

Abstract
Inflammation and coagulation are so tightly linked that the cytokine storm which accompanies the development of sepsis initiates thrombin activation and the development of an intravascular coagulopathy. This review examines the interaction between the inflammatory and coagulation cascades, as well as the role of endogenous anticoagulants in regulating this interaction and dampening the activity of both pathways. Clinical trials attempting to improve outcomes in patients with severe sepsis by inhibiting thrombin generation with heparin and or endogenous anticoagulants are reviewed. In general, these trials have failed to demonstrate that anticoagulant therapy is associated with improvement in mortality or morbidity. While it is possible that selective patients who are severely ill with a high expected mortality may be shown to benefit from such therapy, at the present time none of these anticoagulants are neither approved nor can they be recommended for the treatment of sepsis.
Keywords: Inflammation, Protein C, Heparin, Tissue factor pathway inhibitor, Thrombomodulin, Antithrombin, Sepsis, Coagulation, Neutrophil extracellular traps
Core tip: The interaction between the coagulation and inflammatory cascade contributes to the overall pathophysiology of severe sepsis. These processes become unchecked and thus can lead to significant morbidity and mortality. Many anticoagulants have been studied in clinical trials as a means to modulate the inflammatory and coagulation cascade with the aim to improve outcomes for septic patients via modulation of these cascades. This article outlines the pathophysiology and interaction between inflammation and coagulation in severe sepsis and also reviews the anticoagulants previously studied for modulation.
INTRODUCTION
Sepsis is a leading cause of mortality in the United States responsible for more than 200000 deaths each year. The overall mortality is estimated to be approximately 28.6% for all age groups with mortality increasing in the elderly[1]. Furthermore, the incidence of sepsis is increasing secondary to increased use of immunosuppression therapy, invasive procedures, transplantation, and chemotherapy. Mortality in sepsis is frequently due to organ failure and the risk of mortality increases with the number of failing organs. Individuals with sepsis and three or more failing organs have a 70% mortality rate compared to a mortality rate of 15% without organ failure[2].
Much of the organ failure in sepsis is thought to be caused by microvascular thrombosis. Local thrombus formation is protective when an infection is localized as it works to prevent bacteria spread systemically to other organs. However, once the infection spreads to the blood stream and sepsis develops, the formation of thrombi within the microvasculature acts counterproductively and increases organ damage which may lead to organ failure. Significant coagulation disturbances are thought to complicate approximately 35% of all severe sepsis cases[3]. Small and medium size vessels show fibrin deposition that has been found during autopsy studies of patients with DIC and sepsis. Fibrin is also found in many organs under pathological examination following sepsis[4]. Measures of coagulation activation in patients with sepsis show that some amount of clotting is present in all patients with septic shock regardless of the presence of overt disseminated intravascular coagulation (DIC)[4-6].
Inflammation and coagulation are frequently partners in crime in severe sepsis. Studies in the 1990s showed the complex relationship between these two systems in patients with sepsis or a traumatic insult. The inflammatory mediators were shown to activate coagulation and vice versa as inflammation may be induced by activation of the coagulation cascade[7]. The two are linked through similar activation mechanisms via a variety of pathways. Ultimately it is thought the uncontrolled systemic expression of both systems which plays a key role in the pathogenesis of multi-organ failure in sepsis. In this review article we will outline the role of inflammatory markers and coagulation in sepsis as well as the intricate relationship between the two. Subsequently, we will then review the results of clinical trials attempting to modulate this inflammation in patients with severe sepsis.
INFLAMMATION AND COAGULATION CASCADE RESPONSE TO SEVERE SEPSIS
The innate immune system responds to bacterial infections initiated by cells which detect pathogen associated molecular patterns (PAMPs) that are expressed on invading bacteria. The damaged tissue and cells from the host in sepsis will release intracellular proteins commonly known as alarmins[8]. Together alarmins and PAMPs are termed damage-associated molecular patterns (DAMPs). The initial immune response to a pathogen or DAMPs is driven by pattern recognition receptors (PRRs) that are expressed on immune cells. PRR, however, can also be found on other cells which are primary involved in hemostasis as these are highly conserved receptors. In humans PRRs are mainly reported on platelets as toll-like receptors[9]. This serves as a key link between the immune and coagulation systems as these cells are also then able to recognize and initiate the inflammatory response. Both toll-like receptors and complement receptors are PRRs which can initiate a complex cellular inflammatory response to pathogen invasion. These PRRs further activate the coagulation system through increased production of tissue factor and impairment of anticoagulation and fibrinolysis[10].
Activation of the coagulation cascade and thrombus in sepsis are generally thought of as adverse events occurring as a result of pathogen invasion. However, the recently described process of immunothrombosis suggests that some local thrombosis in response to microorganisms may actually be an independent line of host defense against pathogens[11]. This theory suggests that small amount of clot formation actually is beneficial for the host as bacteria and DAMPs are trapped and kept away from the host circulation, preventing systemic spread of infection and inflammatory cytokines or DAMPs to other organs. The immune system and coagulation systems work closely together via cross signaling to produce immunothrombosis. The innate immune cells, particularly monocytes and neutrophils, are recruited to sites of intravascular thrombosis in response to DAMPs at the site. In turn these cells express activated intravascular tissue factor (TF) which enhances clot formation via the extrinsic pathway of coagulation[11].
Although immunothrombosis may have a beneficial effect for the host in localized infection, this is not true in profound system wide infections. Severe sepsis, septic shock, and DIC occur together when the control mechanisms of inflammation and coagulation and the intricate relationship between these to two breaks down and each proceeds unchecked. The crosstalk between each system may actually perpetuate this process in severe systemic infection. There are several key aspects of the coagulation cascade which, when up-regulated, have a significant impact and are then in turn influenced by the immune system; these include TF, thrombin, and platelets. Likewise, cells which are part of the inflammatory response, particularly neutrophils, and the complement system link inflammation to coagulation during sepsis. Furthermore, the regulation of the coagulation system fails during DIC, endogenous anticoagulant proteins including; tissue factor pathway inhibitor (TFPI), anti-thrombin (AT), and activated protein C (APC) fail to regulate coagulation[9,11].
TF is thought to have a key role in the connection of sepsis and coagulation. It is up-regulated in sepsis through both activation by inflammatory cytokines and failure of control mechanisms like TFPI[10]. The cytokine triggers that cause TF to be expressed include; tumor necrosis factor (TNF), interleukin-1 (IL-1), and other inflammatory mediators including complement[12]. TF is part of the extrinsic pathway of coagulation and in the healthy state, is not exposed on peripheral blood or endothelial cells although it has a significant presence on extravascular cells[13]. The physiologic activation of the extrinsic pathway occurs through disruption of the endothelium and exposure of blood to extravascular cells which express TF or in the intravascular space through triggers that cause circulating monocytes, leukocytes, and neutrophils to expose TF on their cell membranes. The latter is through to be the more common pathway for enhanced expression of TF during pathogen invasion, as the recognition of PAMPs and/or DAMPs by cells of the innate immune system directly enhances TF expression[11] (Figure 1).
Figure 1.
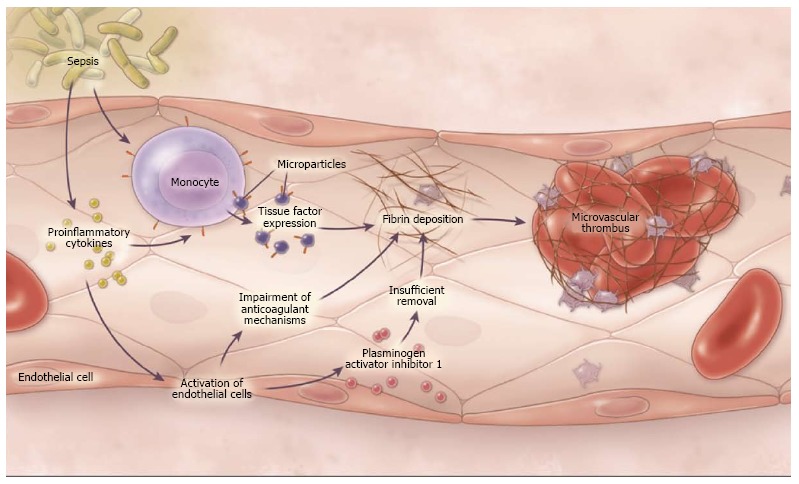
Pathogenesis of disseminated intravascular coagulation in sepsis[10]. Through the generation of proinflammatory cytokines and the activation of monocytes, bacteria cause the up-regulation of tissue factor as well as the release of microparticles expressing tissue factor, thus leading to the activation of coagulation. Proinflammatory cytokines also cause the activation of endothelial cells, a process that impairs anti-coagulant mechanisms and down regulates fibrinolysis by generating increased amounts of plasminogen activator inhibitor. Copyright © 2014 Massachusetts Medical Society (used with permission).
The initiation of coagulation by TF does not always lead to overt DIC in sepsis. Much of the impact of TF on the host response to sepsis may also be due to increasing the inflammatory response. The main inhibitor of TF is TFPI which tightly regulates the interaction of TF with other factors of the extrinsic pathway. TFPI has been studied as a method to modulate the host response to sepsis and through these studies it has become clear that TF has a strong inflammatory contribution during sepsis. Studies have shown that various levels of coagulation are noted in response to sepsis and the effectiveness of TFPI on host survival can be unrelated to differences in coagulation which suggests that much of the beneficial effects of TFPI are from an anti-inflammatory property it can exert on inhibiting TF augmentation of inflammation[14].
Thrombin, like TF, has a large role in coagulation but also exerts some influence on inflammation during sepsis. Thrombin is the central serine protease mediator of hemostasis. It is activated by Factor Xa and, once active, creates an active feedback loop for continued activation of Factor X. Thrombin also converts soluble fibrinogen to insoluble strands of fibrin for clot formation[11,15]. Activated thrombin can promote activation of several pro-inflammatory cytokines including, TNF-α, IL-β, and IL-6 as well as generate C5a (part of the complement response to infection) independent of C3. This activity of thrombin is crucial and demonstrates the complex relationships that exist as it shows the crosstalk that occurs between the coagulation system and components of both the inflammatory and complement systems[15].
Thrombin further actively participates in the inflammatory cascade that occurs during sepsis through specific receptors on platelets, endothelial cells, and white blood cells. These receptors are called protease activated receptors (PARs) and they are responsible for inducing TF emergence from cells and release of plasmin activator inhibitor I or via PARs, which inhibits fibrinolysis as well as decreasing thrombomodulin (TM) (a thrombin co-factor that decreases clot formation)[16]. Thrombin promotes platelet aggregation and activation. The activated platelets express P-selectin after being exposed to thrombin. P-selectin is critical for attachment of white blood cells to endothelial cells and thus initiating white blood cell activation which contributes to diffuse microvascular injury[17].
Thrombin is regulated by AT, which is a serum protein with significant enzymatic activity to prevent coagulation and with its broad spectrum of activity it is considered a central inhibitor of the coagulation system. AT forms a complex with thrombin and inactivates it in addition to inhibiting other factors within the coagulation cascade (including the generation of Factors XIIa, XIa, IXa, and Xa). It acts on both the intrinsic and common coagulation pathways via inhibiting the action of Factors IXa and Xa respectively[7]. AT may also have direct anti-inflammatory actions during sepsis beyond its anti-coagulation properties. AT has the ability to inhibit the function of lipopolysaccharide (LPS) signaling on macrophages. This acts to decrease the level of inflammation through blocking macrophage activation. AT may also compete with bacterial pathogens for binding sites on endothelial surfaces and thus prevent endothelial damage by pathogens[18].
In septic patients with organ failure AT activity is reduced and the degree of reduction is proportional to the severity of sepsis, DIC and organ failure[16]. Part of this fall in AT levels is likely related to the decreased half- life of AT during sepsis. Typically the half-life of AT is between 36-48 h but decreases to less than 18 h during sepsis[7]. Additionally, part of the decrease in half-life for AT is related to neutrophil actions. Activated neutrophils release elastase which destroys AT, and therefore significantly decreases natural regulation of the coagulation system.
APC is another endogenous anticoagulant that has additional anti-inflammatory properties as well. Overall the function of protein C is reduced in sepsis and DIC which results in significant compromise of in regulation of coagulation. APC is down-regulated during the host response to sepsis through cytokines and TM loss on the endothelium as well as a decrease in its primary co-factor protein S[3]. This down-regulation may reflect a conserved part of immunothrombosis and theoretically may be protective in localized tissue infection. However, in sepsis the down-regulation of APC prevents it from actively controlling the host inflammatory response.
APC actually has been found to have an antiinflammatory role during sepsis that may serve partly to keep host response to pathogen invasion in check. APC can decrease apoptosis of endothelial cells and lymphocytes during infection thus reducing DAMPs in the bloodstream which serve to perpetuate further inflammatory reactions. APC also decreases the inflammatory response of key cells; including monocytes, leukocytes and neutrophils. APC decreases NF-κB signaling which decreases monocyte response to inflammation. APC further acts closely with endothelial cells in an anti-inflammation manner. APC specifically decreases TF up-regulation via leukocytes and decreases neutrophils adherence to endothelial cells in response to pathogen invasion[19].
Thrombin is further regulated by TM which is physiologically found in the intravascular space on vessel endothelial walls. TM plays a key role in preventing intravascular clot formation under normal physiological conditions. Thrombin binds with TM in a high affinity complex that prevents thrombin from activating fibrinogen to fibrin. The TM-thrombin complex also works as an anti-coagulant and activates protein C which then inactivates Factors Va and VIIIa[18]. The Thrombin-TM activation of protein C further also acts to decrease the overall inflammatory response to infection by suppressing monocyte-dependent cytokine production[19].
The Thrombin-TM complex has further influence on the anticoagulation and inflammatory systems. Thrombin activatable fibrinolysis inhibitor (TAFI) activity is increased three-fold by the Thrombin-TM complex formation. TAFI enhances fibrin clot stabilization leading to better control of inflammation as part of immunothrombosis. The activity of TAFI also has systemic anti-inflammatory properties. TAFI inactivates endogenous pro-inflammatory mediators, including bradykinin, osteopontin, and some elements of the complement system (C3 and C5)[19].
Platelets, like AT and APC, are also frequently decreased in any host response to insult including pathogen invasion. The decrease in platelets is not fully explainable by overt DIC, but rather more likely related to platelet consumption from immunothrombosis and the role platelets play in the inflammatory response. Thrombocytopenia is in fact, a predictor of poor outcome in sepsis and septic shock. This includes severity of thrombocytopenia as well as overall duration of low platelet count indicates poor outcomes[20].
Platelets have a key role in coagulation and clot formation. Platelets also recognize pathogens via PRRs and therefore, stimulate the host inflammatory response. An important example of cross-linkage between inflammation and coagulation via PRRs is toll-like receptor 4 (TLR4) (a subfamily of PRRs) on platelet cells, which recognize DAMPs and play an intricate role in activating neutrophils in the immune response[15]. TLR4 on platelets recognizes PAMPs of pathogens specifically LPS of gram negative bacteria. These receptors also recognize endogenous DAMPs which include heat shock proteins and fibronectin[21]. Platelets are further able to present microorganisms to neutrophils other immune cells, activating a portion of the innate immune system.
In addition to pathogen recognition, platelets up-regulate and alter the host immune response to pathogens. Platelets, for example, assist in recruiting and enhancing the microbicidal activity of leukocytes by releasing multiple mediators in the bloodstream. Platelets also act to recruit innate immune cells at the site of infection. In addition to recognizing DAMPs, platelets also release DMAPs which promotes additional TF and likely increases thrombosis intravascularly. This process was likely conserved in host immune response as part of the concept of immunothrombosis. Finally, platelets directly bind to neutrophils during the host response to bacterial infection which subsequently stimulates the formation of neutrophil extracellular traps, although the exact methodology behind this is not known[11,22].
Neutrophils are terminally differentiated cells that are involved in the early immune response to invading microbes. Neutrophils are directed by cytokines to infected tissue where the cell is activated and may engulf a pathogen for intracellular killing. Neutrophils may also act though extracellular traps or NETs. NETs are composed of chromatin complexes with granular proteins which bind both Gram negative and positive bacteria and effectively kill microbes in an extracellular matrix. The NET releases cellular components that have antibacterial properties including DNA, myeloperoxidase and elastase and histones. The release of these components into the circulation can have unintended harmful effects on the host during sepsis through activation of the coagulation cascade[8].
NETs can activate coagulation via multiple mechanisms. NETs deliver TF to the extracellular surface and thus activate the extrinsic coagulation pathway. The NET surface is polyanionic and can activate certain contact phase proteins like Factor XII. They further stimulate platelet activation via histones H3 and H4 specifically. These histones also promote the extrinsic pathway through damage to the endothelial wall. Finally, NETs inactive TFPI and TM through a complex enzyme pathway. The inactivity of these endogenous anticoagulants further leads to uncontrolled coagulation during DIC. Much of the coagulation function of NETs is proposed to be protective of the host as immunothrombosis to contain pathogens at an infectious site. However, in systemic infection the protective process becomes destructive through unchecked inflammation and coagulation[11].
The complicated and complex interactions between the traditional coagulation system and the inflammatory system are further complicated by the role of complement during sepsis and DIC. Complement can be viewed as an “alarm system” which can recognize DAMPs and respond to both infectious and non-infectious changes within the host[23]. The complement system is crucial to host defense against invading microbes. It is well known that deficiency in some components of complement can make an individual susceptible to increased risk of infection. For example, deficiency in opsonization contributes to increased susceptibility to infections by pyogenic bacteria including Haemophilus influenza and S. pneumonia. However during multi-organ failure secondary to sepsis, complement is thought to have a detrimental effect on the host and contribute to organ failure[24].
Complement and in particular C5a can have harmful effects during sepsis that act through both inflammation and coagulation dis-regulation. High levels of C5a act to decrease the effectiveness of neutrophils by blocking the NADPH formation of superoxide anions essential for killing gram negative bacteria. Then in turn C5a can increase the function of macrophages causing increased or overproduction of cytokines which contribute to the cytokine storm of sepsis. C5a further enhances the expression of TF therefore disrupting the coagulation balance further. Finally C5a increases the apoptosis of thymocytes decreasing B cell production and leading to an immunodeficiency state in late sepsis[25]. Sepsis and the resulting multi-organ failure are clearly much more complex of a process to be explained by unregulated inflammation or coagulation alone. Instead, it is an intricate web with multiple cross-talk interactions between the various components of each system. This intricate web is the over-riding etiology that leads to sustained multi-organ failure which causes host demise. These are numerous targets within these systems that may be targeted as therapy to augment the overall host response to overwhelming sepsis. Many of the initial clinical trials in sepsis focused on blocking various components of the inflammatory cascade in sepsis. When these trials failed to show a significant improvement in morbidity or mortality it was natural to turn to modifying the coagulant response in an attempt to influence the outcome. Key clinical trials which investigate anticoagulants in an attempt to improve outcomes in septic patients will be reviewed in the following sections.
ANTICOAGULANT EFFECTS ON SEVERE SEPSIS
Antithrombin
AT is an endogenous anticoagulant synthesized by the liver. As its name implies, AT inhibits thrombin as well as factors Xa, IXa, VIIa, XIa, and XIIa. In addition to its anticoagulant effect, AT also has an anti-inflammatory effect at high serum levels. AT binds to glycosaminoglycans on the endothelial cell surface and enhances the microvascular production of prostacyclin I2, a potent vasodilator and inhibitor of platelet function[26].
AT levels are decreased in patients with severe sepsis, and this is associated with a worse prognosis. Several small studies initially suggested that AT administration could have a beneficial effect on organ function and survival in patients with severe sepsis. AT was subsequently studied in the phase 3 KyberSept trial. This study was a large (n = 2314) double blind, placebo controlled, multicenter trial, to determine if high dose AT would provide a survival advantage in patients with severe sepsis[27]. There was no significant effect on the overall mortality at 28 d, 38.9% in the AT group vs 38.7% in the placebo group. However, these results may have been complicated as the concurrent use of low dose heparin for venous thromboembolism prophylaxis was allowed in this study (Table 1).
Table 1.
Previous trials of anticoagulant therapy in severe sepsis
| Ref. | Year | Inclusion criteria | n | Design | Therapy | Primary outcome | Treatment | Control | P value |
| AT | |||||||||
| Warren et al[27] (Kybersept) | 2001 | Severe sepsis | 2314 | Phase 3 RCT | AT 30000 IU × 96 h or placebo | Mortality | 450/115 (38.9%) | 446/1157 (38.7%) | 0.94 |
| Gando et al[29] | 2013 | Sepsis | 60 | Prospective randomized | AT 30 IU/kg × 72 h or placebo | DIC recovery (%) on day 3 | 16/30 (53.3%) | 6/30 (20%) | 0.015 |
| Tagami et al[30] | 2014 | Sepsis-associated DIC in severe pneumonia | 4388 | Retrospective, propensity matched | AT 1500 IU/d | Mortality | 890/2194 (40.6%) | 270/2194 (44.2%) | 0.02 |
| rAPC | |||||||||
| Bernard et al[33] (Prowess trial) | 2001 | Severe sepsis | 1690 | Phase 3 RCT | APC 24 μg/kg per hour × 96 h or placebo | Mortality | 210/850 (24.7%) | 259/840 (30.8%) | 0.005 |
| Vincent et al[35] (Enhance trial) | 2005 | Severe sepsis | 2378 | Prospective single arm multicenter | APC 24 μg/kg per hour × 96 h | Mortality | 25.30% | NA | NA |
| Abraham et al[36] (Address trial) | 2005 | Severe sepsis with APACHE < 25 | 2613 | Phase 3 RCT | APC 24 μg/kg per hour × 96 h or placebo | Mortality | 243/1316 (18.5%) | 220/1297 (17%) | 0.34 |
| Ranieri et al[37] (Prowess shock) | 2012 | Septic shock | 1680 | Phase 3 RCT | APC 24 μg/kg per hour × 96 h or placebo | Mortality | 223/846 (26.4%) | 202/834 (24.2%) | 0.31 |
| Rimmer et al[38] | 2012 | Severe sepsis with septic shock | 933 | Retrospective, 2:1 propensity matched | APC 24 μg/kg per hour × 96 h | Mortality | 180/311 (34.7%) | 254/622 (40.8%) | 0.05 |
| Thrombomodulin | |||||||||
| Saito et al[43] | 2007 | DIC associated with hematologic malignancy or infection | 234 | Phase 3 RCT | Thrombomodulin 0.06 mg/kg × 6 d or heparin 8 U/kg per hour × 6 d | DIC recovery (%) on day 7 | 74/112 (66.1%) (thrombomodulin) | 56 (49.9%) (heparin) | 0.027 |
| Vincent et al[44] | 2013 | Sepsis with DIC | 741 | Phase 2 RCT | Thrombomodulin 0.06 mg/kg × 6 d or placebo | Mortality | 66/371 (17.8%) | 80/370 (21.6%) | 0.273 |
| Heparin | |||||||||
| Jaimes et al[47] (HETRASE study) | 2009 | Sepsis | 317 | RCT | Heparin 500 U/h × 7 d or placebo | LOS | 12 d (median) | 12.5 d (median) | 0.976 |
| rTFPI | |||||||||
| Abraham et al[49] | 2001 | Severe sepsis | 210 | Phase 2 RCT | rTFPI 0.025 or 0.05 mg/kg per hour × 96 h or placebo | Mortality | 43/141 (30%) | 26/69 (38%) | 0.3 |
| Abraham et al[50] (OPTIMIST trial) | 2003 | Severe sepsis | 1754 | Phase 3 RCT | rTFPI 0.025 mg/kg per hour × 96 h or placebo | Mortality | 301/880 (34.2%) | 296/874 (33.9%) | 0.88 |
| Wunderink et al[52] (CAPTIVATE trial) | 2011 | Severe sepsis with community acquired pneumonia | 2102 | Phase 3 RCT | rTFPI 0.025 mg/kg per hour × 96 h or placebo | Severity adjusted 28 d mortality | 185/955 (19.4%) | 178/914 (19.5%) | 0.56 |
RCT: Randomized controlled trial; LOS: Length of stay; APC: Activated protein C; DIC: Disseminated intravascular coagulation; TFPI: Tissue factor pathway inhibitor; AT: Anti-thrombin; NA: Not available.
The simultaneous use of heparin competitively inhibits the binding of AT to other glycosaminoglycans and may have affected the efficacy of AT. Also, AT must bind to glycosaminoglycans on endothelial surfaces to promote local anticoagulant and anti-inflammatory activity[16]. In the subgroup of patients who did not receive concomitant heparin during the treatment phase, the 28 d mortality was lower in the AT group (37.8%) than in the placebo group (43.6%) but this difference did not reach statistical significance (P = 0.08). This mortality trend, however, became significant after 90 d, 44.9% in the AT group vs 52.5% in the placebo group, suggesting that there might be a role for AT therapy in the absence of heparin.
The KyberSept trial examined the results of high dose AT on patient with severe sepsis irrespective of DIC status. Wiedermann et al[28] in a subsequent analysis evaluated patients in the KyberSept trial with DIC and noted an absolute reduction in 28 d mortality of 14.6% compared to placebo (P = 0.02), whereas no effect on 28 d mortality of patients without DIC was seen[28]. A systematic review by Wiedermann suggested that the administration of AT to septic patients with DIC may increase overall survival. AT is approved and is currently being used for the treatment of DIC (including sepsis related DIC) in Japan.
Several studies from Japan have showed positive outcomes from AT use in severe sepsis. A recent small randomized controlled, multicenter trial by Gando et al[29], evaluated the efficacy of a supplemental dose of AT for septic patients with DIC. They enrolled septic patients with DIC and AT levels of 50%-80% normal. The patients were randomly assigned to receive supplemental doses of AT or placebo for 3 d. They noted that AT treatment resulted in significantly decreased DIC scores and better recovery rates from DIC compared with a control group without an increased incidence of bleeding complications. Tagami et al[30] identified 9075 patients with severe pneumonia and sepsis related DIC in a nationwide Japanese database[30]. Using propensity matching to account for confounding factors, they identified 2194 pairs of matched patients who did or did not receive AT treatment. They noted a beneficial effect of AT on 28 d mortality (confidence interval 40.6% vs 44.2%) and adjusted odds ratio favoring AT use. A prospective multicenter study from Japan compared 1500 IU/d vs 3000 IU/d supplemental dose of AT in septic DIC patients. It noted that the AT dose of 3000 IU/d improved survival. AT has been studied extensively and is widely used for the treatment of sepsis related DIC in Japan, however, it remains unavailable for the treatment of severe sepsis or sepsis related DIC in other countries.
Protein C
Protein C has multiple actions within both the coagulation and inflammation pathways. Low levels of protein C are associated with a poor outcome in patients with severe sepsis, suggesting repletion of protein C may benefit these patients. Protein C is converted by thrombin into APC which is an endogenous anticoagulant that inhibits activated cofactors V and VIII, thereby reducing thrombin generation. In addition to its ability to reduce thrombin generation, APC also has anti-inflammatory properties that are independent of its effect on thrombin generation.
Numerous basic science research studies have demonstrated potentially advantageous effects of APC. Extracellular histones released in response to an inflammatory challenge contribute to endothelial dysfunction, organ failure and death during sepsis. APC enhances histone degradation of histones[31], which may account for the cytoprotective activities of APC which include anti-apoptotic activity, ant-inflammatory activity, and endothelial barrier stabilization[32]. Protein C levels are decreased in patients with sepsis and recovery of protein C levels is associated with improved survival. Thus it would seem logical to examine its efficacy as a therapeutic agent.
The Prospective Recombinant Human APC Worldwide Evaluation in Severe Sepsis (PROWESS) trial evaluated the effects of recombinant APC (rhAPC) in patients with severe sepsis[33]. This was the first phase 3 clinical trial to demonstrate improved survival in patients with severe sepsis. PROWESS was a randomized, double blind placebo-controlled multicenter trial which was stopped early because of the improved survival observed in the treated group. The overall mortality rate was 30.8% in the placebo group and 24.7% in the APC group, a reduction of 6.1% (relative risk reduction of 19.4%). The survival benefit was greatest in the sickest patients, those with the most organ failures and highest APACHE II scores. Patients with overt DIC had an absolute reduction in mortality from 40.3% to 30.5%, which is a relative risk reduction of 29.1%[34]. The bleeding event rate was only 3.4%. Subgroup analysis indicated that the mortality benefit was limited to patient with increased illness severity. Based on this study, rhAPC was approved by the Food and Drug Administration in 2001 for the treatment of severe sepsis in patients with an APACHE II of 24 or greater, the median APACHE II score in the PROWESS study.
A subsequent open label trial (ENHANCE) confirmed the mortality rate of approximately 25% in patients with severe sepsis but left open the question of whether rhAPC would be beneficial in less sick patients[35]. The ADDRESS trial randomized 2640 patients with severe sepsis and a single organ failure or APACHE II < 25 to either rhAPC or placebo[36]. There was no difference in survival in these less severely ill patients, but the rate of serious bleeding with rhAPC was 2.4%, double that of the control patients. A subsequent trial focusing on the severely ill patient with septic shock (PROWESS - SHOCK) failed to confirm the improved outcomes noted in the original PROWESS trial, and in 2011 rhAPC was withdrawn from the world market[37].
The mortality rates in the PROWESS-SHOCK trial were substantially lower the expected given the inclusion criteria of septic shock, 26.6% vs 24.2% in the APC and control groups respectively. APC did not reduce mortality at 28 or 90 d, as compared with placebo, but was associated with increased bleeding risks in patients with severe sepsis and septic shock[37]. A subsequent retrospective multicenter cohort study of patients with septic shock, early use of APC was associated with 6.1% absolute reduction in 30 d mortality[38]. Another meta-analysis by Kalil et al[39], noted a significant reduction in hospital mortality (18%), and increased bleeding rate (5.4%) with the real life use of APC compared with controls.
Although rhAPC is no longer accessible, plasma derived APC is available in Japan. A randomized double blind trial compared the efficacy and safety of plasma derived APC with unfractionated heparin in the treatment of DIC by a Japanese group[40]. No significant difference in the rate of complete recovery from DIC was seen between the 2 groups. The rate of death from any cause within 28 d after treatment was 20.4% in the APC group, significantly lower than the 40% death rate observed in the heparin group (P < 0.05). There were no severe adverse events in either group. These findings suggests that plasma derived APC as a remarkably reduced dose compared to rhAPC can improve DIC, without increasing bleeding and its effects should be evaluated in future trials.
TM
TM is an endogenous anticoagulant located on the surface of the endothelial cell that acts by inhibiting thrombin mediated clot formation and enhancing protein C activation at the site of clotting. In addition to its anticoagulant activity, TM also has anti-inflammatory properties, including interfering with the activation of complement and inactivating high-mobility group protein B1, a mediator associated with mortality in late sepsis[41]. TM expression on the endothelial surface is down regulated in patients with sepsis and may contribute to the development of DIC. Replacement of TM, therefore, may offer therapeutic value[42].
Saito et al[43], evaluated the efficacy of recombinant TM in treating DIC in a randomized, multicenter, double blind controlled trial. Recombinant TM or unfractionated heparin was administered to patients with DIC due to either malignancy or sepsis and resolution of DIC was assessed after 7 d[43]. DIC resolved in 66.1% of the group that received recombinant TM, as compared with 49.9% of the heparin groups, respectively (P < 0.05). The incidence of bleeding related adverse events was lower in the recombinant TM group 43.1% as compared to 56.5% in the heparin group. Based on this study, recombinant TM was approved for the treatment of DIC in Japan in 2008.
Subsequently a phase 2 trial evaluated the safety and efficacy of TM in patient with sepsis and DIC[44]. In this trial, DIC was diagnosed by a modified scoring system based on the platelet counts and prothrombin time and international normalized ratio (INR). Patients (371) randomized to TM and patients (370) who received placebo were similar at baseline. Twenty eight day mortality was 17.8% in the TM group and 21.6% in the placebo group. There were no significant differences between the two groups in organ dysfunction, inflammatory markers, bleeding thrombotic events or in the development of new infections during the study. In a post hoc analysis, the greatest benefit from TM was seen in patients with at least one organ dysfunction and an INR of greater than 1.4 at baseline. Based on the results of this encouraging study, a phase 3 trial of recombinant TM in patients with sepsis induced DIC and either shock or acute respiratory failure is currently in progress.
Heparin
Heparin is a sulfated polysaccharide with a heterogeneous structure and complex polymerization (MW, 357 kDa). Heparin binds to AT, causing a conformational change that increases the flexibility of the reactive site loop, activating AT. The activated AT then inactivates thrombin and other proteases, including factor Xa. Heparin also binds platelets to inhibit platelet aggregation, resulting in a strong anticoagulant effect. Heparin at high concentrations prevents histone interactions with platelets, which is a possible therapeutic target to modulate inflammation in severe sepsis. Fuchs et al[45], noted that heparin is highly sulfated and rich in negative charges, and electrostatic interactions with histones, are responsible for its histone-neutralizing effects, which suggests heparin could prevent cytotoxicity and collateral organ damage from histones.
Interest in heparin as a therapeutic agent in sepsis was spurred by the observation of Doshi et al[46], that the use of low dose prophylactic heparin in the placebo arms of KyberSept (phase 3 AT trial) and PROWESS (phase 3 rhAPC trial) was associated with a trend for reduced mortality which was not statistically significant. Heparin use was not randomized in either study and a subsequent study designed to show the safety of concomitant heparin and rhAPC use observed neither benefit nor an increase in bleeding risk. A subsequent retrospective study of 695 propensity matched pairs of patients with septic shock who did or did not receive high dose therapeutic heparin therapy suggested potential reduction in morbidity and mortality with heparin. In the prospective randomized double blind HETRASE Study, 319 patients were randomized to intravenous heparin or placebo. Heparin treatment was safe as there was no increased risk of bleeding. However, there was no significant difference in the primary endpoint, length of hospital stay. Moreover, the heparin treated patients failed to demonstrate a more rapid improvement in organ failure score or increase in survival[47]. Thus, while heparin is readily available, inexpensive, and widely used for DVT prophylaxis in septic patients, its role in the treatment of sepsis remains undetermined.
TFPI
TF is the major initiator of the blood coagulation process during sepsis and TFPI, an endogenous serine protease inhibitor which is synthesized and secreted by endothelial cells, acts to inhibit both the factor VIIa/TF catalytic complex in a Xa dependent fashion as well as factor Xa directly[46]. TFPI may also play a role in maintaining endothelial cell integrity[48].
The initial trial results of recombinant TFPI (rTFPI) administration to patients with severe sepsis were encouraging. A multicenter, randomized placebo controlled, single-blind, dose escalation study enrolled 210 patients who received a continuous infusion of either placebo or rTFPI (dose 0.025 or 0.05 mg/kg per hour) for 4 d. Although the trial was not powered to evaluate mortality, this study noted a trend toward reduction in 28 d all-cause mortality as well as improvement in pulmonary organ dysfunction in the rTFPI group as compared with placebo. Logistic regression modeling suggested a more apparent coagulopathy, manifested by a higher baseline (INR), was associated with more pronounced beneficial TFPI effect[49].
Subsequently Abraham et al[50], examined this concept in a phase 3 randomized, double blind, placebo controlled, multicenter trial which enrolled 1754 patients with severe sepsis and high INR (> 1.2) who were randomly assigned to receive either rTFPI (tifacogin) or placebo. In addition, 201 patients with a low INR (< 1.2) were randomized to receive either rTFPI or placebo. Tifacogin was effective in reducing markers of thrombin activation, but had no effect on 28 d mortality (34.2% with tifacogin vs 33.9% with placebo, P = 0.88). Tifacogin administration was associated with an increase in risk of bleeding, irrespective of baseline INR[50,51]. The most common site of infection in this study was community acquired pneumonia. A post hoc analysis suggested patients, who did not receive concomitant heparin, appeared to benefit from treatment with tifacogin. This led to a large phase 3 trial of rTFPI in patients with severe sepsis from community acquired pneumonia[53]. Again, rTFPI treated patients demonstrated a greater reduction in markers of thrombin activity, but no improvement in mortality was noted.
CONCLUSION
Inflammation and coagulation are tightly linked with each pathway capable of initiating and amplifying the activity of the other. Full blown expression of the coagulation pathway in the septic patient leads to overt DIC, overt but some degree of coagulation activation is apparent in virtually all patients with severe sepsis. Increasing coagulopathy is associated with the development of organ failures and increased mortality[53]. Simultaneous with the development of the coagulopathy there is a fall in the levels of endogenous anticoagulants including APC, AT, and TFPI as a consequence of both increased consumption and impaired production. Survivors of severe sepsis have more rapid return of this endogenous anticoagulant to normal levels. Because these endogenous anticoagulants appear to have anti-inflammatory activity independent of their ability to inhibit thrombin generation, they were administered to patients with severe sepsis in an attempt to improve outcomes. One trial of rhAPC showed an improvement in the survival of the sickest patients, but this benefit could not be replicated in subsequent studies. Phase 3 studies of AT and TFPI failed to demonstrate a clear benefit while a Phase 3 trial of TM is still in progress. Anticoagulant therapy in a patient with an underlying coagulopathy increases the risk of bleeding, which may obscure any potential benefit. At this time future trials of anticoagulant therapy for sepsis should focus on the most severely ill patients with the highest expected mortality, as this is the group in which benefit is most likely to be demonstrated. Until they can be shown to reduce morbidity and mortality, anticoagulants should not be used for the treatment of severe sepsis.
Footnotes
P- Reviewer: Bugaj AM, Feltracco P, Owczarek D S- Editor: Ji FF L- Editor: A E- Editor: Lu YJ
Conflict-of-interest: All authors have no conflicts of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: October 16, 2014
First decision: December 17, 2014
Article in press: February 9, 2015
References
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Dhainaut JF, Shorr AF, Macias WL, Kollef MJ, Levi M, Reinhart K, Nelson DR. Dynamic evolution of coagulopathy in the first day of severe sepsis: relationship with mortality and organ failure. Crit Care Med. 2005;33:341–348. doi: 10.1097/01.ccm.0000153520.31562.48. [DOI] [PubMed] [Google Scholar]
- 3.Levi M. Disseminated intravascular coagulation. Crit Care Med. 2007;35:2191–2195. doi: 10.1097/01.ccm.0000281468.94108.4b. [DOI] [PubMed] [Google Scholar]
- 4.Levi M, Ten Cate H. Disseminated intravascular coagulation. N Engl J Med. 1999;341:586–592. doi: 10.1056/NEJM199908193410807. [DOI] [PubMed] [Google Scholar]
- 5.Dernaika TA, Kinasewitz GT. Heparin in the treatment of severe sepsis: a new look at an old therapy. Crit Care Med. 2008;36:3098–3099. doi: 10.1097/CCM.0b013e31818bdbc6. [DOI] [PubMed] [Google Scholar]
- 6.Kinasewitz GT, Yan SB, Basson B, Comp P, Russell JA, Cariou A, Um SL, Utterback B, Laterre PF, Dhainaut JF. Universal changes in biomarkers of coagulation and inflammation occur in patients with severe sepsis, regardless of causative micro-organism [ISRCTN74215569] Crit Care. 2004;8:R82–R90. doi: 10.1186/cc2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Opal SM. Therapeutic rationale for antithrombin III in sepsis. Crit Care Med. 2000;28:S34–S37. doi: 10.1097/00003246-200009001-00008. [DOI] [PubMed] [Google Scholar]
- 8.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176:231–241. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delvaeye M, Conway EM. Coagulation and innate immune responses: can we view them separately? Blood. 2009;114:2367–2374. doi: 10.1182/blood-2009-05-199208. [DOI] [PubMed] [Google Scholar]
- 10.Hunt BJ. Bleeding and coagulopathies in critical care. N Engl J Med. 2014;370:847–859. doi: 10.1056/NEJMra1208626. [DOI] [PubMed] [Google Scholar]
- 11.Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. 2013;13:34–45. doi: 10.1038/nri3345. [DOI] [PubMed] [Google Scholar]
- 12.Saadi S, Holzknecht RA, Patte CP, Stern DM, Platt JL. Complement-mediated regulation of tissue factor activity in endothelium. J Exp Med. 1995;182:1807–1814. doi: 10.1084/jem.182.6.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Semeraro N, Colucci M. Tissue factor in health and disease. Thromb Haemost. 1997;78:759–764. [PubMed] [Google Scholar]
- 14.Hack CE. Tissue factor pathway of coagulation in sepsis. Crit Care Med. 2000;28:S25–S30. doi: 10.1097/00003246-200009001-00006. [DOI] [PubMed] [Google Scholar]
- 15.Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8:776–787. doi: 10.1038/nri2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iba T, Kidokoro A. High-dose antithrombin therapy for sepsis: mechanisms of action. Shock. 2002;18:389–394. doi: 10.1097/00024382-200211000-00001. [DOI] [PubMed] [Google Scholar]
- 17.Coughlan AF, Hau H, Dunlop LC, Berndt MC, Hancock WW. P-selectin and platelet-activating factor mediate initial endotoxin-induced neutropenia. J Exp Med. 1994;179:329–334. doi: 10.1084/jem.179.1.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duensing TD, Wing JS, van Putten JP. Sulfated polysaccharide-directed recruitment of mammalian host proteins: a novel strategy in microbial pathogenesis. Infect Immun. 1999;67:4463–4468. doi: 10.1128/iai.67.9.4463-4468.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Esmon CT. Inflammation and the activated protein C anticoagulant pathway. Semin Thromb Hemost. 2006;32 Suppl 1:49–60. doi: 10.1055/s-2006-939554. [DOI] [PubMed] [Google Scholar]
- 20.Taylor FB, Wada H, Kinasewitz G. Description of compensated and uncompensated disseminated intravascular coagulation (DIC) responses (non-overt and overt DIC) in baboon models of intravenous and intraperitoneal Escherichia coli sepsis and in the human model of endotoxemia: toward a better definition of DIC. Crit Care Med. 2000;28:S12–S19. doi: 10.1097/00003246-200009001-00004. [DOI] [PubMed] [Google Scholar]
- 21.Ramani V, Madhusoodhanan R, Kosanke S, Awasthi S. A TLR4-interacting SPA4 peptide inhibits LPS-induced lung inflammation. Innate Immun. 2013;19:596–610. doi: 10.1177/1753425912474851. [DOI] [PubMed] [Google Scholar]
- 22.Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13:463–469. doi: 10.1038/nm1565. [DOI] [PubMed] [Google Scholar]
- 23.Markiewski MM, DeAngelis RA, Lambris JD. Complexity of complement activation in sepsis. J Cell Mol Med. 2008;12:2245–2254. doi: 10.1111/j.1582-4934.2008.00504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Markiewski MM, Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. Complement and coagulation: strangers or partners in crime? Trends Immunol. 2007;28:184–192. doi: 10.1016/j.it.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 25.Ward PA. The dark side of C5a in sepsis. Nat Rev Immunol. 2004;4:133–142. doi: 10.1038/nri1269. [DOI] [PubMed] [Google Scholar]
- 26.Wada H, Asakura H, Okamoto K, Iba T, Uchiyama T, Kawasugi K, Koga S, Mayumi T, Koike K, Gando S, et al. Expert consensus for the treatment of disseminated intravascular coagulation in Japan. Thromb Res. 2010;125:6–11. doi: 10.1016/j.thromres.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 27.Warren BL, Eid A, Singer P, Pillay SS, Carl P, Novak I, Chalupa P, Atherstone A, Pénzes I, Kübler A, et al. Caring for the critically ill patient. High-dose antithrombin III in severe sepsis: a randomized controlled trial. JAMA. 2001;286:1869–1878. doi: 10.1001/jama.286.15.1869. [DOI] [PubMed] [Google Scholar]
- 28.Wiedermann CJ, Kaneider NC. A systematic review of antithrombin concentrate use in patients with disseminated intravascular coagulation of severe sepsis. Blood Coagul Fibrinolysis. 2006;17:521–526. doi: 10.1097/01.mbc.0000245302.18010.40. [DOI] [PubMed] [Google Scholar]
- 29.Gando S, Saitoh D, Ishikura H, Ueyama M, Otomo Y, Oda S, Kushimoto S, Tanjoh K, Mayumi T, Ikeda T, et al. A randomized, controlled, multicenter trial of the effects of antithrombin on disseminated intravascular coagulation in patients with sepsis. Crit Care. 2013;17:R297. doi: 10.1186/cc13163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tagami T, Matsui H, Horiguchi H, Fushimi K, Yasunaga H. Antithrombin and mortality in severe pneumonia patients with sepsis-associated disseminated intravascular coagulation: an observational nationwide study. J Thromb Haemost. 2014;12:1470–1479. doi: 10.1111/jth.12643. [DOI] [PubMed] [Google Scholar]
- 31.Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, Taylor FB, Esmon NL, Lupu F, Esmon CT. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15:1318–1321. doi: 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bouwens EA, Stavenuiter F, Mosnier LO. Mechanisms of anticoagulant and cytoprotective actions of the protein C pathway. J Thromb Haemost. 2013;11 Suppl 1:242–253. doi: 10.1111/jth.12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 34.Dhainaut JF, Yan SB, Joyce DE, Pettilä V, Basson B, Brandt JT, Sundin DP, Levi M. Treatment effects of drotrecogin alfa (activated) in patients with severe sepsis with or without overt disseminated intravascular coagulation. J Thromb Haemost. 2004;2:1924–1933. doi: 10.1111/j.1538-7836.2004.00955.x. [DOI] [PubMed] [Google Scholar]
- 35.Vincent JL, Bernard GR, Beale R, Doig C, Putensen C, Dhainaut JF, Artigas A, Fumagalli R, Macias W, Wright T, et al. Drotrecogin alfa (activated) treatment in severe sepsis from the global open-label trial ENHANCE: further evidence for survival and safety and implications for early treatment. Crit Care Med. 2005;33:2266–2277. doi: 10.1097/01.ccm.0000181729.46010.83. [DOI] [PubMed] [Google Scholar]
- 36.Abraham E, Laterre PF, Garg R, Levy H, Talwar D, Trzaskoma BL, François B, Guy JS, Brückmann M, Rea-Neto A, et al. Drotrecogin alfa (activated) for adults with severe sepsis and a low risk of death. N Engl J Med. 2005;353:1332–1341. doi: 10.1056/NEJMoa050935. [DOI] [PubMed] [Google Scholar]
- 37.Ranieri VM, Thompson BT, Barie PS, Dhainaut JF, Douglas IS, Finfer S, Gårdlund B, Marshall JC, Rhodes A, Artigas A, et al. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med. 2012;366:2055–2064. doi: 10.1056/NEJMoa1202290. [DOI] [PubMed] [Google Scholar]
- 38.Rimmer E, Kumar A, Doucette S, Marshall J, Dial S, Gurka D, Dellinger RP, Sharma S, Penner C, Kramer A, et al. Activated protein C and septic shock: a propensity-matched cohort study*. Crit Care Med. 2012;40:2974–2981. doi: 10.1097/CCM.0b013e31825fd6d9. [DOI] [PubMed] [Google Scholar]
- 39.Kalil AC, LaRosa SP. Effectiveness and safety of drotrecogin alfa (activated) for severe sepsis: a meta-analysis and metaregression. Lancet Infect Dis. 2012;12:678–686. doi: 10.1016/S1473-3099(12)70157-3. [DOI] [PubMed] [Google Scholar]
- 40.Aoki N, Matsuda T, Saito H, Takatsuki K, Okajima K, Takahashi H, Takamatsu J, Asakura H, Ogawa N. A comparative double-blind randomized trial of activated protein C and unfractionated heparin in the treatment of disseminated intravascular coagulation. Int J Hematol. 2002;75:540–547. doi: 10.1007/BF02982120. [DOI] [PubMed] [Google Scholar]
- 41.Ito T, Kawahara K, Okamoto K, Yamada S, Yasuda M, Imaizumi H, Nawa Y, Meng X, Shrestha B, Hashiguchi T, et al. Proteolytic cleavage of high mobility group box 1 protein by thrombin-thrombomodulin complexes. Arterioscler Thromb Vasc Biol. 2008;28:1825–1830. doi: 10.1161/ATVBAHA.107.150631. [DOI] [PubMed] [Google Scholar]
- 42.Ito T, Maruyama I. Thrombomodulin: protectorate God of the vasculature in thrombosis and inflammation. J Thromb Haemost. 2011;9 Suppl 1:168–173. doi: 10.1111/j.1538-7836.2011.04319.x. [DOI] [PubMed] [Google Scholar]
- 43.Saito H, Maruyama I, Shimazaki S, Yamamoto Y, Aikawa N, Ohno R, Hirayama A, Matsuda T, Asakura H, Nakashima M, et al. Efficacy and safety of recombinant human soluble thrombomodulin (ART-123) in disseminated intravascular coagulation: results of a phase III, randomized, double-blind clinical trial. J Thromb Haemost. 2007;5:31–41. doi: 10.1111/j.1538-7836.2006.02267.x. [DOI] [PubMed] [Google Scholar]
- 44.Vincent JL, Ramesh MK, Ernest D, LaRosa SP, Pachl J, Aikawa N, Hoste E, Levy H, Hirman J, Levi M, et al. A randomized, double-blind, placebo-controlled, Phase 2b study to evaluate the safety and efficacy of recombinant human soluble thrombomodulin, ART-123, in patients with sepsis and suspected disseminated intravascular coagulation. Crit Care Med. 2013;41:2069–2079. doi: 10.1097/CCM.0b013e31828e9b03. [DOI] [PubMed] [Google Scholar]
- 45.Fuchs TA, Bhandari AA, Wagner DD. Histones induce rapid and profound thrombocytopenia in mice. Blood. 2011;118:3708–3714. doi: 10.1182/blood-2011-01-332676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doshi SN, Marmur JD. Evolving role of tissue factor and its pathway inhibitor. Crit Care Med. 2002;30:S241–S250. doi: 10.1097/00003246-200205001-00012. [DOI] [PubMed] [Google Scholar]
- 47.Jaimes F, De La Rosa G, Morales C, Fortich F, Arango C, Aguirre D, Muñoz A. Unfractioned heparin for treatment of sepsis: A randomized clinical trial (The HETRASE Study) Crit Care Med. 2009;37:1185–1196. doi: 10.1097/CCM.0b013e31819c06bc. [DOI] [PubMed] [Google Scholar]
- 48.Creasey AA, Reinhart K. Tissue factor pathway inhibitor activity in severe sepsis. Crit Care Med. 2001;29:S126–S129. doi: 10.1097/00003246-200107001-00038. [DOI] [PubMed] [Google Scholar]
- 49.Abraham E, Reinhart K, Svoboda P, Seibert A, Olthoff D, Dal Nogare A, Postier R, Hempelmann G, Butler T, Martin E, et al. Assessment of the safety of recombinant tissue factor pathway inhibitor in patients with severe sepsis: a multicenter, randomized, placebo-controlled, single-blind, dose escalation study. Crit Care Med. 2001;29:2081–2089. doi: 10.1097/00003246-200111000-00007. [DOI] [PubMed] [Google Scholar]
- 50.Abraham E, Reinhart K, Opal S, Demeyer I, Doig C, Rodriguez AL, Beale R, Svoboda P, Laterre PF, Simon S, et al. Efficacy and safety of tifacogin (recombinant tissue factor pathway inhibitor) in severe sepsis: a randomized controlled trial. JAMA. 2003;290:238–247. doi: 10.1001/jama.290.2.238. [DOI] [PubMed] [Google Scholar]
- 51.Laterre PF, Opal SM, Abraham E, LaRosa SP, Creasey AA, Xie F, Poole L, Wunderink RG. A clinical evaluation committee assessment of recombinant human tissue factor pathway inhibitor (tifacogin) in patients with severe community-acquired pneumonia. Crit Care. 2009;13:R36. doi: 10.1186/cc7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wunderink RG, Laterre PF, Francois B, Perrotin D, Artigas A, Vidal LO, Lobo SM, Juan JS, Hwang SC, Dugernier T, et al. Recombinant tissue factor pathway inhibitor in severe community-acquired pneumonia: a randomized trial. Am J Respir Crit Care Med. 2011;183:1561–1568. doi: 10.1164/rccm.201007-1167OC. [DOI] [PubMed] [Google Scholar]
- 53.Kidokoro A, Iba T, Fukunaga M, Yagi Y. Alterations in coagulation and fibrinolysis during sepsis. Shock. 1996;5:223–228. doi: 10.1097/00024382-199603000-00010. [DOI] [PubMed] [Google Scholar]