

Abstract
Next-generation sequencing (NGS) technology, with its high-throughput capacity and low cost, has developed rapidly in recent years and become an important analytical tool for many genomics researchers. New opportunities in the research domain of the forensic studies emerge by harnessing the power of NGS technology, which can be applied to simultaneously analyzing multiple loci of forensic interest in different genetic contexts, such as autosomes, mitochondrial and sex chromosomes. Furthermore, NGS technology can also have potential applications in many other aspects of research. These include DNA database construction, ancestry and phenotypic inference, monozygotic twin studies, body fluid and species identification, and forensic animal, plant and microbiological analyses. Here we review the application of NGS technology in the field of forensic science with the aim of providing a reference for future forensics studies and practice.
Keywords: Forensics, Next-generation sequencing, Genomics, Degradation of DNA
Introduction
Since the introduction of the Sanger sequencing method in the 1970s [1], DNA sequencing technology has enabled enormous advances in molecular biology and genetics. Several large projects have been successfully completed using this technology, such as the Human Genome Project, Rice Genome Project and Swine Genome Project, as well as genome projects of many other species. However, disadvantages of the conventional Sanger sequencing technology, including its low throughput, high cost and operation difficulties, have limited its use in deeper and more complex genome analyses [2]. The recent introduction of next-generation sequencing (NGS) technology, with its high-throughput capacity and low cost, has largely overcome these problems, and these technologies have been applied in various fields of life sciences, including forensics [3], disease diagnosis [4], agrigenomics [5] and ancient DNA analysis [6]. In this article, the use of NGS technology in forensic science is reviewed with the aim of providing a reference for future frontier research and application in forensic science.
Overview of NGS technology
NGS technology refers to non-Sanger-based high-throughput DNA sequencing technology. Millions or billions of DNA molecules can be sequenced in parallel, thereby increasing the throughput substantially and minimizing the need for the fragment-cloning method often used in Sanger sequencing. It includes second-generation sequencing technology based on loop array sequencing, which can analyze a large number of samples simultaneously, as well as third-generation sequencing technology, which can determine the base composition of single DNA molecules.
In 2005, Roche introduced the 454 Genome Sequencing System [7], the world’s first pyrosequencing-based high-throughput sequencing system. The first 454 Genome Sequencer was capable of generating approximately 200,000 reads of 110 base pairs (bp) in length (the current maximum read length is 1000 bp). In 2007, Applied Biosystems (ABI) introduced the SOLiD second-generation sequencing system based on the oligonucleotide ligation technique and two-base encoding system, whereas Illumina released Solexa sequencing technology. The Illumina and SOLiD sequencers generated much larger numbers of reads than the 454 system (30 and 100 million reads, respectively); nonetheless, the reads produced were only 35 bp long. In 2010, Ion Torrent, a faster and low-cost sequencer based on semiconductor technology, was introduced. This sequencer does not rely on fluorescence, chemiluminescence, or enzyme cascades for sequencing signal detection. Currently, a maximum read length of up to 400 bp can be obtained using this system.
All these new sequencing methods have led to three major improvements from the conventional technologies. First, these technologies do not require bacterial cloning of DNA fragments; instead, they rely on the preparation of NGS libraries in a cell-free system. Second, instead of hundreds of sequencing reactions, they can parallelize the thousands-to-many-millions of sequencing reaction. Third, the sequencing output is directly detected with no need for electrophoresis. The enormous number of reads generated by NGS enabled the sequencing of entire genomes at an unprecedented speed and thus it came to be widely used in various fields of life sciences. However, one drawback of second-generation sequencing technology is its relatively short read lengths, which has resulted in difficulties in subsequent sequence splicing, assembly, annotation and bioinformatics analysis [8]. Furthermore, standard PCR was used to randomly amplify genomic fragments during library preparation. Due to the complex structure of genomes, factors such as secondary structure and thermal stability will affect the efficiency of PCR amplification. Therefore, the complete genomic sequence may not be represented in the library produced by such amplification. This can be problematic due to the relative deviation between amplified and non-amplified DNA molecules, resulting in potential inaccuracies in gene expression analysis. This concern is particularly relevant for highly-expressed genes [9]. Moreover, these shortcomings have restricted the application and development of second-generation sequencing technology to some extent and have necessitated the development of third-generation single-molecule sequencing technology [10–12].
The third-generation sequencing technology not only allows the detection of single molecules but also enables real-time sequencing. The current leader in this field is the PacBio RS system (Pacific Biosciences), which utilizes the single-molecule, real-time (SMRT) DNA sequencing technology. SMRT sequencing is based on the sequencing-by-synthesis approach; an SMRT chip contains thousands of zero-mode waveguides, in which the DNA polymerase molecules used to synthesize the DNA fragments of interest are attached. Compared to second-generation sequencing, the latest SMRT technology can achieve an average read lengths of 5500−8500 bp. Moreover, it can also directly detect epigenetic modifications such as 4-methylctosine (mC), 5-mC and 6-methyladenine (mA) [13].
Forensic application prospects of NGS technology
The application of DNA technologies in forensic investigations has rendered DNA analysis an important tool in forensic science. Compared to other fields of life sciences, forensic DNA analysis is confronted with template of low copy number, highly-degraded and contaminated samples, the need for high accuracy and reproducibility, as well as time and cost considerations. Today, the majority of forensic DNA tests employ PCR and capillary electrophoresis (CE)-based fragment analysis methods to detect length variation in short tandem repeat (STR) markers.
The CE-based Sanger sequencing has been used to analyze specific regions of mitochondrial DNA (mtDNA) [14]. The development of miniaturized gel electrophoresis and the automation of reaction gel loading and signal detection allowed the Sanger methodology to become the gold standard for DNA sequencing. However, CE-based analysis has its limitations, for example, the inability to analyze multiple genetic polymorphisms in a single reaction using a single workflow, low-resolution genotyping of current markers, loss of useful genomic information from degraded DNA samples, and low-resolution mtDNA and mixture analysis. These limitations of first-generation sequencing prompt the forensic scientists worldwide to explore the usefulness of NGS technology for forensic studies.
STR analysis
STR analysis is likely to remain the most important and commonly-used genetic technique in forensic science for the foreseeable future. It displays multiple advantages, such as rapid and precise allele determination, low DNA template requirement, multiplex amplification and fluorescence-based detection, digitized results and utilization of the abundant genomic element, At present, more than 60 countries worldwide have established forensic DNA databases based on STRs, and these databases continue to grow rapidly. For example, China now has more than 27 million entries in its forensic database [15]. The probability of a random match between unrelated individuals will increase if statistical analyses were based only on the 13 routinely-used Combined DNA Index System (CODIS) STR markers (i.e., CSF1PO, FGA, THO1, TPOX, VWA, D3S1358, D5S818, D7S820, D8S1179, D13S317, D16S539, D18S51 and D21S11) or 15 markers (13 CODIS loci plus D2S1338 and D19S433). To avoid this, incorporation of more STR markers into the common forensic typing assay currently used has been recommended. However, simultaneous detection of more STR markers would be very difficult, due to the technical limitations of fluorescent-based CE sequencers currently in use. Traditional CE-based STR typing using CE is based on the detection of DNA fragment size. Therefore, alleles of identical or similar length but of different sequences cannot be distinguished. Consequently, STR mutations in complex paternity cases often cannot be resolved with traditional CE-based STR analysis. An additional challenge for forensic DNA tests is the analysis of complex DNA mixtures comprising DNA from more than one individual. Contemporary analyses of mixed DNA samples often yield low detection rates, thus are not useful in crime investigations.
When NGS technology was firstly introduced to genomics, it was not suitable for STR testing because the read length was generally too short. With technological advances, the average read length has been continually increasing. Since alleles with similar length can be easily distinguished using NGS technology and digital read count could significantly facilitate the identification of mixed DNA samples and analysis of complex paternity cases, some researchers have recently started using NGS technology for STR testing. For example, a pioneer study was performed by Zajac and colleagues, who analyzed three CODIS STR loci, TPOX, CSF1PO and D18S51, using the trinucleotide threading (TnT) approach by 454 Genome Sequencing System [7,16]. Subsequently, Irwin et al. [17] analyzed 13 CODIS STR loci using the 454 GS Junior system in combination with multiplex identifier technology for single source samples. Bornman et al. [18] went further to show that high-throughput sequencing technology can accurately identify the 13 CODIS STR loci as well as the AMEL gene for not only single source but also mixed samples. To process the forensic NGS data, Warshauer et al. [19] developed STRait Razor, a software that can analyze the NGS data for 44 STRs, including 23 autosomal and 21 Y chromosome STRs. In addition, Van Neste et al. [20] used Illumina’s MiSeq system to establish a reference allele database to detect single source and mixed DNA samples; they observed that most locus genotyping results were stable and reliable.
NGS technology has many potential advantages for STR analysis. These include high throughput, low cost, simultaneous detection of large numbers of STR loci on both autosomes and sex chromosomes, and the ability to distinguish alleles with similar length or digital read count. NGS technology would therefore significantly facilitate the identification of mixed DNA samples and analysis of complex paternity cases, and ultimately greatly increase the efficiency and cost-effectiveness of legal cases.
Mitochondrial genome analysis
mtDNA has proved to be a useful forensic tool in cases involving low amounts of DNA or wherein the maternal lineage needs to be investigated, due to its characteristics of small size, multiple copies, maternal inheritance, high mutation rate and lack of recombination. Currently, forensic mtDNA analyses usually detect only polymorphisms within a hypervariable region. However, for mtDNA to be used as a genetic haplotype marker, additional polymorphic loci are required to increase the discrimination power of identification. Therefore, NGS technology has the potential to greatly assist in the analysis of whole mitochondrial sequences.
With the increased application of NGS technology in various fields, the cost of equipment and reagents has decreased markedly. Parallel sequencing technology, which allows for simultaneous analysis of multiple samples, has also led to cost-effectiveness. For instance, the number of picotiter plates used in the GS-FLX instrument has increased from 2 to 16, and each channel can simultaneously analyze 192 samples using multiplex identifier (MID) technology. Furthermore, Binladen et al. [21] used a primer coding technique and produced 256 tagged primers for use in multiple parallel sequencing, allowing 256 samples to be sequenced in a single run. Moreover, Gunnarsdóttir et al. [22] used NGS technology to sequence whole mitochondrial genomes of 109 Filipino individuals at the same time and obtained on average ∼55 × coverage per sequence, with <1% missing data per sequence.
Human mtDNA heteroplasmy is common and heteroplasmy of cells from different tissues within a single individual has also been observed [23]. mtDNA heteroplasmy is one of the factors affecting the performance of forensic mitochondrial analysis. The detection of heteroplasmy at the whole mitochondrial genome level has been reported [24], supporting the advantages of using NGS to detect mitochondrial heteroplasmy, including high accuracy and sensitivity, high throughput, low cost, and simple operation [25]. In a separate study, multiple mitochondrial hypervariable regions, an autosomal STR locus (D18S51) and a Y chromosome STR locus (DYS389I/II) were simultaneously examined using the 454 GS Junior system. The results demonstrated that a mixing ratio of two DNA sources as low as 1:250 can be detected, and the authors concluded that by increasing the sequencing coverage, a mixing ratio of 1:1000 might be detectable as well [26]. To compare the haplotypes defined by using NGS technology at the whole mitochondrial genome level with conventional Sanger sequencing, 64 whole mitochondrial genome sequences were analyzed. The results showed differences in <0.02 % of nucleotides using these two methods and that approximately two-thirds of the differences were observed in or around homopolymeric stretches, since these areas were prone to sequencing errors [27]. To evaluate the reproducibility between samples that were sequenced twice with NGS, Mikkelsen et al. [28] reported that using the 454 NGS method, 95% of the reads was sequenced correctly in homopolymers of up to 6 bases if the results were carefully and visually inspected. Previously-unreported heteroplasmy in the GM9947A component of the National Institute of Standards and Technology (NIST) human mtDNA SRM-2392 standard reference was detected in this study.
Y chromosome analysis
Genetic markers on the Y chromosome have assumed a valuable role within forensic molecular biology. Most commonly, Y-STRs are used to unambiguously resolve the male component of DNA mixtures when a high female background is present, or to reconstruct paternal relationships between male individuals. Using NGS technology, more than 10 million nucleotides of the Y chromosome were compared between two male individuals who shared the same ancestor 13 generations ago [29]. Four genetic differences were detected, suggesting that Y chromosome sequencing could solve the problem of distinguishing between mixed male samples from the same parent. In addition, Van Geystelen et al. [30,31] developed AMY-tree using Y chromosome single nucleotide polymorphisms (SNPs) and successfully verified the differences between 118 unrelated male individuals from 109 different geographical regions. This study demonstrated that AMY-tree can determine Y chromosome pedigrees and identify unknown Y-SNPs from different geographical regions.
Forensic microbiological analysis
Microbial forensics is a new discipline developed by the Federal Bureau of Investigation (FBI) after the Anthrax attack on 18 September 2001 in the USA. It is based on the fast and accurate detection and identification of microorganisms founded at biological crime, with the aim of tracing the source of the microbe [32]. Because microbiological terrorist attack could lead to serious consequences, forensic microbiological analysis has been attracting considerable attention [33]. Using whole-genome sequencing by the SOLiD system in a real investigative case, Cummings et al. [34] identified suspects by sequencing four strains each of Bacillus anthracis and Yersinia pestis at a cost of only $1000 and reported that this would be reduced to less than $50 if the HiSeq 2000 system were used.
Brenig et al. [35] used the 454 sequencing system to identify biological traces using deep sequencing and metagenomic analysis and indicated that the method can be used for the forensic identification of biological traces. Fierer et al. [36] examined the bacteria left by human skin on the surface of contact objects by using an NGS-based metagenomic method and showed that the bacteria left by human skin possess sufficient DNA information for forensic analysis.
A study by Lilje et al. [37] investigated the criteria for soil metagenome data management and database searching. Eleven samples collected from different environments (forests, fields, grasslands and an urban park) with different microbial flora were sequenced using the Roche/454 platform. The results demonstrated that 18S rRNA gene marker analysis could be used to create and run a filtered database, which was very computationally efficient and flexible. Similarly, Giampaoli et al. [38] successfully applied a metabarcoding approach to forensic and environmental soil samples, allowing accurate and sensitive analysis of the DNA of microflora, plants, metazoa and protozoa.
All the studies described above demonstrate that NGS has the advantages of high throughput, multiplexing capability and accuracy, which makes it suitable for rapid whole-genome typing of microbial pathogens during forensic or epidemiological investigation. Rare polymorphisms can be reliably detected by analyzing every base of the genome, thus giving forensic data higher resolution and greater accuracy. It is expected that a high-quality forensic microbial database will soon become a reality and aid in the fast and accurate identification of criminals and biological terrorists.
Animal and plant DNA analysis
Species identification is one of most important components of forensic practice. For example, in some cases of poaching [39] and trading of endangered species [40], it has been used to provide important information and assist in police investigations. In the food industry, identification of the species present in meat products can be achieved [41], and in archeology, human remains can be distinguished from non-human remains [42]. At present, most DNA typing methods for species determination are based on PCR amplification using species-specific primers for single species. However, forensic scientists are often faced with situations in which no a priori species information is available. The development of NGS technology has allowed DNA typing to be used in more projects involving species identification [43,44]. For example, Cheng et al. recently identified plants and animals in traditional Chinese medicines using a cost-effective and efficient next-generation deep sequencing method [45].
Ancestry studies and phenotypic inferences
Information embedded within the human genome may provide insights into personal characteristics such as ethnicity [46], physical and physiological characteristics and age [47,48]. In forensic studies, characteristics inferred from DNA analysis make it possible for criminal investigations to evolve from the “passive comparison” into the “active search” stages. In criminal cases where a possible suspect and database information are unavailable, it is possible to rapidly narrow down the potential suspects by using ancestry studies and phenotypic inference derived from a DNA sample. For example, in the 2004 Madrid train bombings, source population of the suspects was inferred by using 34 autosomal SNPs related to the ancestry of population [49]. Other studies reported SNPs closely related to colors of the iris [50] and hair [51] with an accuracy of 90%. Klimentidis et al. [52] investigated facial features using DNA test and association analysis and validated their results using facial reconstruction (molecular photo fitting). The results demonstrated that there is a relationship between social and biological measures of race/ethnicity but that it is far from perfect. In all these studies, only commercial SNP chip scanning were used. If NGS technology for whole-genome sequencing were applied in these cases, more information and accuracy would be obtained.
Epigenetic analysis
DNA sequencing analysis is a powerful tool in forensic identification [53]. Recently, a number of studies have suggested that epigenetic markers can also have various applications in forensic science. For example, evidence supports that epigenetic markers can be used to distinguish monozygotic (MZ) twins [54], predict tissue type [55] and accurately determine the age of a DNA donor [56]. Epigenetic approaches based on NGS technology include whole-genome bisulfite sequencing [57], methylation beadchips, reduced representation bisulfite sequencing [58] and methylated DNA immunoprecipitation sequencing [59]. These sequencing methods require large amounts of DNA; their ability to use trace DNA samples will therefore be crucial to the success of forensic epigenetic analysis. Interestingly, extremely low amounts of starting DNA (100 pg) were successfully analyzed through genome-wide amplification of a bisulfite-modified DNA template, followed by quantitative methylation detection using pyrosequencing [60]. Additionally, another encouraging study performed bisulfite genomic DNA sequencing with micro-volume blood spot samples [61].
MZ twin studies continue to be a hot topic in the field of forensic science. As both individuals have exactly the same DNA sequence, conventional genotyping approaches such as STR, SNP, sex chromosome STR, and mtDNA analyses cannot tell them apart. In 2014, Weber-Lehmann et al. [62] described how identification of extremely-rare mutations by ultra-deep NGS can differentiate between MZ twins, suggesting a solution to paternity and forensic cases involving MZ twins. Li et al. [54] used Illumina Human Methylation BeadChip technology to examine the DNA methylation status of 27,578 CpG sites from 22 MZ twins. As a result, they filtered 92 significantly-methylated CpG sites, representing potential targets for epigenetic studies aimed at distinguishing between MZ twins. In 2010, the BGI and twin research group TwinsUK at King’s College, London, co-sponsored epigenetic research projects using NGS technology to conduct an in-depth study aimed at capturing the subtle differences in epigenetic signals from 5000 pairs of twins [63]. The research outcomes are likely to be highly applicable in the forensic identification of MZ twins.
MicroRNA analysis
Although mRNA analysis has become a well-established technique in many forensic laboratories, microRNAs (miRNAs) have only recently been introduced to forensic science. miRNAs are a class of endogenous small RNA molecules 18–24 nucleotides in length. Owing to their small size, resistance to degradation and tissue-specific or highly tissue-divergent expression, they are suitable for forensic body fluid identification, species identification and post-mortem interval (PMI) inference analysis [64]. Currently, analysis of miRNA is mostly achieved by real-time PCR and biochip technology, whereby only known miRNA sequences can be analyzed. In 2009, Hanson et al. [65] introduced miRNA profiling to forensic science and showed that 452 miRNAs were genotyped via the quantitative PCR method from forensic samples. In another study, the expression levels of 718 miRNAs in semen, saliva, venous blood, menstrual blood and vaginal secretions were profiled on a microarray [66]. Among them, 14 differentially-expressed miRNAs were identified, which could serve as potential candidates for body fluid identification. Using NGS technology, millions of miRNA sequences can be rapidly analyzed to identify organ- and developmental stage-specific expression, as well as miRNA expression in different disease states, thus providing a powerful tool for forensic analysis.
Conclusion
In practical forensic science, DNA samples are usually limited and often cannot fulfill the requirements of simultaneously analyzing multiple loci on different chromosomes in mitochondrial genome [67–69]. This may result in difficulties in providing sufficient information and can limit their use as legal evidence. In addition, mixed stain identification and complex paternity cases cannot be solved with traditional STR genotyping strategies. NGS technology not only meets these requirements but can also potentially be applied in many areas of research, including DNA database construction, ancestry and phenotypic inference, MZ twin studies, body fluid and species identification, and forensic microbiological analysis (Figure 1).
Figure 1.
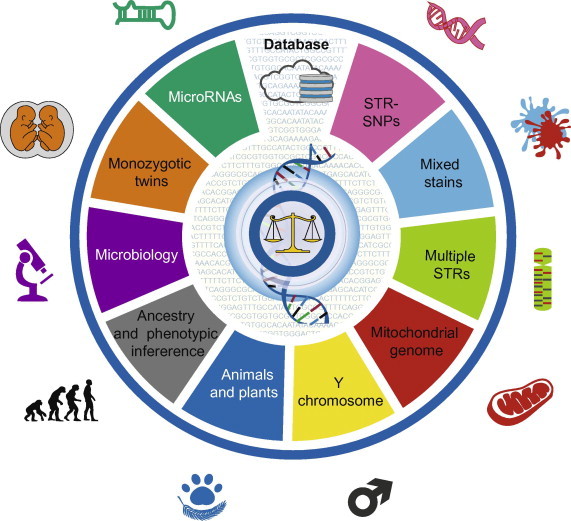
Forensic analysis by next-generation sequencing
The introduction of next-generation sequencing (NGS) technology that is much cheaper and more rapid than the classic Sanger sequencing method has revolutionized our thinking about scientific strategies in forensic research. NGS will potentially influence many aspects of forensic science, including short tandem repeats (STRs) and microRNA analysis, monozygotic twin and mixed stain recognition, Y chromosome and mitochondrial whole-genome studies, forensic microbiological analysis, multiple species identification, and ancestry and phenotype inference. More importantly, high-throughput screening techniques have generated large amounts of data, facilitating a systematic understanding of relationships between molecular components. Therefore, comprehensive genome-wide analysis, in combination with the techniques of genomics, proteomics, transcriptomics and epigenomics, will provide new insights in the field of applied forensics.
In forensic science, standard STR typing provides sufficient discrimination power for most applications, and most countries have already established large-scale forensic DNA databases for resolving crimes based on STR technology. Although the use of whole-exome or whole-genome sequencing could provide more information for forensic analyses, considering the compatibility and cost, NGS technology would not likely soon replace conventional STR typing. Forensic scientists performed custom-designed target-enrichment panels to analyze STR loci. However, these methods only covered some of the currently-common used loci in forensic studies [16–18]. With the development of NGS technology, it is likely that its cost will rapidly decrease and NGS kits for forensic application will soon be commercially available. This will allow the simultaneous detection of multiple STR loci on both autosomes and sex chromosomes, analysis of mitochondrial genome polymorphisms and analysis of SNPs related to ancestry and physical and psychological characteristics, providing important information for forensic investigations (Figure 2). Using these methods, NGS is capable of providing data on loci across the genome. As law enforcement agencies have started to share information, multiple international databases have recognized the need to analyze additional loci in a single run and have expanded their locus database. These expanded locus sets have improved the efficiency of law enforcement investigations, where a suspect was identified or the database yielded a match. When neither a suspect nor a hit exists, additional information from evidence samples might provide valuable clues regarding the phenotype of the offender, who may be out of custody and at risk of re-offending.
Figure 2.
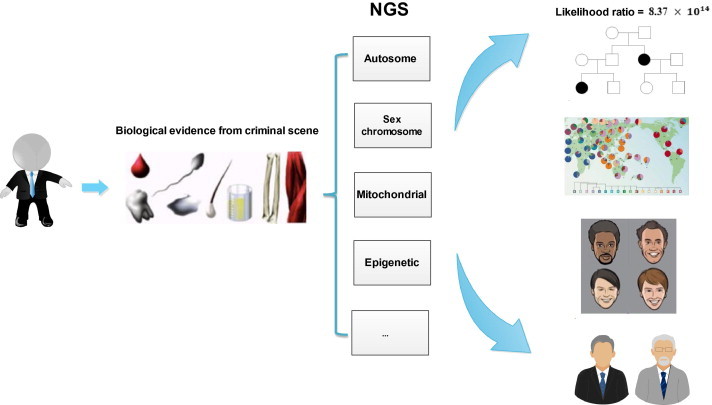
Diverse range of information can be obtained by NGS of biological evidence samples collected from crime scenes
Through applying NGS technology, multiple results can be obtained simultaneously from biological evidence samples collected from crime scenes, such as STRs, single nucleotide polymorphisms (SNPs) of autosomes, sex chromosomes and mitochondrial genomes, as well as epigenetic information. By integrating all the information, the evidence samples can be used not only for suspect identification but also for inferring the criminal suspects’ physical, psychological and geographical characteristics, as well as the source population.
Although NGS technology appears to have an important role in future forensic studies, more work is required to fully achieve this goal, which includes overcoming problems with low-template library preparation, error rate, type estimations and issues with NGS data processing and mining. Guidelines for the application of NGS in forensic science also need to be generated. With the technical advances of NGS technology and continuous translational efforts of forensic scientists, we believe that NGS technology is likely to become an easily accessible routine method in forensic practice.
Competing interests
The authors have declared that no competing interests exist.
Acknowledgements
The authors thank Dr. Hongzhu Qu and Dr. Xiangdong Fang for critical reading and Miss Nan Ding for figure preparation. This study was supported by the National Natural Science Foundation of China (Grant Nos. 81172909 and 81330073).
Footnotes
Peer review under responsibility of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China.
References
- 1.Sanger F., Nicklen S., Coulson A.R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fullwood M.J., Wei C.L., Liu E.T., Ruan Y. Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses. Genome Res. 2009;19:521–532. doi: 10.1101/gr.074906.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weber-Lehmann J., Schilling E., Gradl G., Richter D.C., Wiehler J., Rolf B. Finding the needle in the haystack: differentiating “identical” twins in paternity testing and forensics by ultra-deep next generation sequencing. Forensic Sci Int Genet. 2014;9:42–46. doi: 10.1016/j.fsigen.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 4.McCarthy J.J., McLeod H.L., Ginsburg G.S. Genomic medicine: a decade of successes, challenges, and opportunities. Sci Transl Med. 2013;5:189sr4. doi: 10.1126/scitranslmed.3005785. [DOI] [PubMed] [Google Scholar]
- 5.Goddard M.E., Hayes B.J. Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat Rev Genet. 2009;10:381–391. doi: 10.1038/nrg2575. [DOI] [PubMed] [Google Scholar]
- 6.Poinar H.N., Schwarz C., Qi J., Shapiro B., Macphee R.D., Buigues B. Metagenomics to paleogenomics: large-scale sequencing of mammoth DNA. Science. 2006;311:392–394. doi: 10.1126/science.1123360. [DOI] [PubMed] [Google Scholar]
- 7.Margulies M., Egholm M., Altman W.E., Attiya S., Bader J.S., Bemben L.A. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Dijk E.L., Auger H., Jaszczyszyn Y., Thermes C. Ten years of next-generation sequencing technology. Trends Genet. 2014;30:418–426. doi: 10.1016/j.tig.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 9.Karn T. High-throughput gene expression and mutation profiling: current methods and future perspectives. Breast Care (Basel) 2013;8:401–406. doi: 10.1159/000357461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Derrington I.M., Butler T.Z., Collins M.D., Manrao E., Pavlenok M., Niederweis M. Nanopore DNA sequencing with MspA. Proc Natl Acad Sci U S A. 2010;107:16060–16065. doi: 10.1073/pnas.1001831107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luan B., Peng H., Polonsky S., Rossnagel S., Stolovitzky G., Martyna G. Base-by-base ratcheting of single stranded DNA through a solid-state nanopore. Phys Rev Lett. 2010;104:238103. doi: 10.1103/PhysRevLett.104.238103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eid J., Fehr A., Gray J., Luong K., Lyle J., Otto G. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 13.Murray I.A., Clark T.A., Morgan R.D., Boitano M., Anton B.P., Luong K. The methylomes of six bacteria. Nucleic Acids Res. 2012;40:11450–11462. doi: 10.1093/nar/gks891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rizzi E., Lari M., Gigli E., De Bellis G., Caramelli D. Ancient DNA studies: new perspectives on old samples. Genet Sel Evol. 2012;44:21. doi: 10.1186/1297-9686-44-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Proceedings of the 4th national symposium on forensic DNA inspection technology & 2012 international symposium on new advances in forensic genetics. Fuzhou, China. December 9–12, 2012.
- 16.Zajac P., Oberg C., Ahmadian A. Analysis of short tandem repeats by parallel DNA threading. PLoS One. 2009;4:e7823. doi: 10.1371/journal.pone.0007823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Irwin J., Just R., Scheible M., Loreille O. Assessing the potential of next generation sequencing technologies for missing persons identification efforts. Forensic Sci Int Genet Suppl Ser. 2011;3:447–448. [Google Scholar]
- 18.Bornman D.M., Hester M.E., Schuetter J.M., Kasoji M.D., Minard-Smith A., Barden C.A. Short-read, high-throughput sequencing technology for STR genotyping. Biotechniques. 2012:1–6. [PMC free article] [PubMed] [Google Scholar]
- 19.Warshauer D.H., Lin D., Hari K., Jain R., Davis C., LaRue B. STRait Razor: a length-based forensic STR allele-calling tool for use with second generation sequencing data. Forensic Sci Int Genet. 2013;7:409–417. doi: 10.1016/j.fsigen.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 20.Van Neste C., Vandewoestyne M., Van Criekinge W., Deforce D., Van Nieuwerburgh F. My-Forensic-Loci-queries (MyFLq) framework for analysis of forensic STR data generated by massive parallel sequencing. Forensic Sci Int Genet. 2014;9:1–8. doi: 10.1016/j.fsigen.2013.10.012. [DOI] [PubMed] [Google Scholar]
- 21.Binladen J., Gilbert M.T., Bollback J.P., Panitz F., Bendixen C., Nielsen R. The use of coded PCR primers enables high-throughput sequencing of multiple homolog amplification products by 454 parallel sequencing. PLoS One. 2007;2:e197. doi: 10.1371/journal.pone.0000197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gunnarsdottir E.D., Li M., Bauchet M., Finstermeier K., Stoneking M. High-throughput sequencing of complete human mtDNA genomes from the Philippines. Genome Res. 2011;21:1–11. doi: 10.1101/gr.107615.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao Y., Wan L.H., Gu L.G., Huang Y.X., Xiu C.X., Hu S.H. Heteroplasmy in human mtDNA control region. Fa Yi Xue Za Zhi. 2006;22:190–192. [PubMed] [Google Scholar]
- 24.Li M., Schonberg A., Schaefer M., Schroeder R., Nasidze I., Stoneking M. Detecting heteroplasmy from high-throughput sequencing of complete human mitochondrial DNA genomes. Am J Hum Genet. 2010;87:237–249. doi: 10.1016/j.ajhg.2010.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang S., Huang T. Characterization of mitochondrial DNA heteroplasmy using a parallel sequencing system. Biotechniques. 2010;48:287–296. doi: 10.2144/000113389. [DOI] [PubMed] [Google Scholar]
- 26.Holland M.M., McQuillan M.R., O’Hanlon K.A. Second generation sequencing allows for mtDNA mixture deconvolution and high resolution detection of heteroplasmy. Croat Med J. 2011;52:299–313. doi: 10.3325/cmj.2011.52.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parson W., Strobl C., Huber G., Zimmermann B., Gomes S.M., Souto L. Evaluation of next generation mtGenome sequencing using the Ion Torrent Personal Genome Machine (PGM) Forensic Sci Int Genet. 2013;7:543–549. doi: 10.1016/j.fsigen.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mikkelsen M., Frank-Hansen R., Hansen A.J., Morling N. Massively parallel pyrosequencing of the mitochondrial genome with the 454 methodology in forensic genetics. Forensic Sci Int Genet. 2014;12:30–37. doi: 10.1016/j.fsigen.2014.03.014. [DOI] [PubMed] [Google Scholar]
- 29.Xue Y., Wang Q., Long Q., Ng B.L., Swerdlow H., Burton J. Human Y chromosome base-substitution mutation rate measured by direct sequencing in a deep-rooting pedigree. Curr Biol. 2009;19:1453–1457. doi: 10.1016/j.cub.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Geystelen A., Decorte R., Larmuseau M.H. Updating the Y-chromosomal phylogenetic tree for forensic applications based on whole genome SNPs. Forensic Sci Int Genet. 2013;7:573–580. doi: 10.1016/j.fsigen.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 31.Van Geystelen A., Decorte R., Larmuseau M.H. AMY-tree: an algorithm to use whole genome SNP calling for Y chromosomal phylogenetic applications. BMC Genomics. 2013;14:101. doi: 10.1186/1471-2164-14-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McEwen S.A., Wilson T.M., Ashford D.A., Heegaard E.D., Kournikakis B. Microbial forensics for natural and intentional incidents of infectious disease involving animals. Rev Sci Tech. 2006;25:329–339. [PubMed] [Google Scholar]
- 33.Beecher D.J. Forensic application of microbiological culture analysis to identify mail intentionally contaminated with Bacillus anthracis spores. Appl Environ Microb. 2006;72:5304–5310. doi: 10.1128/AEM.00940-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cummings CA, Bormann Chung CA, Fang R, Barker M, Brzoska P, Williamson PC, et al. Accurate, rapid and high-throughput detection of strain-specific polymorphisms in Bacillus anthracis and Yersinia pestis by next-generation sequencing. Investig Genet 2010;1:5. [DOI] [PMC free article] [PubMed]
- 35.Brenig B., Beck J., Schutz E. Shotgun metagenomics of biological stains using ultra-deep DNA sequencing. Forensic Sci Int Genet. 2010;4:228–231. doi: 10.1016/j.fsigen.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 36.Fierer N., Lauber C.L., Zhou N., McDonald D., Costello E.K., Knight R. Forensic identification using skin bacterial communities. Proc Natl Acad Sci U S A. 2010;107:6477–6481. doi: 10.1073/pnas.1000162107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lilje L., Lillsaar T., Rätsep R., Simm J., Aaspõllu A. Soil sample metagenome NGS data management for forensic investigation. Forensic Sci Int Genet. 2013;4:e35–e36. [Google Scholar]
- 38.Giampaoli S., Berti A., Di Maggio R.M., Pilli E., Valentini A., Valeriani F. The environmental biological signature: NGS profiling for forensic comparison of soils. Forensic Sci Int. 2014;240:41–47. doi: 10.1016/j.forsciint.2014.02.028. [DOI] [PubMed] [Google Scholar]
- 39.An J., Lee M.Y., Min M.S., Lee M.H., Lee H. A molecular genetic approach for species identification of mammals and sex determination of birds in a forensic case of poaching from South Korea. Forensic Sci Int. 2007;167:59–61. doi: 10.1016/j.forsciint.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 40.Eurlings M.C., Lens F., Pakusza C., Peelen T., Wieringa J.J., Gravendeel B. Forensic identification of Indian snakeroot (Rauvolfia serpentina Benth. ex Kurz) using DNA barcoding. J Forensic Sci. 2013;58:822–830. doi: 10.1111/1556-4029.12072. [DOI] [PubMed] [Google Scholar]
- 41.Ali M.E., Hashim U., Kashif M., Mustafa S., Che Man Y.B., Abd Hamid S.B. Development of swine-specific DNA markers for biosensor-based halal authentication. Genet Mol Res. 2012;11:1762–1772. doi: 10.4238/2012.June.29.9. [DOI] [PubMed] [Google Scholar]
- 42.Malmstrom H., Stora J., Dalen L., Holmlund G., Gotherstrom A. Extensive human DNA contamination in extracts from ancient dog bones and teeth. Mol Biol Evol. 2005;22:2040–2047. doi: 10.1093/molbev/msi195. [DOI] [PubMed] [Google Scholar]
- 43.Hajibabaei M., Shokralla S., Zhou X., Singer G.A., Baird D.J. Environmental barcoding: a next-generation sequencing approach for biomonitoring applications using river benthos. PLoS One. 2011;6:e17497. doi: 10.1371/journal.pone.0017497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hancock-Hanser B.L., Frey A., Leslie M.S., Dutton P.H., Archer F.I., Morin P.A. Targeted multiplex next-generation sequencing: advances in techniques of mitochondrial and nuclear DNA sequencing for population genomics. Mol Ecol Resour. 2013;13:254–268. doi: 10.1111/1755-0998.12059. [DOI] [PubMed] [Google Scholar]
- 45.Cheng X., Chen X., Su X., Zhao H., Han M., Bo C. DNA extraction protocol for biological ingredient analysis of Liuwei Dihuang Wan. Genomics Proteomics Bioinformatics. 2014;12:137–143. doi: 10.1016/j.gpb.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shriver M.D., Smith M.W., Jin L., Marcini A., Akey J.M., Deka R. Ethnic-affiliation estimation by use of population-specific DNA markers. Am J Hum Genet. 1997;60:957–964. [PMC free article] [PubMed] [Google Scholar]
- 47.Eiberg H., Troelsen J., Nielsen M., Mikkelsen A., Mengel-From J., Kjaer K.W. Blue eye color in humans may be caused by a perfectly associated founder mutation in a regulatory element located within the HERC2 gene inhibiting OCA2 expression. Hum Genet. 2008;123:177–187. doi: 10.1007/s00439-007-0460-x. [DOI] [PubMed] [Google Scholar]
- 48.Wei Y.L., Li C.X., Li S.B., Liu Y., Hu L. Association study of monoamine oxidase A/B genes and schizophrenia in Han Chinese. Behav Brain Funct. 2011;7:42. doi: 10.1186/1744-9081-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Phillips C., Prieto L., Fondevila M., Salas A., Gomez-Tato A., Alvarez-Dios J. Ancestry analysis in the 11-M Madrid bomb attack investigation. PLoS One. 2009;4:e6583. doi: 10.1371/journal.pone.0006583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walsh S., Liu F., Ballantyne K.N., van Oven M., Lao O., Kayser M. IrisPlex: a sensitive DNA tool for accurate prediction of blue and brown eye colour in the absence of ancestry information. Forensic Sci Int Genet. 2011;5:170–180. doi: 10.1016/j.fsigen.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 51.Han J.L., Kraft P., Nan H., Guo Q., Chen C., Qureshi A. A genome-wide association study identifies novel alleles associated with hair color and skin pigmentation. PLoS Genet. 2008;4:e1000074. doi: 10.1371/journal.pgen.1000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klimentidis Y.C., Shriver M.D. Estimating genetic ancestry proportions from faces. PLoS One. 2009;4:e4460. doi: 10.1371/journal.pone.0004460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jeffreys A.J., Brookfield J.F., Semeonoff R. Positive identification of an immigration test-case using human DNA fingerprints. Nature. 1985;317:818–819. doi: 10.1038/317818a0. [DOI] [PubMed] [Google Scholar]
- 54.Li C., Zhao S., Zhang N., Zhang S., Hou Y. Differences of DNA methylation profiles between monozygotic twins’ blood samples. Mol Biol Rep. 2013;40:5275–5280. doi: 10.1007/s11033-013-2627-y. [DOI] [PubMed] [Google Scholar]
- 55.Frumkin D., Wasserstrom A., Budowle B., Davidson A. DNA methylation-based forensic tissue identification. Forensic Sci Int Genet. 2011;5:517–524. doi: 10.1016/j.fsigen.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 56.Bocklandt S., Lin W., Sehl M.E., Sanchez F.J., Sinsheimer J.S., Horvath S. Epigenetic predictor of age. PLoS One. 2011;6:e14821. doi: 10.1371/journal.pone.0014821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grunau C., Clark S.J., Rosenthal A. Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. Nucleic Acids Res. 2001;29:E65–5. doi: 10.1093/nar/29.13.e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meissner A., Gnirke A., Bell G.W., Ramsahoye B., Lander E.S., Jaenisch R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33:5868–5877. doi: 10.1093/nar/gki901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weber M., Davies J.J., Wittig D., Oakeley E.J., Haase M., Lam W.L. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37:853–862. doi: 10.1038/ng1598. [DOI] [PubMed] [Google Scholar]
- 60.Paliwal A., Vaissiere T., Herceg Z. Quantitative detection of DNA methylation states in minute amounts of DNA from body fluids. Methods. 2010;52:242–247. doi: 10.1016/j.ymeth.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 61.Xu H., Zhao Y., Liu Z., Zhu W., Zhou Y., Zhao Z. Bisulfite genomic sequencing of DNA from dried blood spot microvolume samples. Forensic Sci Int Genet. 2012;6:306–309. doi: 10.1016/j.fsigen.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 62.Weber-Lehmann J., Schilling E., Gradl G., Richter D.C., Wiehler J., Rolf B. Finding the needle in the haystack: Differentiating “identical” twins in paternity testing and forensics by ultra-deep next generation sequencing. Forensic Sci Int Genet. 2014;9:42–46. doi: 10.1016/j.fsigen.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 63.Yang X., Jiao R., Yang L., Wu L.P., Li Y.R., Wang J. New-generation high-throughput technologies based ‘omics’ research strategy in human disease. Yi Chuan. 2011;33:829–846. [PubMed] [Google Scholar]
- 64.Courts C., Madea B. Micro-RNA – A potential for forensic science? Forensic Sci Int. 2010;203:106–111. doi: 10.1016/j.forsciint.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 65.Hanson E.K., Lubenow H., Ballantyne J. Identification of forensically relevant body fluids using a panel of differentially expressed microRNAs. Anal Biochem. 2009;387:303–314. doi: 10.1016/j.ab.2009.01.037. [DOI] [PubMed] [Google Scholar]
- 66.Zubakov D., Boersma A.W., Choi Y., van Kuijk P.F., Wiemer E.A., Kayser M. MicroRNA markers for forensic body fluid identification obtained from microarray screening and quantitative RT-PCR confirmation. Int J Legal Med. 2010;124:217–226. doi: 10.1007/s00414-009-0402-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hawkins T.L., Detter J.C., Richardson P.M. Whole genome amplification-applications and advances. Curr Opin Biotechnol. 2002;13:65–67. doi: 10.1016/s0958-1669(02)00286-0. [DOI] [PubMed] [Google Scholar]
- 68.Kwok P.Y. Making ‘random amplification’ predictable in whole genome analysis. Trends Biotechnol. 2002;20:411–412. doi: 10.1016/s0167-7799(02)02034-6. [DOI] [PubMed] [Google Scholar]
- 69.Wells D., Sherlock J.K., Handyside A.H., Delhanty J.D. Detailed chromosomal and molecular genetic analysis of single cells by whole genome amplification and comparative genomic hybridisation. Nucleic Acids Res. 1999;27:1214–1218. doi: 10.1093/nar/27.4.1214. [DOI] [PMC free article] [PubMed] [Google Scholar]