages and limitations [8]. The users can choose the most appropriate mapping tool for their specific data.
Input and output
Meta-QC-Chain can take NGS reads in FASTA or FASTQ formats as input. The output format can be defined to be either FASTA or FASTQ. In addition, Meta-QC-Chain provides an option to keep the paired reads by checking the quality of both ends of the paired reads simultaneously in every QC procedure. Moreover, for backward compatibility, the file format related component provides application programming interfaces (APIs) as open portals to accept more file formats and to adapt for other features of sequences such as meta-pair reads’ approximate insert size distribution (which can be used by some QC pipelines).
Parallel computing
Meta-QC-Chain optimizes both the read quality trimming and contamination screening procedures based on parallel computation. As the processing of each read (or paired reads for pair-end input data) is independent, Meta-QC-Chain appoints weighted and balanced tasks, each including a suitable number of reads (which is dependent on both the total read number and the assigned CPU core number), to different threads, then processes different CPU cores simultaneously and in parallel. A parameter (−t) can be used to assign the number of CPU cores to be used. In addition, all procedures are conducted with only one disk I/O operation, which significantly improves the efficiency of analysis, especially for a huge dataset.
All of the experiments were performed on a rack server with an Intel dual Xeon E5-2650 CPU (2.0 GHz, 16 cores in total, supporting 32 threads), 64GB DDR3 ECC RAM and 2TB HDD.
Results and discussions
We tested the performance of Meta-QC-Chain using three metagenomic data sets including two real datasets and one simulated dataset. All sequences can be downloaded from the website of Meta-QC-Chain (http://computationalbioenergy.org/meta-qc-chain.html).
Two DNA samples from human saliva (R1 and R2) were sequenced by Illumina GAIIx with an average read length of 100 bp and pair end insert size of 400 bp. Firstly, a technical test report and a GC distribution plot were generated in the output directory, showing multiple statistics of the input data. The GC distribution plot was exported in a PNG graph (Figure 2). Users can compare this information to the expected GC content of their samples/species. Using the sequencing-quality trimming step, all reads were trimmed to 50 bp, reads with 90% of high quality bases of score >20 were kept, while duplicated and tag sequence reads were filtered out. In the end, 49% and 63% of the raw reads were kept as good reads for R1 and R2, respectively (Table 1). In the contamination screening step, 18S rRNA reads of each dataset were extracted and mapped to SILVA 18S database [6]. Human was identified as the dominant possible contaminating species for both R1 and R2 datasets (Figure 3A and B). This is consistent with the sample source environment of human saliva, which has a high possibility of containing human DNA. Other species shown in the screening results were identified by random alignment since there is great similarity in 18S rRNA sequences. It is clear that each of the randomly identified species represented a very small percentage and can be easily manually filtered. Meta-QC-Chain completed all of the QC processes within 12 and 20 min on the R1 and R2 datasets (5 GB and 8.3 GB), respectively (Table 2).
Figure 2.
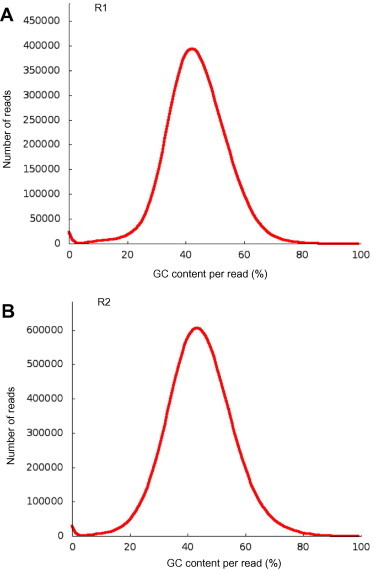
GC distribution plots generated by Meta-Parallel-QC for two human saliva metagenomic datasets Shown in the graph is the read GC content distribution of two real human saliva metagenomic dataset R1 (A) and R2 (B). Detailed information about these two datasets is listed in Table 1.
Table 1.
Summary of the three datasets examined in the current study
| Dataset |
Raw data |
Data after quality trimming |
||
|---|---|---|---|---|
| No. of reads | Data size (GB) | No. of reads | Data size (GB) | |
| R1 | 19,185,960 | 5.0 | 9,414,926 | 1.2 |
| R2 | 33,134,512 | 8.3 | 20,951,704 | 2.8 |
| S1 | 22,127,714 | 2.2 | 22,127,714 | 2.2 |
Figure 3.
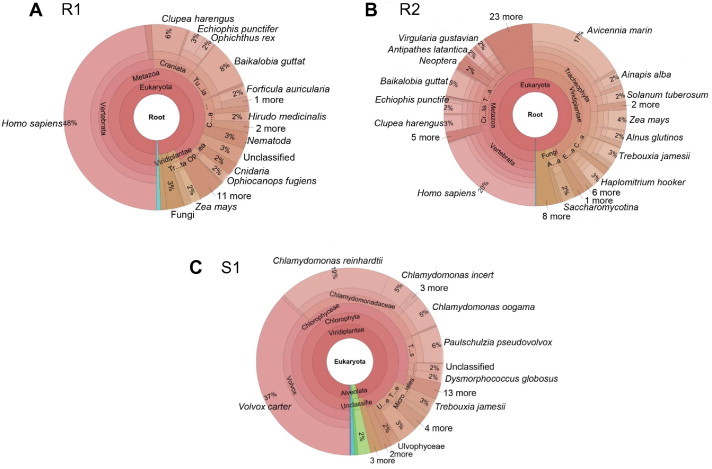
Contaminating species identified from the three metagenomic datasets by Meta-QC-Chain Human was identified as the largest contaminating species in real sequenced human saliva datasets R1 (A) and R2 (B). Chlorophyta algae species were identified as possible contaminations in simulated dataset S1 (C). “1 more” or “11 more” means more species identified with very low proportion of 18S rRNAs, which can be neglected here.
Table 2.
Running time of Meta-QC-Chain and Prinseq on the three datasets
| Dataset |
Meta-QC-Chain |
Prinseq |
|||
|---|---|---|---|---|---|
| Technical tests | Read quality trimming | Contamination screening | Total | Total | |
| R1 | 1 min 02 s | 8 min 33 s | 1 min 53 s | 11 min 28 s | 50 min 43 s |
| R2 | 1 min 37 s | 14 min 07 s | 4 min 04 s | 19 min 48 s | 76 min 03 s |
| S1 | 2 min 38 s | 4 min 19 s | 10 min 14 s | 17 min 01 s | 64 min 48 s |
Note: R1 and R2 are the two metagenomic datasets generated from human saliva sequenced in-house, whereas S1 is a simulated dataset for test.
A simulated metagenomic dataset named S1 was also created using DWGSIM 0.1.8 (https://github.com/nh13/DWGSIM) to test the performance of Meta-QC-Chain in contamination reorganization. The simulated data were designed with high read quality and all reads were kept after sequencing-quality trimming (Table 1). Simulated reads of algae (Chlamydomonas reinhardtii) were mixed with reads simulated from ten bacterial genomes to create a known source of contamination. For S1, results have also shown that Meta-QC-Chain successfully identified the Chlorophyta algal species as possible eukaryotic contamination (Figure 3C). Contamination was identified from both Chlamydomonadaceae and Volvox species, likely because of the high 18S sequence similarity of these algal groups. Some other species were also identified and presented, which may result from random alignment of 18S rRNA and high similarity of 18S rRNA reads [5].
We compared the performance of Meta-QC-Chain to another publicly available metagenomic QC tool Prinseq [3]. Both tools can accomplish technical tests and sequencing-quality trimming functions, however, the total running time of Prinseq is significantly longer than that of Meta-QC-Chain (Table 2). By grouping metagenomes from similar environments, Prinseq can help identify potentially incorrect samples. However, as samples might be processed using different protocols or sequenced using different techniques, this feature should be used with caution. Moreover, Prinseq cannot provide accurate and detailed information of the contaminating species, thus compromising the QC effect on downstream analysis.
Conclusion
Meta-QC-Chain provides a de novo and parallel-computing solution for quality control of metagenomic NGS data, including technical tests, sequencing-quality trimming and contamination screening. It can check and process raw metagenomic read quality and is able to detect and characterize contaminating species de novo with high speed and accuracy. Parallel computation is applied on all of the QC processes. QC results are generated in text report and graphic view. Therefore, Meta-QC-Chain is a useful and efficient quality control tool for metagenomic data, which can support and facilitate metagenomic study for researchers and analysts.
Meta-QC-Chain certainly has some limitations. For example, currently it is difficult to identify virus contaminations, and it is not able to be embedded into other analysis pipelines as a component. Therefore, we are working on improving Meta-QC-Chain to be more comprehensive and compatible.
Authors’ contributions
QZ and KN conceived and designed the experiments; QZ performed the experiments; QZ and XS analyzed the data. XS and GJ contributed reagents/materials/analysis tools. QZ and KN wrote the paper. All authors read and approved the final manuscript.
Completing interests
The authors declare no completing interests.
Acknowledgements
This work was supported by the National High-tech R&D Program (863 Program; Grant Nos. 2009AA02Z310 and 2012AA02A707) funded by Ministry of Science and Technology of China, Natural Science Foundation of China (Grant Nos. 61103167 and 31271410) and Chinesisch-Deutschen Zentrum für Wissenschaftsförderung (Grant No. GZ 878).
Footnotes
Peer review under responsibility of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China.
References
- 1.Mardis E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008;24:133–141. doi: 10.1016/j.tig.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 2.Bokulich N.A., Subramanian S., Faith J.J., Gevers D., Gordon J.I., Knight R. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods. 2013;10:57–59. doi: 10.1038/nmeth.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schmieder R., Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel R.K., Jain M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PloS One. 2012;7:e30619. doi: 10.1371/journal.pone.0030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou Q., Su X., Wang A., Xu J., Ning K. QC-Chain: fast and holistic quality control method for next-generation sequencing data. PLoS One. 2013;8:e60234. doi: 10.1371/journal.pone.0060234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song B., Su X., Xu J., Ning K. MetaSee: an interactive and extendable visualization toolbox for metagenomic sample analysis and comparison. PLoS One. 2012;7:e48998. doi: 10.1371/journal.pone.0048998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fonseca N.A., Rung J., Brazma A., Marioni J.C. Tools for mapping high-throughput sequencing data. Bioinformatics. 2012;28:3169–3177. doi: 10.1093/bioinformatics/bts605. [DOI] [PubMed] [Google Scholar]