

Abstract
Charcot–Marie–Tooth disease type 4B2 with early-onset glaucoma (CMT4B2, OMIM 604563) is a genetically-heterogeneous childhood-onset neuromuscular disorder. Here, we report the case of a 15-year-old male adolescent with lower extremity weakness, gait abnormalities, foot deformities and early-onset glaucoma. Since clinical diagnosis alone was insufficient for providing pathogenetic evidence to indicate that the condition belonged to a consanguineous family, we applied whole-exome sequencing to samples from the patient, his parents and his younger brother, assuming that the patient’s condition is transmitted in an autosomal recessive pattern. A frame-shift mutation, c.4571delG (P.Gly1524Glufs∗42), was revealed in the CMT4B2-related gene SBF2 (also known as MTMR13, MIM 607697), and this mutation was found to be homozygous in the proband and heterozygous in his parents and younger brother. Together with the results of clinical diagnosis, this case was diagnosed as CMT4B2. Our finding further demonstrates the use of whole-exome sequencing in the diagnosis and treatment of rare diseases.
Keywords: Whole-exome sequencing, Charcot–Marie–Tooth disease, Early-onset glaucoma, SBF2
Introduction
Determination of the pathogenesis of inherited diseases is a challenging endeavor because of the large number of known and unknown factors involved, although some causative factors can be revealed by traditional clinical diagnostic methods such as magnetic resonance imaging (MRI) [1]. To date, there are only a few effective cures for genetic diseases. For instance, some symptomatic therapies for specific diseases such as genetic-based cholestatic disorders, Wilson’s disease and tyrosinemia can be cured by orthotopic liver transplantation [2].
Here, we report a case of a 15-year-old male adolescent who initially manifested weakness in both lower limbs at the age when he learned to walk, and then gradually developed obvious gait abnormalities, muscle atrophy, and talipes equinovarus, accompanied with early-onset glaucoma. The parents of the proband are consanguineous, and he has a younger brother aged 2 with normal phenotype. To further investigate this inherited disease, we performed whole-exome sequencing on the proband, his parents and younger brother, assuming that the condition was inherited in an autosomal recessive pattern.
Results
A 15-year-old male adolescent with unconfirmed Charcot–Marie–Tooth disease-like symptoms
The case in question relates to a 15-year-old male adolescent. He was born after a full-term pregnancy and caesarian section due to fetal distress with normal Apgar score, followed by normal growth and intelligence development. His consanguineous parents and the 2-year-old younger brother exhibited normal phenotype. However, he presented lower extremity weakness, gait abnormalities, foot deformities, and early-onset glaucoma, but no complaint of obvious paresthesia, numbness or pain. Biochemical examination showed that metabolites in urinary and blood samples of the probands were within normal range. In addition, his cranial MRI showed no abnormality and karyotype analyses also appeared normal. We further performed electromyography (EMG) examination on motor and sensory nerves, and found that the current nerve conduction velocity (NCV) and EMG finding is consistent with a primary demyelinating motor and sensory polyneuropathy, with moderate-to-severe slowing of conduction velocities and absence of sensory nerve action potentials (SNAPs). Besides, the clinical diagnosis mostly matches Charcot–Marie–Tooth disease type 1 (CMT1). However, mutation analysis of the main gene PMP22 of CMT1 was negative. Therefore, the exact cause of the condition remains unclear.
CMT is a clinically and genetically heterogeneous group of inherited motor and sensory peripheral neuropathies. In general, this syndrome has an infantile or juvenile onset, with motor and sensory polyneuropathy semiology and pes cavus [3–6]. On the basis of mode of inheritance, clinical phenotypes, electrophysiological and pathological features, CMT can be classified into the following types: CMT1, CMT2, CMT4, autosomal recessive (AR)-CMT2 and dominant intermediate (DI)-CMT (Table S1). Each type can be further divided into several subtypes, depending on the underlying causative molecular defect. Based on the aforementioned EMG results and primary complaints from the patient [7], we hypothesized that this proband suffered from CMT4B2 [8–10], although idiopathic congenital talipes equinovarus [11] and other neuromuscular disorders such as spinal muscular atrophy [12] cannot be excluded.
Targeted next-generation sequencing (NGS) of the four family members
About 265,000 exonic regions from the genomic DNA of the 4 family members were captured. On an average, 12 Gb of sequencing data were generated from each individual, resulting in a mean depth of more than 110 × per base per targeted region. The coverage of the target region was over 99% (Table 1). In total, we identified 107,122 single-nucleotide polymorphisms (SNPs) from the exome of the proband (Table 2) by using SOAPsnp based on Bayes’ theorem, by filtering out low-quality mutations according to the criteria (reads < 20, depths < 4, estimated copy number > 2 and the interval in between < 5). Numbers and distribution of SNPs and insertion-deletions (indels) of the four family members are listed in Table 2 and Table 3, respectively. Next, we filtered the SNPs and indels according to the filtering strategy as delineated in Figure 1. Based on the initial screening criteria with variant location and frequency ⩽ 0.005 in dbSNP, Hapmap and 1000 Genomes Database, and mutation type, we identified 3592 SNPs and indels. The subsequent restrictive filtering control further narrowed down the list and only 542 SNPs and indels were retained.
Table 1.
Exon coverage and depth using whole-exome sequencing for the four family members
| Exome capture statistics | Father | Mother | Son-case | Son-control |
|---|---|---|---|---|
| Target region (bp) | 44,234,141 | 44,131,461 | 44,234,141 | 44,234,141 |
| Raw reads | 132,038,330 | 143,078,102 | 123,878,094 | 156,397,584 |
| Raw data yield (Mb) | 11,883 | 12,877 | 11,149 | 14,076 |
| Mean depth of target region (×) | 126.08 | 136.35 | 115.18 | 145.29 |
| Coverage of target region (%) | 99.40 | 99.41 | 99.41 | 99.05 |
| CCDS exons (read coverage) | 265,204 | 264,392 | 265,247 | 264,808 |
| CCDS genes (read coverage) | 18,386 | 18,335 | 18,386 | 18,381 |
Note: CCDS, Consensus Coding Sequence Dataset.
Table 2.
Detailed whole-exome sequencing data for SNPs detected in the four family members
| Type | Father | Mother | Son-case | Son-control |
|---|---|---|---|---|
| Total number of SNPs from each member | 112,238 | 111,822 | 107,122 | 111,669 |
| Missense | 13,246 | 13,221 | 12,748 | 12,748 |
| Readthrough | 92 | 88 | 86 | 85 |
| Nonsense | 177 | 155 | 158 | 157 |
| Splice site | 2798 | 2732 | 2647 | 2731 |
| Synonymous-coding | 10,876 | 10,763 | 10,318 | 10,386 |
| 5′-UTR | 9211 | 9055 | 8741 | 9040 |
| 3′-UTR | 3387 | 3421 | 3238 | 3425 |
| Intronic | 70,013 | 69,952 | 66,830 | 70,645 |
| Intergenic | 2438 | 2435 | 2356 | 2452 |
| Homozygous | 45,121 | 44,639 | 49,047 | 48,643 |
| Heterozygous | 67,117 | 67,183 | 58,075 | 63,026 |
| Total number of SNPs from all members | 154,336 |
Table 3.
Detailed whole-exome sequencing data for indels detected in the four family members
| Type | Father | Mother | Son-case | Son-control |
|---|---|---|---|---|
| Total number of indels from each member | 7174 | 7073 | 7024 | 7352 |
| Frameshift | 308 | 309 | 289 | 260 |
| CDS-indel | 184 | 175 | 183 | 212 |
| Splice site | 410 | 387 | 380 | 421 |
| 5′-UTR | 664 | 631 | 642 | 637 |
| 3′-UTR | 256 | 228 | 247 | 264 |
| Intron | 5233 | 5221 | 5149 | 5440 |
| Promoter | 35 | 38 | 33 | 35 |
| Intergenic | 84 | 84 | 101 | 83 |
| Homozygous | 2761 | 2570 | 2863 | 3275 |
| Heterozygous | 4413 | 4503 | 4161 | 4077 |
| Total insertions | 3223 | 3218 | 3220 | 3542 |
| Total deletions | 3951 | 3855 | 3804 | 3810 |
| Insertion-coding | 208 | 217 | 206 | 229 |
| Deletion-coding | 284 | 267 | 266 | 243 |
| Total number of indels from all members | 11,502 |
Figure 1.
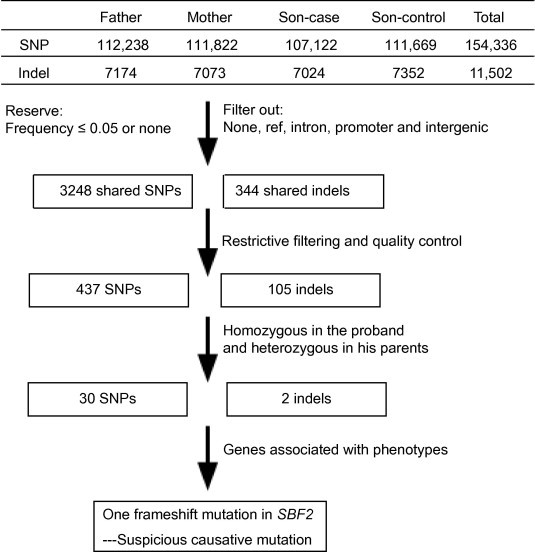
Bioinformatic analysis pipeline for whole-exome sequencing data in the current study
The mutations detected by the whole-exome sequencing were filtered through a series of steps. First, the low-quality mutations were filtered out (criteria: reads < 20, depths < 4, estimated copy number > 2 and the interval in between < 5). Second, the SNPs and indels were filtered based on the frequency and location mutation type. Third, candidate SNPs and indels were further filtered considering the database frequency and mutation types by adopting the pedigree analysis procedures developed in house (only 542 SNPs and indels were retained). Finally, we took into account the inheritance mode and phenotype and the candidate gene SBF2 was found to be related to the phenotype. None in SNP category indicate that the mutation was detected in the SOAPsnp but was filtered out at the first step, whereas none in indel category indicate lack of indel in the tested sample. Ref indicates identity with the reference sequence. Intergenic, intron and promoter indicate that SNP or indel is located in the intergenic region, intron or promoter region, respectively.
Identification of a novel SBF2 mutation, c.4571delG, in CMT4B2
Based on the family history and clinical results (Figure 2A), we determined that the condition is transmitted in an autosomal recessive pattern. As a result, only homozygous variations identified in the patient were retained for further analysis, since heterozygous variations carried by the parents demonstrated normal phenotype. We therefore obtained 32 SNPs and indels from 29 genes (Table S2). Among them, only the gene SBF2 was found to be related to the phenotype.
Figure 2.
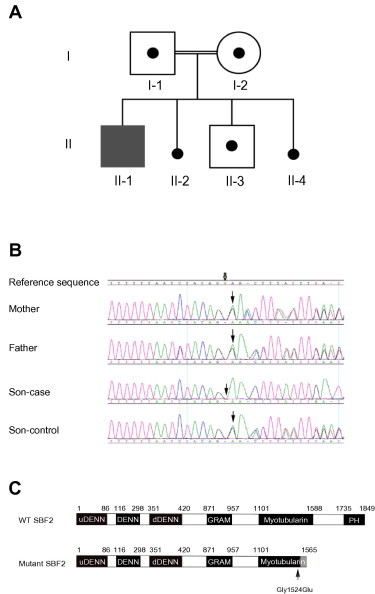
Pedigree diagram of the family
A. The proband (II-1, Son-case) showed clinical manifestations, but not his consanguineous parents (I-1, Father; I-2, Mother) and younger brother (II-3, Son-control). II-2 and II-4 were subjected to active abortion. B. Diagonal arrow indicates the SBF2 c. 4571del; the proband carried c. 4571delG, a 1-bp deletion in exon 34 of SBF2 in homozygous form; his parents and younger brother carried this mutation in the heterozygous form. The GenBank accession number for the reference sequence is NG_008074.1. C. Schematic representation of wild type and the potential mutated SBF2 protein. The c. 4571delG deletion causes amino acid change at codon 1524 (Gly1524Glu), leads to the frame shift onward and thus creates a stop codon at codon 1566 (p.Gly1524Glufs∗42). This premature termination may result in a truncated SBF2 protein product comprising 1565 amino acid residues or lack of expression due to the nonsense-mediated decay. The gray box represents the 42 C-terminal missense-mutated residues including the Gly1524Glu mutation. DENN, differentially expressed in neoplastic versus normal cells; uDENN, upstream DENN; dDENN, downstream DENN. Myotubularin indicates the myotubularin-like phosphatase domain. PH, pleckstrin homology domain.
SBF2 (also called MTMR13 or DENND7B, MIM 607697) is located on chromosome 11, base pairs 9,800,213–10,315,753. It contains 40 exons and spans over 600 kb. SBF2 encodes SET binding factor 2, a novel inactive phosphatase of the myotubularin family [8,13], which is present in various tissues such as the cord and the peripheral nerve. Mutations in SBF2 may cause CMT with early-onset glaucoma (CMT4B2) [7]. In humans, myotubularin-related proteins (MTMRs) constitute a large family with 14 members and have been suggested to work in phosphoinositide-mediated signaling events that may also convey the control of myelination [8]. Recessive mutations in SBF2 are typically found in patients with redundant myelin loops or myelin outfoldings in sural nerve biopsies [8]. Remarkably, our sequencing data revealed one novel mutation, c.4571delG, in exon 34 of the SBF2 gene, which was detected as homozygous in the proband and heterozygous in the other three family members. This was also validated by the Sanger sequencing method (Figure 2B). The c.4571delG (p.Gly1524Glufs∗42) of SBF2 is a frameshift deletion mutation that causes a mutation in the myotubularin-like phosphatase domain of the protein. The deletion of a single guanine residue at codon 1524 within exon 34 produces a frameshift mutation, which generates a stop codon at codon 1566 (p.Gly1524Glufs∗42) and results in a premature termination codon. The parents carried this mutation in heterozygous form and were considered obligate carriers. The proband harbored the c.4571_4571delG in the homozygous form, while his younger brother with normal phenotype carried this mutation in its heterozygous form. The same mutation is also inherited from one of their consanguineous parents. The mutation was not present in the dbSNP and 1000 Genomes Database.
The catalytically-inactive SBF2 associates with MTMR2, which is an active lipid phosphatase also belonging to the MTMR family. MTMR2 is found in the mutated form in CMT4B1 [9,10]. Previous studies revealed a possible scenario in which MTMR phosphatases play roles in regulating endosomal trafficking of phosphatidylinositol 3-phosphate (PI(3)P) and bisphosphate PI(3,5)P2 [11,12]. However, molecular and cellular mechanisms addressing the correlation between the catalytic activity of MTMR phosphatases and their mutation in CMT4B remain to be elucidated. Our study showed that the c.4571delG mutation generated a truncated SBF2 protein (Figure 2C), which may disrupt the soluble and membrane-associated protein complex formed by SBF2 and MTMR2, leading to defects in PI(3)P and PI(3,5)P2 regulation. Because this mutation causes a premature termination, the production and function of the protein would be altered and such alteration was inherited from the consanguineous parents who carried the heterozygous deletion mutation but exhibited normal phenotype. Therefore, we considered the homozygous deletion mutation to be “pathogenic”. However, to verify this possibility, further experimental evidence would be required.
Discussion
Here we describe a novel mutation of the CMT-associated gene SBF2 in a Chinese patient. CMT is a clinically and genetically heterogeneous group of inherited motor and sensory peripheral neuropathies, with a prevalence of 17–40 per 100,000 inhabitants [14,15]. In general, this syndrome has an infantile or juvenile onset, with motor and sensory polyneuropathy semiology and pes cavus [3–6]. Mendelian segregation in families may follow either autosomal dominant, autosomal recessive or X-linked patterns. Many previous studies have shown that molecular genetics can be used in the typing of CMTs such as autosomal dominant and X-linked inheritance [16]. It is now used in case of rarer types of CMTs, such as CMT4 with autosomal recessive pattern, that are similar to CMT1 with dominant forms, but are usually more severe with an earlier onset age [17,18]. According to autosomal recessive inheritance and both axonal and demyelinating neuropathies, CMT4 can be differentiated from other forms of CMT. Nerve conduction velocities in these individuals are often below 40 m/s. The affected individuals exhibit the typical CMT phenotype of distal muscle weakness and atrophy associated with sensory loss, and frequently, pes cavus foot deformity. These individuals tend to present these symptoms at earlier ages and with more severe disease progression, although many CMT4 subtypes exist with extensive clinical overlap [13,19–21].
Up till now, 9 genes associated with CMT4 subtypes have been discovered: GDAP1 (CMT4A), MTMR2 (CMT4B1), SBF2 (CMT4B2), SH3TC2 (CMT4C), NDRG1 (CMT4D), EGR2 (CMT4E), PRX (CMT4F), FGD4 (CMT4H) and FIG4 (CMT4J) [22]. These subtypes can be differentiated through clinical features, ethnic background and neuropathological features. However, most of these subtypes can only be differentiated through genetic tests. For instance, CMT4B1 is caused by mutations in the gene encoding MTMR2 on chromosome 11q22 [23–25], and mutations in the SBF2 gene have been identified in CMT4B2 on chromosome 11p15 [8,26]. At present, there are still no effective cures for CMT4; however, we can administer symptomatic treatments by the neurologist, physiatrist and orthopedic surgeon, such as special shoes for foot drop correction and walking assistance, surgery for severe pes cavus, exercise for recovery, and symptomatic treatments for pain, depression and other symptoms [4,27–29].
In this study, we detected 32 homozygous mutations in 29 genes in a patient with unconfirmed CMT disease-like symptoms and the homozygous deletion mutation c.4571delG in CMT4B2-related gene SBF2 was considered pathogenic. Our protein domain organization analysis suggests that the c.4571delG mutation introduces a premature stop codon, which, if homozygous, may result in the loss of gene expression due to the nonsense-mediated decay or production of a truncated SBF2 protein lacking pleckstrin homology (PH) domain. The PH domain is present in more than 100 cellular signaling proteins, e.g., protein kinases and GTPases, and characteristically binds to proteins such as phosphatidylinositol within biological membranes [30]. It plays multiple roles in navigating proteins to different types of host membranes and recruiting them to appropriate subcellular compartments. In addition, PH domains are believed to bind to the heterotrimeric Gβγ protein complex: G protein, protein kinase C and small GTPases [31]. However, it appears that PH domains, despite sharing low-sequence homology and non-conserved protein function, have a conserved core structurally composed of seven β-strands and one C-terminal α-helix [32]. Here, our finding of the SBF2 c.4571delG mutant may introduce the possibility of disruption or dysfunction of SBF2-MTMR2 complex formation, which may, in turn, alter PI(3)P and PI(3,5)P2 regulation due to the PH domain deletion. Several studies also showed that SBF2 was identified as the disease-causing gene of CMT4B1 and CMT4B2 by molecular cloning [8,13,33]. Nonsense or splice-site mutations affecting the N-terminus part of the SBF2 protein have been identified in patients with CMT4B2 [13,34]. Irregular myelin structure (outfolding) is a characteristic sign of CMT4B2, and the mutations reported in this study may result in nonfunctional proteins. In addition, studies found that mutations of SBF2 can lead to thrombocytopenia [35], pancreatic adenocarcinoma [36] and hereditary neuropathies [8] as well.
NGS has significantly facilitated genome analysis. Whole-exome genome sequencing (WES) of all protein-coding DNA is well justified as an efficient strategy to search for alleles underlying rare Mendelian disorders [19]. Protein-coding genes constitute only approximately 1% of the human genome, but harbor ∼85% genetic mutations that determine disease-related traits. In contrast to the laborious approach of SNP homozygosity mapping, the exome sequencing approach is more time-saving, is independent from shared allelic heritage and can be done in the presence of allelic heterogeneity [37]. When WES was initially introduced, its utility was highly debated, with respect to high cost, incomplete coverage of exome and low sensitivity for structural variation. However, WES will likely be employed much more commonly in the future because of the practical advantages of the technology [38]. In addition to being applied in the diagnosis of Mendelian disorders, WES can also be used in the analysis of complex diseases, such as Parkinson’s disease, hypertension and autism, and will be helpful for individualized medical treatments [39].
In summary, we found a novel frameshift mutation, c.4571delG(P. Gly1524Glufs∗42), in the SBF2 gene, which likely leads to the CMT4B2 in the proband. Furthermore, our study shows that whole-exome sequencing can provide fast and accurate sequencing information for clinic diagnosis. Therefore, when a genetic disorder is strongly suspected, even in the case when traditional clinical genetic testing has difficulty in making a definite diagnosis, the application of NGS should be considered. This approach, together with continuing improvement of sequencing technology, allows medical genomics to be a routine diagnostic method for rare Mendelian disorders in the near future.
Materials and methods
Blood samples
According to the institutional ethical procedures, the peripheral blood samples of the proband, his parents and younger brother were obtained with their informed consents.
DNA extraction
Genomic DNA was extracted from 200 μl of peripheral venous blood by standard procedures using Qiamp Blood DNA mini Kit (Qiagen, Hilden, Germany) as instructed by the manufacturer. DNA integrity was evaluated by 2% agarose gel electrophoresis. All DNA samples were stored at −20 °C after analysis with NanoDrop.
Whole-exome sequencing
Whole-exome sequencing was carried out using the SeqCap EZ Human Exome Library v3.0 (64 M). Prior to sequencing, 3 μg of qualified genomic DNA from each sample was randomly fragmented to 200–300 bp in size on a Covaris Acoustic System, followed by end-repair, A-tailing and pair-end index adapter ligation. The adapter-ligated templates were purified using AgencourtAM Pure SPRI beads and amplified by 4 cycles of ligation-mediated PCR (LM-PCR): 2 min at 94 °C; 4 cycles of 10 s at 94 °C, 30 s at 62 °C and 30 s at 72 °C; followed by 5 min extension at 72 °C. LM-PCR products were hybridized to the SeqCap EZ Oligo pool for enrichment. After the hybridized fragments were bound to the streptavidin beads in 24 h, non-hybridized fragments were removed. Next, captured LM-PCR products were amplified by PCR (2 min at 98 °C; 15 cycles of 10 s at 98 °C, 30 s at 60 °C and 30 s at 72 °C; then 5 min at 72 °C), and analyzed using the Agilent 2100 Bioanalyzer and quantitative PCR to estimate the enrichment magnitude. The captured library was sequenced using Illumina HiSeq2000 Analyzers. We conducted 90 cycles per read to generate paired-end reads and 8 bp of the index tag following standard sequencing protocols from the manufacturer.
Sanger sequencing
Mutations identified by whole-exome sequencing were validated by Sanger sequencing. Primers flanking the candidate loci were designed based on reference genomic sequences of the Human Genome from GenBank in NCBI [GenBank ID: reference GRCh37(hg19)] and were synthesized by Invitrogen (Shanghai, China). PCR amplification was carried out using ABI 9700 Thermal Cycler with the primers (sense 5′-GGACTCCTCTTGTGCATTCTG-3′ and antisense 5′-GATAGACTGCTGGCTGCTTAG-3′). Subsequently, all PCR products were sequenced on ABI PRISM 3730 automated sequencer (Applied Biosystems). Genomic sequence data analysis was performed by DNASTAR SeqMan.
Authors’ contributions
MQ, XY and MC conceived and designed the experiments. YC performed the experiments and analyzed the data. YC, HH and NL contributed reagents/materials/analysis tools. MC, JW, LT, JW, WW wrote the paper. MQ, XY, HH, MC, HC and TW revised the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors have declared that no competing interests exist.
Acknowledgements
We are sincerely grateful to Mr. Jiang and his family for providing samples and offering us enough trust, understanding and partial funds. This work was supported by the National Natural Science Foundation of China (Grant No. 81172681).
Footnotes
Peer review under responsibility of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China.
Contributor Information
Xin Yi, Email: yix@genomics.cn.
Ming Qi, Email: qiming@genomics.cn.
Supplementary material
The classification of CMT diseases
List of 32 mutations homozygous in the proband but heterozygous in his parents
References
- 1.Ghosh P.S., Shah S.N., Mitra S. Knee MRI showing thickened peripheral nerves in Charcot–Marie–Tooth disease. Acta Neurol Belg. 2012;112:315–316. doi: 10.1007/s13760-012-0036-y. [DOI] [PubMed] [Google Scholar]
- 2.Fagiuoli S., Daina E., D’Antiga L., Colledan M., Remuzzi G. Monogenic diseases that can be cured by liver transplantation. J Hepatol. 2013;59:595–612. doi: 10.1016/j.jhep.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 3.Harding A.E., Thomas P.K. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain. 1980;103:259–280. doi: 10.1093/brain/103.2.259. [DOI] [PubMed] [Google Scholar]
- 4.Pareyson D., Marchesi C. Diagnosis, natural history, and management of Charcot–Marie–Tooth disease. Lancet Neurol. 2009;8:654–667. doi: 10.1016/S1474-4422(09)70110-3. [DOI] [PubMed] [Google Scholar]
- 5.Shy M.E., Garbern J.Y., Kamholz J. Hereditary motor and sensory neuropathies: a biological perspective. Lancet Neurol. 2002;1:110–118. doi: 10.1016/s1474-4422(02)00042-x. [DOI] [PubMed] [Google Scholar]
- 6.Dyck P.J. Inherited neuronal degeneration and atrophy affecting peripheral motor, sensory, and autonomic neurons. In: Dyck P.J., Thomas P.K., Lambert E.M., editors. Peripheral neuropathy. WB Saunders; Filadelfia: 1984. pp. 1600–1655. [Google Scholar]
- 7.Hirano R., Takashima H., Umehara F., Arimura H., Michizono K., Okamoto Y. SET binding factor 2 (SBF2) mutation causes CMT4B with juvenile onset glaucoma. Neurology. 2004;63:577–580. doi: 10.1212/01.wnl.0000133211.40288.9a. [DOI] [PubMed] [Google Scholar]
- 8.Senderek J., Bergmann C., Weber S., Ketelsen U.P., Schorle H., Rudnik-Schoneborn S. Mutation of the SBF2 gene, encoding a novel member of the myotubularin family, in Charcot–Marie–Tooth neuropathy type 4B2/11p15. Hum Mol Genet. 2003;12:349–356. doi: 10.1093/hmg/ddg030. [DOI] [PubMed] [Google Scholar]
- 9.Bolino A., Muglia M., Conforti F.L., LeGuern E., Salih M.A., Georgiou D.M. Charcot–Marie–Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nat Genet. 2000;25:17–19. doi: 10.1038/75542. [DOI] [PubMed] [Google Scholar]
- 10.Robinson F.L., Dixon J.E. The phosphoinositide-3-phosphatase MTMR2 associates with MTMR13, a membrane-associated pseudophosphatase also mutated in type 4B Charcot–Marie–Tooth disease. J Biol Chem. 2005;280:31699–31707. doi: 10.1074/jbc.M505159200. [DOI] [PubMed] [Google Scholar]
- 11.Ikonomov O.C., Sbrissa D., Foti M., Carpentier J.L., Shisheva A. PIKfyve controls fluid phase endocytosis but not recycling/degradation of endocytosed receptors or sorting of procathepsin D by regulating multivesicular body morphogenesis. Mol Biol Cell. 2003;14:4581–4591. doi: 10.1091/mbc.E03-04-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Odorizzi G., Babst M., Emr S.D. Phosphoinositide signaling and the regulation of membrane trafficking in yeast. Trends Biochem Sci. 2000;25:229–235. doi: 10.1016/s0968-0004(00)01543-7. [DOI] [PubMed] [Google Scholar]
- 13.Azzedine H., Bolino A., Taieb T., Birouk N., Di Duca M., Bouhouche A. Mutations in MTMR13, a new pseudophosphatase homologue of MTMR2 and Sbf1, in two families with an autosomal recessive demyelinating form of Charcot–Marie–Tooth disease associated with early-onset glaucoma. Am J Hum Genet. 2003;72:1141–1153. doi: 10.1086/375034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Combarros O., Calleja J., Polo J.M., Berciano J. Prevalence of hereditary motor and sensory neuropathy in Cantabria. Acta Neurol Scand. 1987;75:9–12. doi: 10.1111/j.1600-0404.1987.tb07882.x. [DOI] [PubMed] [Google Scholar]
- 15.Skre H. Genetic and clinical aspects of Charcot–Marie–Tooth’s disease. Clin Genet. 1974;6:98–118. doi: 10.1111/j.1399-0004.1974.tb00638.x. [DOI] [PubMed] [Google Scholar]
- 16.Barisic N., Claeys K.G., Sirotkovic-Skerlev M., Lofgren A., Nelis E., De Jonghe P. Charcot–Marie–Tooth disease: a clinico-genetic confrontation. Ann Hum Genet. 2008;72:416–441. doi: 10.1111/j.1469-1809.2007.00412.x. [DOI] [PubMed] [Google Scholar]
- 17.Harding A.E., Thomas P.K. Autosomal recessive forms of hereditary motor and sensory neuropathy. J Neurol Neurosurg Psychiatry. 1980;43:669–678. doi: 10.1136/jnnp.43.8.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas P.K. Autosomal recessive hereditary motor and sensory neuropathy. Curr Opin Neurol. 2000;13:565–568. doi: 10.1097/00019052-200010000-00010. [DOI] [PubMed] [Google Scholar]
- 19.Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M. Exome sequencing identifies the cause of a Mendelian disorder. Nat Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelis E., Erdem S., Van Den Bergh P.Y., Belpaire-Dethiou M.C., Ceuterick C., Van Gerwen V. Mutations in GDAP1: autosomal recessive CMT with demyelination and axonopathy. Neurology. 2002;59:1865–1872. doi: 10.1212/01.wnl.0000036272.36047.54. [DOI] [PubMed] [Google Scholar]
- 21.Otagiri T., Sugai K., Kijima K., Arai H., Sawaishi Y., Shimohata M. Periaxin mutation in Japanese patients with Charcot–Marie–Tooth disease. J Hum Genet. 2006;51:625–628. doi: 10.1007/s10038-006-0408-3. [DOI] [PubMed] [Google Scholar]
- 22.Bird T.D. Charcot–Marie–Tooth neuropathy type 4. In: Pagon R.A., Adam M.P., Ardinger H.H., Bird T.D., Dolan C.R., Fong C.T., editors. Gene reviews. University of Washington; Seattle (WA): 1998. updated 2014 April 17. [Google Scholar]
- 23.Bolino A., Brancolini V., Bono F., Bruni A., Gambardella A., Romeo G. Localization of a gene responsible for autosomal recessive demyelinating neuropathy with focally folded myelin sheaths to chromosome 11q23 by homozygosity mapping and haplotype sharing. Hum Mol Genet. 1996;5:1051–1054. doi: 10.1093/hmg/5.7.1051. [DOI] [PubMed] [Google Scholar]
- 24.Kim S.A., Taylor G.S., Torgersen K.M., Dixon J.E. Myotubularin and MTMR2, phosphatidylinositol 3-phosphatases mutated in myotubular myopathy and type 4B Charcot–Marie–Tooth disease. J Biol Chem. 2002;277:4526–4531. doi: 10.1074/jbc.M111087200. [DOI] [PubMed] [Google Scholar]
- 25.Houlden H., King R.H., Wood N.W., Thomas P.K., Reilly M.M. Mutations in the 5′ region of the myotubularin-related protein 2 (MTMR2) gene in autosomal recessive hereditary neuropathy with focally folded myelin. Brain. 2001;124:907–915. doi: 10.1093/brain/124.5.907. [DOI] [PubMed] [Google Scholar]
- 26.Othmane K.B., Johnson E., Menold M., Graham F.L., Hamida M.B., Hasegawa O. Identification of a new locus for autosomal recessive Charcot–Marie–Tooth disease with focally folded myelin on chromosome 11p15. Genomics. 1999;62:344–349. doi: 10.1006/geno.1999.6028. [DOI] [PubMed] [Google Scholar]
- 27.Guyton G.P., Mann R.A. The pathogenesis and surgical management of foot deformity in Charcot–Marie–Tooth disease. Foot Ankle Clin. 2000;5:317–326. [PubMed] [Google Scholar]
- 28.Ward C.M., Dolan L.A., Bennett D.L., Morcuende J.A., Cooper R.R. Long-term results of reconstruction for treatment of a flexible cavovarus foot in Charcot–Marie–Tooth disease. J Bone Joint Surg Am. 2008;90:2631–2642. doi: 10.2106/JBJS.G.01356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Young P., De Jonghe P., Stogbauer F., Butterfass-Bahloul T. Treatment for Charcot–Marie–Tooth disease. Cochrane Database Syst Rev. 2008:CD006052. doi: 10.1002/14651858.CD006052.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wackerhage H., Del Re D.P., Judson R.N., Sudol M., Sadoshima J. The Hippo signal transduction network in skeletal and cardiac muscle. Sci Signal. 2014;7:re4. doi: 10.1126/scisignal.2005096. [DOI] [PubMed] [Google Scholar]
- 31.Zhu G., Chen J., Liu J., Brunzelle J.S., Huang B., Wakeham N. Structure of the APPL1 BAR-PH domain and characterization of its interaction with Rab5. EMBO J. 2007;26:3484–3493. doi: 10.1038/sj.emboj.7601771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rebecchi M.J., Scarlata S. Pleckstrin homology domains: a common fold with diverse functions. Annu Rev Biophys Biomol Struct. 1998;27:503–528. doi: 10.1146/annurev.biophys.27.1.503. [DOI] [PubMed] [Google Scholar]
- 33.Bolino A., Levy E.R., Muglia M., Conforti F.L., LeGuern E., Salih M.A. Genetic refinement and physical mapping of the CMT4B gene on chromosome 11q22. Genomics. 2000;63:271–278. doi: 10.1006/geno.1999.6088. [DOI] [PubMed] [Google Scholar]
- 34.Conforti F.L., Muglia M., Mazzei R., Patitucci A., Valentino P., Magariello A. A new SBF2 mutation in a family with recessive demyelinating Charcot–Marie–Tooth (CMT4B2) Neurology. 2004;63:1327–1328. doi: 10.1212/01.wnl.0000140617.02312.80. [DOI] [PubMed] [Google Scholar]
- 35.Abuzenadah A.M., Zaher G.F., Dallol A., Damanhouri G.A., Chaudhary A.G., Al-Sayes F. Identification of a novel SBF2 missense mutation associated with a rare case of thrombocytopenia using whole-exome sequencing. J Thromb Thrombolysis. 2013;36:501–506. doi: 10.1007/s11239-012-0864-x. [DOI] [PubMed] [Google Scholar]
- 36.Franks I. Genetics: variants in SBF2 gene associated with survival in pancreatic adenocarcinoma. Nat Rev Gastroenterol Hepatol. 2013;10:4. doi: 10.1038/nrgastro.2012.244. [DOI] [PubMed] [Google Scholar]
- 37.Majewski J., Schwartzentruber J., Lalonde E., Montpetit A., Jabado N. What can exome sequencing do for you? J Med Genet. 2011;48:580–589. doi: 10.1136/jmedgenet-2011-100223. [DOI] [PubMed] [Google Scholar]
- 38.Goh G., Choi M. Application of whole exome sequencing to identify disease-causing variants in inherited human diseases. Genomics Inform. 2012;10:214–219. doi: 10.5808/GI.2012.10.4.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nuytemans K., Vance J.M. Whole exome sequencing. Rinsho Shinkeigaku. 2010;50:952–955. doi: 10.5692/clinicalneurol.50.952. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The classification of CMT diseases
List of 32 mutations homozygous in the proband but heterozygous in his parents