

Abstract
MicroRNAs (miRNAs) were discovered two decades ago, yet there is still a great need for further studies elucidating their genesis and targeting in different phyla. Since experimental discovery and validation of miRNAs is difficult, computational predictions are indispensable and today most computational approaches employ machine learning. Toxoplasma gondii, a parasite residing within the cells of its hosts like human, uses miRNAs for its post-transcriptional gene regulation. It may also regulate its hosts’ gene expression, which has been shown in brain cancer. Since previous studies have shown that overexpressed miRNAs within the host are causal for disease onset, we hypothesized that T. gondii could export miRNAs into its host cell. We computationally predicted all hairpins from the genome of T. gondii and used mouse and human models to filter possible candidates. These were then further compared to known miRNAs in human and rodents and their expression was examined for T. gondii grown in mouse and human hosts, respectively. We found that among the millions of potential hairpins in T. gondii, only a few thousand pass filtering using a human or mouse model and that even fewer of those are expressed. Since they are expressed and differentially expressed in rodents and human, we suggest that there is a chance that T. gondii may export miRNAs into its hosts for direct regulation.
Keywords: Toxoplasma gondii, MicroRNA, Regulation, Host interaction, Parasite
Introduction
Mature microRNAs (miRNAs) are short, single-stranded RNAs of 18–24 nucleotides (nt) in length. They essentially represent a short RNA sequence which is somewhat complementary to their target mRNAs, leading to either destabilization of the mRNAs or inhibition of their translation [1,2]. These posttranscriptional regulators were initially discovered in Caenorhabditis elegans about two decades ago [3]. Since then, miRNAs have been discovered in many species from viruses to human, in which they play various roles that are still under investigation [4,5]. Many such studies succeeded in creating links between miRNA dysregulation and human diseases like cancer and neurodegeneration [4,6,7]. Thus, it is not surprising that it has been estimated that 30% of all protein-coding genes are controlled by one or more miRNAs [2]. Although miRNAs are found in multicellular organisms ranging from sponges [8] to animals, the plant miRNA pathway may have evolved distinctly [9].
Many mammalian miRNA loci are found in close proximity to each other and such clustered miRNAs are transcribed from a single polycistronic transcription unit (TU) [10]; conversely, some miRNAs originate from distinct gene promoters [8] or are part of other transcription units, for example, genes. MicroRNAs seem to be located in most parts of a genome. Some can arise from non-coding TUs, others come from protein-coding TUs [8]. Approximately 40% of miRNAs are located in intronic regions of non-coding transcripts and 10% can be placed into exonic regions. Most of the remaining miRNAs are found within introns of protein-coding TUs [8], although alternative splicing might produce miRNAs that can be equally well labeled as exonic or intronic according to our observation.
T. gondii can produce and utilize miRNAs and these miRNAs show metazoan-like features, in terms of its own regulation [11]. Unfortunately, a limited body of knowledge about miRNAs in T. gondii is available and no miRNAs from Apicomplexa have been recorded in miRBase. We established a miRNA regulatory network in T. gondii [12]. Our prediction was based on filtration of detected hairpins and targets using different features [12]. Due to the little knowledge about the actual biological miRNA pathway in Apicomplexa existing at that time, this network should be recomputed once enough experimental evidence becomes available.
In contrast to our approach used to establish a preliminary T. gondii miRNA regulatory network, to date, most miRNA hairpin detection approaches are based on machine learning [13]. Despite the popularity of data mining approaches, there are two major drawbacks with current miRNA gene identification approaches [13]. The first concerns class imbalance during learning [14], which is due to the assumption that there are few true miRNAs in a genome (currently about 1881 hairpins for human in miRBase [15]), while millions of hairpins are expected to exist in a genome that are not miRNAs (11 million for human [16]). We have investigated the impact of class imbalance on learning for miRNA prediction and found that during learning the positive and negative examples should be balanced for best performance [14]. Among the positive data, another problem arises since most of the validation of miRNAs is not at the protein level but at the transcription level [17]. The second major problem resides in feature selection and filtering. Features like stem length and minimum free energy are used for filtering data such that candidates outside of predefined ranges are discarded, which may lead to poor performance of trained models and a low prediction accuracy [18]. In addition to these issues, we have shown that the quality of positive examples for miRNA gene predictions, which is usually obtained from miRBase, may confer convoluting artifacts [19]. We took these issues into consideration and found that filtering of questionable miRNAs from miRBase improves prediction accuracy [19].
Not much is known about miRNAs in T. gondii, but the influence of T. gondii on its host’s miRNAs has been studied. It has been shown that T. gondii causes dysregulation of miRNA expression profiles within its host cells and, for example, plays a role in brain cancer [20]. Other studies showed upregulation of specific human miRNAs in infected cells, compared to the non-infected cells [21–23]. Since T. gondii contains miRNAs [11], we hypothesize that T. gondii may be able to process and export hairpins into its host cells. We are thus interested whether human or rodent analogous miRNAs are produced by T. gondii, which could be leaked into the host cells to modulate miRNA levels in the human cells similarly as described for brain cancer [20] but by directly increasing the expression levels of selected miRNAs through export into the host cells. To investigate this matter, we folded the genome of T. gondii, extracted all possible hairpins and investigated them with learned models for human and rodents. We further investigated if the hairpins in T. gondii are actually expressed and checked sequence homology of the mature sequences in human and rodent examples from miRBase against the predicted hairpins. From ∼4.5 million hairpins in the genome of T. gondii, ∼28,000 (0.6%) and ∼65,000 (1.4%) were found plausible in respect to the human and rodent models, respectively, with ∼9000 (0.2%) for both. Depending on the similarity cutoff, some of the putative pre-miRNAs show a good homology to either human and/or rodent mature miRNAs with well-conserved seed sequences (1977; 0.04% at a cutoff >0.9). Furthermore, we confirmed expression for ∼8000 putative hairpins in T. gondii, using publicly-available next generation sequencing datasets (see Materials and methods). Based on the sum of these findings we believe that T. gondii produces human/rodent like pre-miRNAs and with a leap of faith, we conclude that it may transport these hairpins into its host cells for regulation purposes.
Results
Establishing of human and rodent models via data mining
We used the human and the combined rodent miRNAs from miRBase for training of two models for miRNA prediction with the widely-applied pseudo dataset serving as the negative examples (see Materials and methods). The best model for human achieved an F-measure of 0.87 and the best for rodents was 0.92 (Table 1). These results are not sufficient if the aim is to experimentally validate a large number of hairpin candidates. For the current study, however, they suffice to provide evidence for whether a predicted T. gondii miRNA hairpin could be functional in a rodent or human host. In the future, better models will increase the confidence in the results of this study and a selection of good candidates could be experimentally validated.
Table 1.
Statistics for the best models for various learners
| Positive data | Learner | Recall | Precision | Sensitivity | Specificity | F-measure | Accuracy |
|---|---|---|---|---|---|---|---|
| Human | SVM | 1.000 | 0.501 | 1.000 | 1.000 | 0.668 | 0.501 |
| DT | 0.840 | 0.839 | 0.840 | 0.840 | 0.835 | 0.835 | |
| MLP | 0.835 | 0.835 | 0.835 | 0.835 | 0.835 | 0.835 | |
| NB | 0.845 | 0.843 | 0.845 | 0.845 | 0.838 | 0.837 | |
| BLR | 0.840 | 0.800 | 0.840 | 0.840 | 0.765 | 0.741 | |
| RF | 0.872 | 0.872 | 0.872 | 0.872 | 0.872 | 0.872 | |
| Rodent | SVM | 1.000 | 0.501 | 1.000 | 1.000 | 0.668 | 0.501 |
| DT | 0.859 | 0.851 | 0.859 | 0.859 | 0.840 | 0.837 | |
| MLP | 0.907 | 0.904 | 0.907 | 0.907 | 0.890 | 0.888 | |
| NB | 0.897 | 0.890 | 0.897 | 0.897 | 0.871 | 0.866 | |
| BLR | 0.864 | 0.812 | 0.864 | 0.864 | 0.761 | 0.728 | |
| RF | 0.918 | 0.918 | 0.918 | 0.918 | 0.916 | 0.916 | |
Note: The best results for each learner given either human or rodent positive data with pseudo hairpins as negative data. RF performs best for this dataset (F-measure bolded). General performance among classifiers does not differ greatly even without parameter optimization. SVM, support vector machine; DT, decision tree; MLP, multi-layer perceptron; NB, naïve Bayes; BLR, Bayesian logistic regression; RF, random forest.
Extraction of hairpins from the T. gondii genome
The folded T. gondii genome contains about 5 million hairpin structures, but this number is inflated twofold since both the template and the reverse strand have been used. Furthermore, the folds of overlaps between two consecutive overlapping 500 nt fragments are rarely identical. Therefore, it is difficult to integrate hairpins from overlaps. Thus, we decided to abstain from integration and accepted all hairpin models which inflates the number of results (at maximum doubles it). We were able to calculate features that define a pre-miRNA for about 4.5 million of extracted hairpins while the remaining 500,000 hairpins had certain not applicable structures for some feature calculations (e.g., having no bonds after extraction and refolding). These were discarded as it is highly unlikely that they can be true hairpins.
Application of learned models to putative hairpins
The learners that were trained in the Konstanz Information Miner (KNIME, http://www.knime.org/) provide a score for a candidate hairpin (being miRNA class or pseudo class) and assign it to the miRNA class if with a score ⩾0.5 (Figure 1). To see the impact of this assignment on the number of hairpins that are assigned as being a miRNA, we plotted the miRNA score versus the number of candidates with equal or higher miRNA score at different cutoff values (Figure 2). As expected, with more stringent scoring, the number of candidate hairpins assigned to be a miRNA candidate drops sharply. At a cutoff of 0.9, there are about 65,000 miRNA candidates for rodents and ∼28,000 for human, respectively (Figures 1 and 2). 9000 putative pre-miRNAs from T. gondii pass the expectation of both models. These numbers are quite large compared to about 2000 human miRNAs listed in miRBase, but given 5 million hairpins and an accuracy of around 0.9, we could expect to find 500,000 candidate hairpins passing the model. Instead, only ∼0.6% of T. gondii hairpins passing the human model (1.4% for rodent) remained after filtering, suggesting that using the trained models was quite successful, although still too many hairpins may have passed this filtering step. Such practice most likely causes the inclusion of false positive candidates, which is not very problematic for this study, but would cause problems if the aim is experimental validation of miRNAs.
Figure 1.
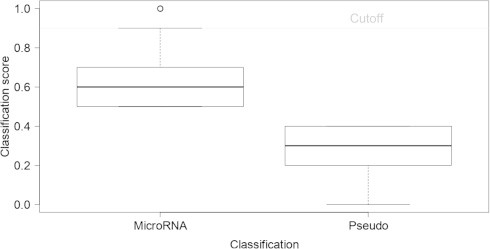
MicroRNA and pseudo classification score distribution
All hairpins in T. gondii were classified as either microRNA class (score ⩾ 0.5) or pseudo (score < 0.5). The distribution of the microRNA score is shown in the left box and whisker plot, whereas the pseudo prediction is shown in the right box. The horizontal line indicates the cutoff value (score > 0.9) we used for selecting microRNAs and call them putative microRNA hairpins.
Figure 2.
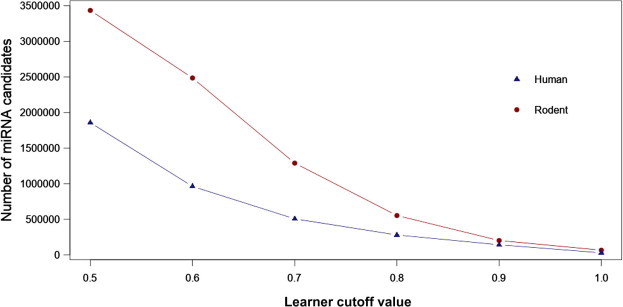
Hairpin counts at different learner threshold
Number of T. gondii hairpins that pass the human model (blue) and rodent model (red) in respect to cutoff values for accepting a hairpin as a putative pre-microRNA. The learners were applied to all potential hairpins in the T. gondii genome. Rodents have roughly twice as many hairpins that pass the filtering as compared to human at all cutoff values.
Analysis of filtered hairpins
After applying the models, the distribution of stem length (SL) and base pairing propensity (BPP) for the hairpin candidates from the T. gondii genome and the corresponding distribution for the hairpins that pass the 90% threshold for being a miRNA according to the human model and rodent model were calculated (Figure 3). By using the learned models to filter the results, the distribution of SL and BPP is changed such that it better fits the expectations for a pre-miRNAs for human (Figure 3C). This is well seen for the SL distribution for both human and rodents. SL tends to be in an acceptable range around 20–30 for miRNAs but tends to be more variable and generally larger for the hairpins classified as pseudo. Also, SL distribution for miRNAs is similar between rodents and human. BPP normalized to the hairpin length (HPL) is well distributed for human but not for rodent miRNAs. BPP/HPL ratios among accepted miRNAs have a smaller inter quartile range but larger values, which translates to more bonds within their hairpins for human, but interestingly not for rodent models, possibly due to the usage of a mixture rodent dataset for training. Human hairpins classified as miRNAs fit the general expectation for pre-miRNAs in respect to normalized BPP distribution. Although stems can be of varying size to produce mature miRNAs of 18–24 nt in length, they should contain a large number of bonds in order to be recognized by the various enzymes in the canonical miRNA genesis pathway.
Figure 3.
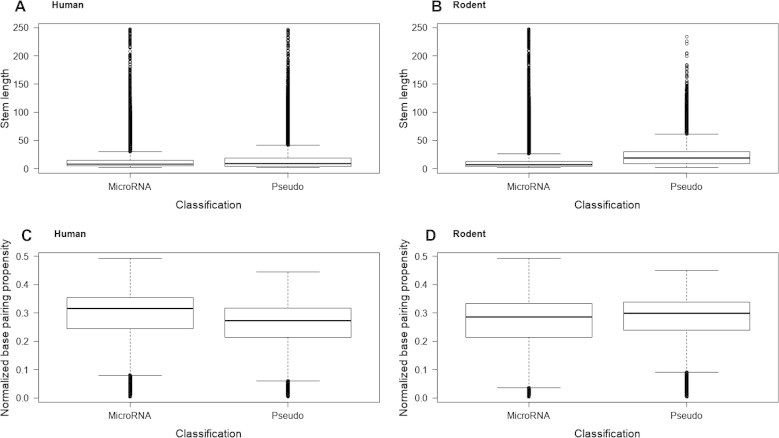
Hairpin length and base pairing propensity distribution
Distribution of stem length (A and B) and base pairing propensity normalized to hairpin length (C and D) for all hairpins that were extracted from the T. gondii genome, according to the human and rodent models, respectively. Pseudo refers to all hairpins that have been named pseudo microRNA using the trained models and microRNA refers to the ones that were named microRNA (score > 0.9).
Comparison to known human and rodent mature miRNAs
We then asked the question as to whether any of the known mature miRNAs from human or rodent match to any of the predicted hairpins in T. gondii. We scored the mature matches (see Materials and methods) and plotted them against different miRNA score thresholds as provided by the trained models (Figure 4). It is well seen for human 5′ mature miRNAs (Figure 4A) and somewhat less clear for rodent 5′ and 3′ mature miRNAs (Figure 4D and E), that with an increase in mature MatchScore, a better miRNA score can be expected and vice versa. Interestingly, this trend was not observed for the 3′ mature human miRNAs. For rodents, the correlation between high MatchScore and high miRNA score is not as strong as that for human, which is expected due to the mixture model of multiple rodent species used. Possibly, the 3′ mature miRNAs in human are not as reliable as the 5′ miRNAs, since many 3′ mature miRNAs may only be predictions. Some of the mature miRNAs in miRBase are not assigned a 5′ or 3′ status and they are summarized in Figure 4C and F. It can be seen that for human, no trend can be discerned, whereas for rodents the expected trend is observable. The number of matches changes with the applied MatchScore threshold. We decided to go with a conservative cutoff of 40 at which we find around 150 viable hairpins (Table S1).
Figure 4.
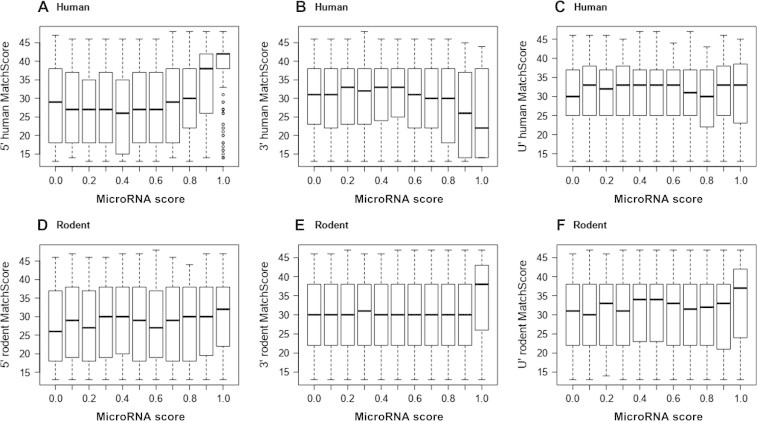
MatchScore at different microRNA score threshold
Box and whisker plots for the distribution of our calculated MatchScore (see Materials and methods), separate for 5′ (A and D), 3′ (B and E) and unspecified prime (C and F) mature sequences matching to T. gondii hairpins. The distribution is calculated for different microRNA cutoff values provided by the learned models for human and rodents. Panels A−C show human mature microRNAs matching to predicted T. gondii hairpins, while panels D−F show the same information for rodents. Unspecified prime (U′) refers to mature sequences for which 5′ or 3″ were not specified in miRBase.
Expression of T. gondii hairpins
In order to see whether any of these putative hairpins are expressed in T. gondii, we examined the expression profile in several publicly-available next-generation sequencing data collections (see Materials and methods). About 26,000 T. gondii hairpins are expressed within a human host and about 300,000 in a rodent one. About 11,000 are shared between the two hosts (Figure S1). For most hairpins at a model threshold of 0.9, we cannot find evidence of expression and when increasing the expression level threshold, the number of remaining hairpins decreases further (Figure 5). At a model threshold of 0.9, 1165 human, 7369 rodent, and 340 shared matches remain (Figure S1). For humans, less data was available to us so the expression counts and the hairpin numbers are much lower than those in rodents (scaled by factor 10 in Figure 5). The dataset for rodents was targeted to detect small RNAs, which is additionally in its favor. A representative selection of miRNAs that are expressed in human and/ or rodents is presented in Table 2. The comprehensive list of all miRNAs and their expression within the SRA collections we used is provided in Table S2.
Figure 5.
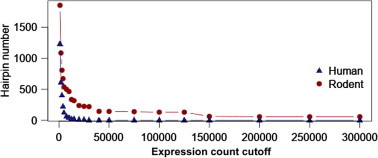
Hairpin count at different expression threshold
The number of expressed predicted T. gondii hairpins found were plotted against different cutoff values of expression (summed across replicates) for human (blue, scaled by a factor of 10 for better visualization) and rodents (red, not scaled).
Table 2.
T. gondii miRNAs expressed in human and rodent hosts
| T. gondii hairpin | Human model winner | Rodent model winner | Human expression summed | Rodent expression summed | Human miRNA match | Rodent miRNA match |
|---|---|---|---|---|---|---|
| TGME49_chrIV| 1013750|226 | miRNA | miRNA | 352 | 4 | hsa-let-7i-3p | mmu-let-7i-3p |
| TGME49_chrV| 869500|99 | miRNA | miRNA | 246 | 1 | hsa-miR-6758-3p | NA |
| TGME49_chrXI |5274000|264 | miRNA | miRNA | 16,368 | 62 | NA | NA |
| tgme49_asmbl. 431|0|426 | miRNA | miRNA | 0 | 296 | NA | rco-miR408 |
| tgme49_asmbl. 1387|250|438 | miRNA | miRNA | 0 | 199 | NA | NA |
| TGME49_chrVIIb| 800750|118 | miRNA | miRNA | 2 | 1 | hsa-miR-1277-5p | mmu-miR-466 m-3p |
| TGME49_chrIV| 1013750|226 | miRNA | miRNA | 352 | 4 | hsa-let-7i-3p | mmu-let-7i-3p |
| TGME49_chrVIIb| 3618000|195 | miRNA | miRNA | 146 | 5 | hsa-miR-297 | mmu-miR-297c-5p |
| TGME49_chrIX| 305250|185 | miRNA | miRNA | 106 | 36 | hsa-miR-877-3p | mmu-miR-877-3p |
| TGME49_chrVIIa| 327000|0 | miRNA | miRNA | 70 | 9 | hsa-miR-1281 | mmu-miR-7011-3p |
Note: Selection of 10 representative putative hairpins from Toxoplasma which pass the model threshold of 0.9 and are expressed according to the sample data we used in either or both of the hosts. A more comprehensive list of results can be found in Table S2. NA: not applicable.
T. gondii miRNAs might regulate host gene expression
MicroRNAs are derived from a hairpin structure and the mature sequences can be located on either arm of the stem and should not extend into the loop. We earlier pointed out that there are possibly wrong entries in miRBase [19], which do not adhere to these rules. In this analysis we also found such examples and discarded them, but we also found suitable miRNA candidates with match to rodent and human mature miRNAs (Figure 6). The mature miRNA seems capable of regulating its targets if the hairpin is transported into the host cell, since the complete seed sequence is identical and even some of the downstream sequence has a good match with the putative T. gondii mature miRNA. Table 3 provides the 5 best hairpin candidates from T. gondii that pass the human or rodent model, respectively. The remaining candidates that we believe are viable (∼150) are listed in Table S1, while Table S3 provides a comprehensive list of all such results.
Figure 6.
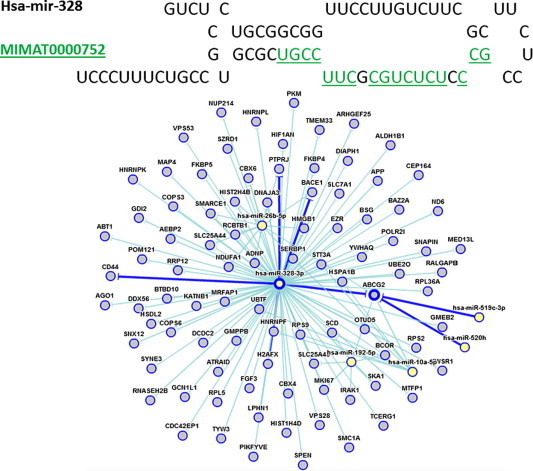
Hairpin and mature microRNA example and regulative network
A good match between a predicted hairpin from T. gondii (Supercontig tgme49_asmbl.1884, start: 5568) and the mature sequence MIMAT0000752 from the human pre-miRNA hsa-mir-328. The identities between the mature sequence and the putative hairpin are highlighted in green and underlined within the fold. The overall match length of the putative mature sequence is 19. miRTarBase contains the targets of hsa-mir-328-3p and the network (http://tinyurl.com/network3283p). Both are shown below the hairpin. The microRNA (yellow nodes) is centered in the network. Links to genes (blue nodes), indicating regulation with strong experimental evidence, are in dark blue otherwise in light blue.
Table 3.
Hairpins from T. gondii that may act as miRNAs in human or rodents
| Species | Hairpin accession | Mature miRNA accession | Toxoplasma matches | MatchScore | Alignment length | miRNA length |
|---|---|---|---|---|---|---|
| Human | hsa-miR-7107-3p | MIMAT0028112 | ChrVIIa|1970500|299 ChrVIIa|1970250|48 ChrVIIb|1456750|305 | 48 | 24 | 27 |
| hsa-miR-6873-3p | MIMAT0027647 | ChrXI|6456500|87 | 47 | 23 | 23 | |
| hsa-miR-6821-5p | MIMAT0027542 | ChrXI|1196000|234 | 47 | 23 | 23 | |
| hsa-miR-4644 | MIMAT0019704 | ChrXII|4149750|338 | 47 | 23 | 23 | |
| hsa-miR-3149 | MIMAT0015022 | ChrVIIb|1780250|75 ChrVIIb|1780000|308 | 47 | 23 | 23 | |
| Rodent | mmu-miR-6981-5p | MIMAT0027864 | ChrXII|2084000|351 | 48 | 24 | 26 |
| mmu-miR-669p-5p | MIMAT0014889 | ChrIX|6207250|158 | 47 | 23 | 24 | |
| mmu-miR-5131 | MIMAT0020642 | ChrIb|1049750|335 ChrIb|1049750|42 ChrIb|1049500|80 ChrIb|1049500|297 | 47 | 23 | 23 | |
| mmu-miR-669a-5p | MIMAT0003477 | ChrIX|6207250|158 | 47 | 23 | 24 | |
| mmu-miR-7033-5p | MIMAT0027970 | ChrXI|3948000|293 | 47 | 23 | 23 | |
Note: Hairpin accession and mature miRNA accession are referenced from miRBase. The T. gondii match is in the given chromosome and the 500-nt extracted fraction starts at the position specified through the number following it. Within that fragment the hairpin starts at the position specified thereafter. The score is biased to maximize similarity in the seed sequence (see Materials and methods). Alignment length and miRNA length are given to show that they are quite comparable. More comprehensive lists can be found in Tables S1 and S3. mmu, Mus musculus; hsa, Homo sapiens.
Discussion
We have mined the T. gondii genome for hairpins which fit a human or a rodent model and have found many fitting instances of hairpins (∼28,000 for human and ∼65,000 for rodents). Additionally, we examined whether there are mature sequences matching to human or rodent mature sequences within these hairpins. There were high quality matches at a conservative MatchScore cutoff of 40 between T. gondii hairpins and human (∼60) or rodent (∼90) mature miRNAs. Taken together, these data show that there may actually be miRNAs encoded in T. gondii, which, if exported into the host, could regulate target gene expression. We then checked whether we could find any evidence of expression of these hairpins in T. gondii and considered a number of available datasets from the Sequence Read Archive (SRA). About 8000 of the hairpins which pass the human or rodent model at a cutoff of 0.9 are found in the expressome of T. gondii cultivated in a human host or in a rodent host (Tables 2 and S2). Other putative hairpins may not be reported as expressed in the datasets we assessed, but could well be reported as expressed in other datasets. Thus, there is a need to increase the amount of expression data to map against in order to validate the lowly-expressed candidates and identify more hairpins.
We pointed out earlier, that one motivation for this study was our hypothesis that T. gondii may excrete miRNAs into the host to regulate protein expression levels. We found evidence for expression of some of the putative T. gondii hairpins and we identified well-conserved mature miRNAs in suitable hairpins according to the learned models. This taken together suggests that there is a possibility for hairpins to be transported into the host, where they could be processed by Dicer and incorporated into RISC and then regulate their targets. The accuracy of models for both human and rodent miRNAs must be further increased in the future to reduce the number of false predictions, but this is a field with ongoing research and we expect better models to appear soon [13].
Nonetheless, among the currently available candidates that we found, many may function as miRNAs in the host. For instance, Figure 6 shows the currently-known regulatory network involving hsa-mir-328, one of the potential miRNAs with a close match among the putative hairpins in the T. gondii genome. This single human miRNA has around 90 validated targets [32]. Considering the existence of many candidate miRNAs in T. gondii and the multitude of functions they could regulate in the host cell, the size of the network becomes very large and signifies the importance of studying this gene regulation mechanism.
In summary, we believe that there is some evidence suggesting that T. gondii may produce hairpins similar to its hosts and use them to regulate the hosts transcriptionally. Recent studies indicate that T. gondii infection can effectively alter the levels of host miRNAs [21]. Therefore, we suggest that some targeted experimental studies should be designed, which would monitor the expression levels of these miRNAs in infected cells and enable the backtracking of the origin of the miRNAs by, for example, differential labeling. This in turn may enable new predictive models to further investigate the occurrence of hairpins in T. gondii that can regulate its various hosts.
Conclusion
Toxoplasma gondii contains hairpins that are similar to human and rodent hairpins in terms of generally-used features such as HPL, BPP, and hairpin minimum free energy. We found that some of the hairpins are expressed as reported by the publicly-available data. The hairpins show good matches to human and rodent mature sequences, which could potentially bind the same mRNA as their respective human or rodent counterparts. It could be difficult to differentiate among human mature sequences and T. gondii mature sequences experimentally. Therefore it seems plausible that what looks like an increase in miRNA content in the infected host may be due to export of hairpins from T. gondii. It is also possible that other actions by T. gondii increase the human miRNA count via a different route. We conclude that export of miRNAs from T. gondii to its hosts is theoretically possible. However, more computational studies and follow-up experimental studies are necessary to provide further supporting evidence to this claim.
Materials and methods
Datasets
Although we have shown previously, that not all entries in miRBase are trustworthy [19], positive examples (1829 filtered hairpins with mature sequences) for human miRNAs were obtained from miRBase (http://www.mirbase.org, release 20) since it is the most comprehensive repository. In order to analyze rodent data, miRNA hairpin sequences of all rodentia species available in miRBase (Cricetulus griseus, Mus musculus and Rattus norvegicus) were combined and used as positive data. This amounted to 1888 hairpins containing 2987 mature sequences. As negative data, the pseudo hairpin dataset as used by Ng and Mishra [24] was obtained, since we have shown earlier, that this dataset is the one currently best suited for data mining approaches [14]. The genome of T. gondii strain ME49 was obtained from NCBI Genome Assembly (Genome ID: 30, Genome Assembly ID: 22622 and RefSeq accession No.: NZ_ABPA00000000.1). Four next-generation sequencing datasets for the T. gondii transcriptome grown in human host cells (database accession Nos: SRR1408847, SRR1408858, SRR1408861, and SRR1407792) and 5 miRNA-sequencing datasets for T. gondii grown in Kunming mice were downloaded from the SRA database [25] (database accession Nos: SRR771607, SRR771608, SRR771609, SRR771610 and SRR771611). The outcome of all calculations performed in this study is made available as File S1.
Numerical features for pre-miRNA description
Lopes et al. recently compared six ab initio hairpin detection algorithms and the features used by the tools [26]. They propose a feature set, SELECT, which they claim should be used for hairpin detection. Unfortunately, they reported sensitivity and specificity for their analysis which may be misleading (we are often able to achieve a sensitivity of 1 at a specificity at 1, but a low F-measure at the same time, see Table 1). In our previous studies, we also proposed that these two measures are not trustworthy and that recall, precision, and F-measure should be used when reporting miRNA prediction accuracy measures [27]. Furthermore, other advice for the proper use of machine learning in miRNA research, concerning positive data acquisition, class balance and feature selection, were also ignored, so that we elected not to use their SELECT feature set.
Previously, we ranked all features (approximately 700) that were explicitly or implicitly proposed to define a miRNA hairpin [14]. We also showed that 30 features may be enough for an efficient classification analysis. For this study, we implemented a subset of features including SL, HPL and a number of previously-published features normalized to either SL or HPL. These 32 features are shown in Table S4 and some of them are more extensively described in [27], but it is not within the scope of this study to perform feature selection or discuss features for miRNA prediction. The selected features were implemented in Java and calculated using our computer cluster at the Izmir Institute of Technology.
Data mining
KNIME is a workflow management system and a platform for data mining. In this study, we performed classification for model generation and predictions on T. gondii datasets by using KNIME. The overall strategy for data mining is summarized in Figure 7. The model establishment is shown on the top and the application of the model to T. gondii candidate hairpins is shown in the bottom panel.
Figure 7.
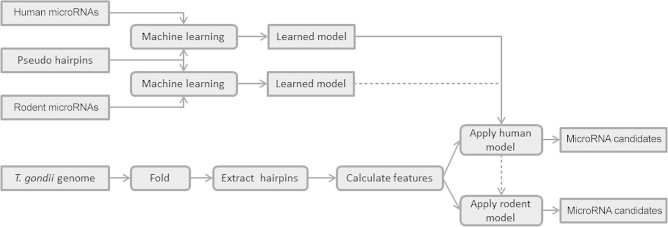
Candidate hairpin generation workflow from T. gondii genome
Workflow overview of how microRNA candidates were generated from the T. gondii genome. Two workflows were performed in parallel: model learning on top and candidate generation from the T. gondii genome on bottom.
For classification, positive and negative datasets were loaded on the platform. The negative dataset was randomly sampled, so that it had the same size with the positive data. Then, the combination of these datasets was divided into training (90%) and testing (10%) datasets using stratified sampling. Different classifiers, including support vector machine (SVM), decision tree (DT), multi-layer perceptron (MLP), naïve Bayes (NB), Bayesian logistic regression (BLR) and random forest (RF), were trained on these data. Following 100 repetitions of the random sampling and learning procedure, we obtained the highest and most consistent accuracy, F-measure, precision and recall values for the RF classifier. For further predictions, we used the model obtained from this learner (Table 1). The workflow created in KNIME is available as File S2 and can be used to repeat the learning.
The model obtained was then loaded into another KNIME workflow, which merely applies the model to all input data and associates scores with how well the prediction fits to a miRNA or a pseudo hairpin (Figure S2). The KNIME workflow to apply the model is available as File S3.
Extraction of hairpins from the T. gondii genome
The T. gondii genome was cut into 500-nt-long sequences with 250 nt overlap. All sequences were folded using RNAFold from the Vienna package [28]. All hairpins that at minimum contained a stem with three consecutive bonds and a loop with at least one nucleotide were extracted. The extracted candidates were then re-folded using RNAFold without the 500-nt context. All features were calculated for each candidate hairpin. After calculation, all candidates containing NA values were removed before further analysis. The remaining hairpin candidates were checked with the human and the rodent model using KNIME; and the classification assignment and score were added to the dataset.
Hairpin candidates stemming from overlaps may inflate the counts presented in this manuscript. However, in many cases the folds of overlapping candidates are not very similar so that they could not be combined into one candidate. Therefore, they could not be removed and were left in place during downstream analyses.
Mapping of expression data to the predicted hairpins
Figure 8 provides an overview of how the expression of miRNAs in T. gondii was estimated. This is a crude estimate and its sole purpose is to establish whether human and mouse analogous miRNAs are expressed in T. gondii.
Figure 8.
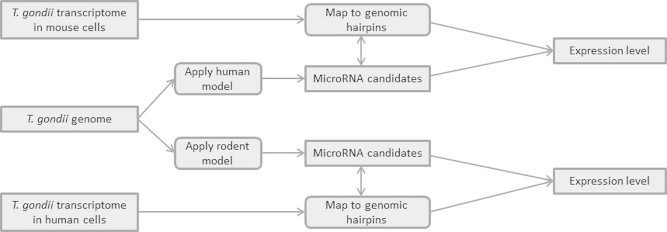
Assigning expression levels to hairpin candidates
Overview over the workflow generating an insight into how much of a given hairpin in the T. gondii genome is expressed in a mouse and in a human model. Filtering of candidates is indicated in the middle and mapping to mouse expression data (on top) and human expression data (on bottom) are shown.
The reads were pre-processed for adapter and quality trimming by using in-house scripts (Bagcı and Allmer, manuscript in preparation) and Sickle (http://omictools.com/sequencing/common-tools/quality-control/adapter-trimming/sickle-s714.html) to trim low-quality regions and reads from paired-end sequencing data. The remaining reads were mapped to the T. gondii genome by STAR aligner [29] and mapped reads were extracted from the alignment files in FASTQ format in order to remove any read coming from the host transcriptome. The reads were then aligned to the predicted T. gondii hairpins by the Bowtie short read aligner [30] and the number of hits to each hairpin sequence in the T. gondii genome was calculated for each SRA dataset separately. Mismatches in the seed region (15 nt) were not allowed and only the best hits with minimum number of mismatches were reported as the alignment.
Aligning human and rodent mature miRNAs against T. gondii hairpins
To find homologous regions between T. gondii predicted hairpins and human, as well as rodent, mature miRNA sequences from miRBase, we used blastn-short of the BLAST package (version 2.2.29+) with default settings [31]. A BLAST database of predicted T. gondii hairpins was created and mature miRNA sequences from human and rodents were blasted against it separately. We then calculated an alignment score in order to find the best match for each hairpin with the formula:
where SM refers to the number of matches in the seed region and M to the number of matches in the rest of the sequence. SMM represents the number of mismatches and gaps in seed region and MM the number of mismatches and gaps in the rest of the alignment. This formula introduces a bias toward alignments with matching seed regions.
The matches for 5′ and 3′ mature sequences were separated and ranked accordingly to their alignment score and only the one with the highest score was extracted for further analysis. Among matches with equal scores, the winning candidate was chosen arbitrarily.
Authors’ contributions
MDS and JA conceived the study and designed the project. CB participated in the study design and performed next-generation sequencing data analysis, sequence alignment and statistical analyses. JA extracted the pre-miRNAs from the T. gondii genome and calculated the features. MDS performed data mining and applied the learned models. All authors participated in writing, read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgements
This work was supported by the Scientific and Technological Research Council of Turkey (Grant No. 113E326) awarded to JA.
Footnotes
Peer review under responsibility of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China.
Supplementary material
Supplementary Figure S1.
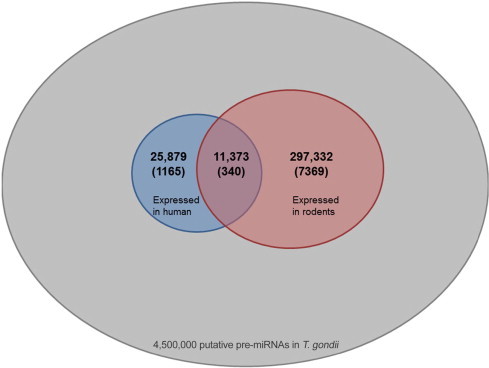
Distribution of expressed hairpins in T. gondii. The number of putative T. gondii hairpins expressed in a human host (blue) largely intersects with the number of hairpins expressed in a rodent host (red). Numbers given in parentheses show the hairpins that are expressed and pass the 0.9 threshold of the respective models.
Supplementary Figure S2.
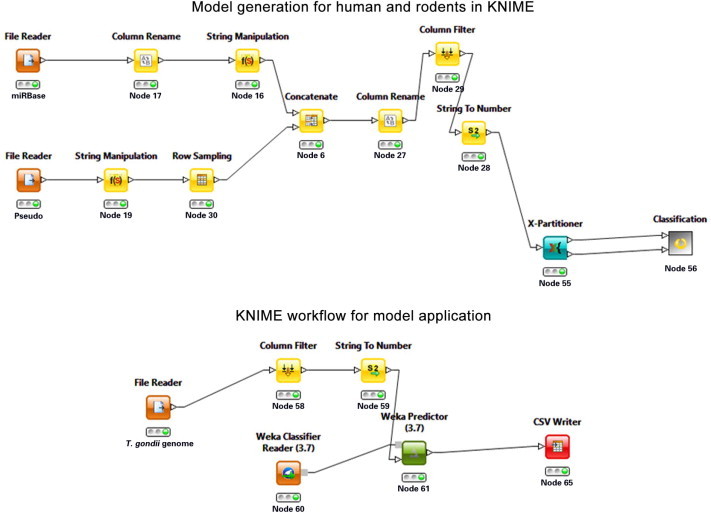
KNIME workflows for model generation and application. The workflow across the top shows the model generation and the workflow on bottom shows how the trained model above was applied to the hairpin candidates. The workflows are available as supplements (Files S2 and S3).
Candidate microRNAs that may regulate host genes. Note: Extension of results is presented in Table 3. The list of candidates is extended, but the score is kept high in order to ensure that most candidates may actually be viable.
T. gondii microRNAs expressed in human and rodents. Note: List of all putative hairpins in T. gondii that are either expressed in human, rodents or both.
Candidate microRNAs that may regulate host genes (comprehensive). Note: This table provides the same information as Table 3 and Table S1, but is more comprehensive and lists all candidates that are expressed.
Feature explanation and references. Note: The table lists the 32 features used in this study. It first provides our acronym, then the type that we classified it into followed by a more detailed description and the reference to the study that introduced it.
Data file. This file contains all data except for the calculated features. We elected not to present the calculated features since the file would be too big for us to host (>1GB).
Training and testing workflow for KNIME. This file can be directly applied to use the workflow in KNIME if training and testing data is available (not provided).
Model application workflow for KNIME. This file can be directly used in KNIME to predict whether a hairpin is of class microRNA or pseudo if the features as described in Materials and methods are calculated accordingly.
References
- 1.Ambros V., Lee R.C., Lavanway A., Williams P.T., Jewell D. MicroRNAs and other tiny endogenous RNAs in C. elegans. Curr Biol. 2003;13:807–818. doi: 10.1016/s0960-9822(03)00287-2. [DOI] [PubMed] [Google Scholar]
- 2.Filipowicz W., Bhattacharyya S.N., Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 3.Lee R.C., Feinbaum R.L., Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 4.Bushati N., Cohen S.M. MicroRNA functions. Annu Rev Cell Dev Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- 5.Jones-Rhoades M.W., Bartel D.P., Bartel B. MicroRNAs and their regulatory roles in plants. Annu Rev Plant Biol. 2006;57:19–53. doi: 10.1146/annurev.arplant.57.032905.105218. [DOI] [PubMed] [Google Scholar]
- 6.Hébert S.S., Horré K., Nicolaï L., Bergmans B., Papadopoulou A.S., Delacourte A. MicroRNA regulation of Alzheimer’s amyloid precursor protein expression. Neurobiol Dis. 2009;33:422–428. doi: 10.1016/j.nbd.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 7.Wang G., van der Walt J.M., Mayhew G., Li Y.J., Züchner S., Scott W.K. Variation in the miRNA-433 binding site of FGF20 confers risk for Parkinson disease by overexpression of alpha-synuclein. Am J Hum Genet. 2008;82:283–289. doi: 10.1016/j.ajhg.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim V.N., Han J., Siomi M.C. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009;10:126–139. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- 9.Chapman E.J., Carrington J.C. Specialization and evolution of endogenous small RNA pathways. Nat Rev Genet. 2007;8:884–896. doi: 10.1038/nrg2179. [DOI] [PubMed] [Google Scholar]
- 10.Lee Y., Kim M., Han J., Yeom K.H., Lee S., Baek S.H. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braun L., Cannella D., Ortet P., Barakat M., Sautel C.F., Kieffer S. A complex small RNA repertoire is generated by a plant/fungal-like machinery and effected by a metazoan-like Argonaute in the single-cell human parasite Toxoplasma gondii. PLoS Pathog. 2010;6:e1000920. doi: 10.1371/journal.ppat.1000920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cakir MV, Allmer J. Systematic computational analysis of potential RNAi regulation in Toxoplasma gondii. In: 5th international symposium on health informatics and bioinformatics, Ankara, Turkey: IEEE XPlorer; 2010. pp. 31–8.
- 13.Saçar M.D., Allmer J. Machine learning methods for microRNA gene prediction. Methods Mol Biol. 2014;1107:177–187. doi: 10.1007/978-1-62703-748-8_10. [DOI] [PubMed] [Google Scholar]
- 14.Sacar MD, Allmer J. Data mining for microrna gene prediction: On the impact of class imbalance and feature number for microrna gene prediction. In: 8th international symposium on health informatics and bioinformatics: IEEE XPlorer; 2013. pp. 1–6.
- 15.Griffiths-Jones S. miRBase: microRNA sequences and annotation. Curr Protoc Bioinformatics. 2010 doi: 10.1002/0471250953.bi1209s29. [chapter 12:Unit 12.9.1–10] [DOI] [PubMed] [Google Scholar]
- 16.Bentwich I., Avniel A., Karov Y., Aharonov R., Gilad S., Barad O. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet. 2005;37:766–770. doi: 10.1038/ng1590. [DOI] [PubMed] [Google Scholar]
- 17.Saçar M.D., Allmer J. Current limitations for computational analysis of miRNAs in cancer. Pakistan J Clin Biomed Res. 2013;1:3–5. [Google Scholar]
- 18.Ding J., Zhou S., Guan J. MiRenSVM: towards better prediction of microRNA precursors using an ensemble SVM classifier with multi-loop features. BMC Bioinformatics. 2010;11:S11. doi: 10.1186/1471-2105-11-S11-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saçar M.D., Hamzeiy H., Allmer J. Can miRBase provide positive data for machine learning for the detection of miRNA hairpins? J Integr Bioinform. 2013;10:215. doi: 10.2390/biecoll-jib-2013-215. [DOI] [PubMed] [Google Scholar]
- 20.Thirugnanam S., Rout N., Gnanasekar M. Possible role of Toxoplasma gondii in brain cancer through modulation of host microRNAs. Infect Agent Cancer. 2013;8:8. doi: 10.1186/1750-9378-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao J., Li Y., Prandovszky E., Karuppagounder S.S., Talbot C.C., Dawson V.L. MicroRNA-132 dysregulation in Toxoplasma gondii infection has implications for dopamine signaling pathway. Neuroscience. 2014;268:128–138. doi: 10.1016/j.neuroscience.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cai Y., Chen H., Mo X., Tang Y., Xu X., Zhang A. Toxoplasma gondii inhibits apoptosis via a novel STAT3-miR-17-92-Bim pathway in macrophages. Cell Signal. 2014;26:1204–1212. doi: 10.1016/j.cellsig.2014.02.013. [DOI] [PubMed] [Google Scholar]
- 23.Cannella D., Brenier-Pinchart M.P., Braun L., van Rooyen J.M., Bougdour A., Bastien O. MiR-146a and miR-155 delineate a microRNA fingerprint associated with Toxoplasma persistence in the host brain. Cell Rep. 2014;6:928–937. doi: 10.1016/j.celrep.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ng K.L.S., Mishra S.K. De novo SVM classification of precursor microRNAs from genomic pseudo hairpins using global and intrinsic folding measures. Bioinformatics. 2007;23:1321–1330. doi: 10.1093/bioinformatics/btm026. [DOI] [PubMed] [Google Scholar]
- 25.Wheeler D.L., Barrett T., Benson D.A., Bryant S.H., Canese K., Chetvernin V. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2008;36:D13–D21. doi: 10.1093/nar/gkm1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lopes Ide O., Schliep A., de Carvalho A.C. The discriminant power of RNA features for pre-miRNA recognition. BMC Bioinformatics. 2004;15:124. doi: 10.1186/1471-2105-15-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saçar MD, Allmer J. Comparison of four ab initio microRNA prediction tools. In: Proceedings of the international conference on bioinformatics models, methods and algorithms. Barcelona: SciTePress – Science and Technology Publications; 2013. pp. 190–5.
- 28.Hofacker I.L. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 32.Hsu S.D., Tseng Y.T., Shrestha S., Lin Y.L., Khaleel A., Chou C.H. miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2014;42:D78–D85. doi: 10.1093/nar/gkt1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Candidate microRNAs that may regulate host genes. Note: Extension of results is presented in Table 3. The list of candidates is extended, but the score is kept high in order to ensure that most candidates may actually be viable.
T. gondii microRNAs expressed in human and rodents. Note: List of all putative hairpins in T. gondii that are either expressed in human, rodents or both.
Candidate microRNAs that may regulate host genes (comprehensive). Note: This table provides the same information as Table 3 and Table S1, but is more comprehensive and lists all candidates that are expressed.
Feature explanation and references. Note: The table lists the 32 features used in this study. It first provides our acronym, then the type that we classified it into followed by a more detailed description and the reference to the study that introduced it.
Data file. This file contains all data except for the calculated features. We elected not to present the calculated features since the file would be too big for us to host (>1GB).
Training and testing workflow for KNIME. This file can be directly applied to use the workflow in KNIME if training and testing data is available (not provided).
Model application workflow for KNIME. This file can be directly used in KNIME to predict whether a hairpin is of class microRNA or pseudo if the features as described in Materials and methods are calculated accordingly.