

 This work is licensed under a
This work is licensed under a Abstract
Background
Neonatal diabetes mellitus (NDM) is a rare form of monogenic diabetes and usually presents in the first 6 months of life. We aimed to describe the clinical characteristics and molecular genetics of a large Turkish cohort of NDM patients from a single centre and estimate an annual incidence rate of NDM in South-Eastern Anatolian region of Turkey.
Design and methods
NDM patients presenting to Diyarbakir Children State Hospital between 2010 and 2013, and patients under follow-up with presumed type 1 diabetes mellitus, with onset before 6 months of age were recruited. Molecular genetic analysis was performed.
Results
Twenty-two patients (59% males) were diagnosed with NDM (TNDM-5; PNDM-17). Molecular genetic analysis identified a mutation in 20 (95%) patients who had undergone a mutation analysis. In transient neonatal diabetes (TNDM) patients, the genetic cause included chromosome 6q24 abnormalities (n=3), ABCC8 (n=1) and homozygous INS (n=1). In permanent neonatal diabetes (PNDM) patients, homozygous GCK (n=6), EIF2AK3 (n=3), PTF1A (n=3), and INS (n=1) and heterozygous KCNJ11 (n=2) mutations were identified. Pancreatic exocrine dysfunction was observed in patients with mutations in the distal PTF1A enhancer. Both patients with a KCNJ11 mutation responded to oral sulphonylurea. A variable phenotype was associated with the homozygous c.-331C>A INS mutation, which was identified in both a PNDM and TNDM patient. The annual incidence of PNDM in South-East Anatolian region of Turkey was one in 48 000 live births.
Conclusions
Homozygous mutations in GCK, EIF2AK3 and the distal enhancer region of PTF1A were the commonest causes of NDM in our cohort. The high rate of detection of a mutation likely reflects the contribution of new genetic techniques (targeted next-generation sequencing) and increased consanguinity within our cohort.
Introduction
Neonatal diabetes mellitus (NDM) is a rare form of monogenic diabetes which presents in the first 6 months of life (1). There are two main clinical subtypes of NDM: permanent neonatal diabetes (PNDM) and the remitting and frequently relapsing, transient neonatal diabetes (TNDM) (2, 3). The underlying genetic defect in TNDM can be ascertained in more than 90% cases. The majority of cases of TNDM are linked to an imprinted region on chromosome 6q24, the abnormality either being paternal uniparental disomy, paternal duplication or defective methylation of the maternal allele (4). The latter can sometimes be due to biallelic mutations in ZFP57, a gene involved in the regulation of DNA methylation (5). Heterozygous mutations in genes encoding the KIR6.2 and SUR1 subunits of the pancreatic ATP-sensitive potassium (KATP) channels (KCNJ11 and ABCC8) are common causes of TNDM (6).
PNDM is genetically heterogeneous with mutations in 20 different genes described to date: KCNJ11, ABCC8, FOXP3, GCK, PDX1, PTF1A, EIF2AK3, SLC2A2, GATA6, SLC19A2, WFS1, NEUROD1, NEUROG3, RFX6, WFS1, NKX2-2, MNX1, IER3IP1, INS and GL IS3 (1, 7). Heterozygous mutations in the KCNJ11, ABCC8 and INS gene are the most common causes of PNDM. However, in populations with a high degree of consanguinity, rare genetic causes of PNDM such as homozygous mutations in EIF2AK3, INS and GCK account for the majority of cases (8).
The incidence of NDM in different countries has been calculated from the referral rate of new cases to the diagnostic laboratory or referral endocrine centres. Thus reported incidences are quite variable (one in 21 196 to one in 215 417) not only depending on the calculation method but also the population characteristics (e.g. prevalence of consanguinity) (9, 10, 11). In general, the incidence in western countries is lower than the eastern countries with a high prevalence of consanguinity. Turkey, particularly South-Eastern Anatolian region, is an area with a high prevalence (>40%) of consanguineous marriage (12).
To the best of our knowledge, the regional and nationwide annual incidence of NDM has not been reported from Turkey. We describe the clinical characteristics, molecular genetics and long-term follow-up of a large Turkish cohort of NDM patients from a single paediatric endocrine centre. We also estimate the incidence of NDM in South-Eastern Anatolian region of Turkey based on referrals to a regional paediatric endocrine centre.
Subjects and methods
Patients
Patients referred/presenting with NDM to the paediatric endocrine department at Diyarbakir Children State Hospital, Turkey between January 2010 and December 2013 were phenotyped in detail. The paediatric endocrinology department at Diyarbakir Children State Hospital is the only paediatric endocrine centre in Diyarbakir and also receives NDM referrals from four neighbouring cities (Siirt, Sirnak, Mardin and Batman) in the South-Eastern Anatolian region.
NDM was defined as onset of diabetes mellitus diagnosed before 6 months of age. Details of clinical presentation, birth and family history, clinical phenotype, biochemical data, pancreatic imaging and management were collected using a standardised proforma.
Molecular genetic analysis was undertaken as described below. In addition, genetic analysis was undertaken on patients with presumed type 1 diabetes mellitus under follow-up where the clinical phenotype was consistent with monogenic diabetes.
Incidence of NDM
The annual incidence of NDM was calculated for the South-Eastern Anatolian region of Turkey between 2010 and 2013 inclusive, based on referral rates of NDM to the tertiary paediatric endocrine centre in Diyarbakir. The annual live birth rate data for this region was supplied by Turkish Statistical Institute (TSI; http://tuikapp.tuik.gov.tr/demografiapp/dogum.zul).
Genetic analysis
Genomic DNA was extracted from peripheral leukocytes of 19 patients using standard procedures, and the coding regions and intron/exon boundaries of the ABCC8, KCNJ11, INS and EIF2AK3 genes were amplified by PCR (primers available on request). The amplicons were sequenced using the Big Dye Terminator Cycler Sequencing Kit v3.1 (Applied Biosystems) according to manufacturer's instructions, and reactions were analysed on an ABI 3730 Capillary sequencer (Applied Biosystems). The sequences were compared with the reference sequences (NM_000525.3, NM_000352.3 (U63421 and L78208), NM_000207.2 and AF110146.1) using Mutation Surveyor v3.24 Software (SoftGenetics, State College, PA, USA).
For one patient, Sanger sequencing of the coding regions of GCK (NM_000162.3) was undertaken. This was prompted by the presence of mild-fasting hyperglycaemia in both parents. In a second patient with pancreatic exocrine insufficiency, sequence analysis of a distal PTF1A regulatory enhancer region was performed by methods described previously (13).
All 20 known neonatal diabetes genes were subsequently screened on the Illumina HiSeq2000 targeted next-generation sequencing platform in six patients where Sanger sequencing, or methylation analysis, had not identified a causative mutation. Details of the methodology have been reported previously (7).
Chromosome 6q24 methylation analysis was performed on three patients with TNDM, in whom ABCC8, KCNJ11, INS and EIF2AK3 mutations had been excluded. Methylation-specific PCR was used to detect hypomethylation of the 6q24 locus, followed by microsatellite analysis to discriminate uniparental disomy of chromosome 6 (UPD6pat) from isolated hypomethylation at 6q24, as described (14). The samples with 6q24 hypomethylation but not UPD6pat were tested for hypomethylation at other imprinted loci and for ZFP57 mutations, as described (5).
The study was performed according to the principles of the Declaration of Helsinki with written informed consent given by the patients' parents.
Statistical analyses
Statistical analysis was performed using IBM SPSS 21.0 for Windows statistical software. Shappiro–Wilk test was used to test the normality of distribution of data. The ratios were compared using χ 2 test (or Fisher's exact test). The means were compared using ‘independent sample t-test’ in normally distributed data and medians using Mann–Whitney U test for non-normally distributed data. Data were expressed as median (interquartile range) or mean±s.d. (range). A P value ≤0.05 was considered to be statistically significant.
Results
Presenting characteristics
Twenty-two patients were diagnosed with NDM. Seventeen patients were born between January 2010 and December 2013. Five patients were born before January 2010 but were not tested for mutations at the time of diagnosis of NDM. Three of them had been misdiagnosed as type 1 diabetes mellitus, whereas two died in early infancy.
NDM resolved in five patients (TNDM), whereas 15 patients had PNDM. Two patients died in early infancy due to unrelated illnesses and hence could not be clearly classified as TNDM or PNDM. However, due to a strong indication of GCK-NDM, we classified these patients into the PNDM group. Comparison of the two subtypes revealed significant difference in the age at presentation of diabetes, with TNDM patients presenting earlier than PNDM patients (Table 1). Incidentally, all TNDM patients in our cohort were females, whereas for all types of NDM, the male-to-female ratio was 13:9.
Table 1.
Comparison of clinical characteristics for patients with TNDM and PNDM.
| TNDM (n=5) | PNDM (n=17) | P | |
|---|---|---|---|
| Age at presentation (weeks) | 1.4±0.9 | 6.2±5.5 | 0.005 |
| Gestational age (weeks) | 38.8±2.2 | 38.2±2.7 | 0.499 |
| Birth weight (g) | 2388±526 | 2213±626 | 0.596 |
| Plasma glucose (mmol/l) | 31.1±9.4 | 28.3±10.6 | 0.884 |
| C peptide (pmol/l) | 0.7±0.8 | 0.3±0.3 | 0.690 |
| Serum insulin (mU/l) | 5.2±1.5 | 4.2±3.2 | 0.345 |
| Female/male | 5/0 | 4/13 | 0.006 |
| DKA at presentation (n, %) | 1/4 (25) | 2/5 (40) | 0.166 |
| Monogenic diabetes in FH (n, %) | 2/5 (40) | 10/14 (71) | 0.126 |
| Consanguinity in FH, n (%) | 5/5 (100) | 12/14 (86) | 0.544 |
DKA, diabetic ketoacidosis; FH, family history.
Patients with EIF2AK3 and KATP channel mutations were appropriate for gestational age (birth weight between −2 s.d. and +2 s.d.), whereas patients with mutations in GCK, PTF1A, INS and 6q24 methylation abnormality were small for gestational age (birth weight <−2 s.d.) (Table 2).
Table 2.
Genotype–phenotype analysis and follow-up characteristics of mutation positive NDM patients.
| Family and patient no. | Gene | Age a (weeks) | GW/BW (weeks/g) | Current age (years) | Site of mutation | DNA (protein) description | Zygosity/novelty | NDM subtype | F/H MD | EPI | Pancreas imaging | SU-R | Associated disease and follow-up |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1.1 | GCK | 4 | 38/1600 | 2.4 | Exon 5 | c.506A>G (p.K169R) | HM/novel | Permanent | Yes | No | Normal | No | Thalassemia major |
| 2.2 | GCK | 1 | 40/1900 | 6.3 | Exon 5 | c.506A>G (p.K169R) | HM | Permanent | Yes | No | Normal | No | Two siblings (patient 2.3 and 2.4) also had NDM |
| 2.3 | GCK | 2 | 40/1700 | Died | Exon 5 | c.506A>G (p.K169R) b | HM | Permanent b | Yes | NA | Normal | NA | Died due to sepsis at 3 months |
| 2.4 | GCK | 4 | 39/1600 | Died | Exon 5 | c.506A>G (p.K169R) b | HM | Permanent b | Yes | NA | Normal | NA | Died due to intestinal obstruction (post surgery) at 3 months |
| 3.5 | GCK | 2 | 40/2400 | 2.7 | Exon 6 | c.658T>C (p.C220R) | HM/novel | Permanent | Yes | No | Normal | No | Patient 3.5 and 3.6 are siblings in a large consanguineous family with a number of individuals with monogenic diabetes |
| 3.6 | GCK | 1 | 36/1600 | 1.2 | Exon 6 | c.658T>C (p.C220R) | HM | Permanent | Yes | No | Normal | No | |
| 4.7 | 6q24 (ZFP57) | 1 | 40/3150 | 3.0 | Exon 6 | c.682C>T (p.R228C) | HM/novel | Transient | Yes | No | Normal | NA | Remission at 3 months |
| 5.8 | 6q24 (ZFP57) | 1 | 40/2200 | 1.0 | Exon 6 | c.964delC (p.Q322RfsX13) | HM/novel | Transient | No | No | Normal | NA | Macroglossia, remission at 5 months |
| 6.9 | 6q24 | 1 | 39/1980 | Died | Complete loss of methylation | UPDPat6 | Transient | No | No | Normal | NA | Remission at 3 months, died when she was 5 months | |
| 7.10 | PTF1A | 1 | 31/1500 | 3.1 | Promoter | g.23508437A>G | HM | Permanent | Yes | Yes | Agenesis | NA | Developmental delay |
| 8.11 | PTF1A | 10 | 39/2400 | 3.1 | Promoter | g.23508437A>G | HM | Permanent | No | Yes | Agenesis | NA | |
| 9.12 | PTF1A | 3 | 32/1200 | 2.4 | Promoter | g.23508365A>G | HM | Permanent | No | Yes | Agenesis | NA | Neonatal cholestasis |
| 10.13 | EIF2AK 3 | 14 | 40/3000 | Died | Exon 5 | c.997C>T (p.Q333X) | HM | Permanent | Yes | No | Hypoplasia | NA | A hepatic failure observed at 12 months old, skeletal dysplasia on X-rays, died at the age of 3 years due to second attack of hepatic failure |
| 11.14 | EIF2AK3 | 12 | 40/2800 | 3.3 | Exon 9 | c.1562G>A (p.W521X) | HM/novel | Permanent | No | No | Normal | NA | No liver dysfunction observed, severe skeletal dysplasia |
| 12.15 | EIF2AK3 | 10 | 40/3050 | 1.0 | Intron 11 | c.1884-1G>C (p.?) | HM/novel | Permanent | Yes | No | Normal | NA | Transient elevation of liver enzymes, hypoalbuminaemia, no skeletal dysplasia at the 1 year of age |
| 13.16 | INS | 1 | 35/1910 | 1.2 | Promoter | c.-331C>A (p.?) | HM | Transient | No | No | Normal | NA | Remission at the age of 2 months |
| 14.17 | INS | 1 | 37/1400 | 0.7 | Promoter | c.-331C>A (p.?) | HM | Permanent | Yes | No | Normal | NA | No remission |
| 15.18 | KCNJ11 | 2 | 39/3000 | 4.8 | Exon 1 | c.602G>A (p.R201H) | HT | Permanent | Yes | No | Normal | Yes | Successful transfer to SU therapy and weaned off insulin therapy |
| 16.19 | KCNJ11 | 13 | 40/2800 | 6.6 | Exon 1 | c.602G>A (p.R201H) | HT | Permanent | No | No | Normal | Yes | Developmental delay, epilepsy, successful transfer to SU therapy and weaned off insulin therapy |
| 17.20 | ABCC8 | 3 | 40/2700 | 2.4 | Exon 10 | c.1594A>G (p.S532G) | HT/novel | Transient | Yes | No | Normal | NA | Remission at the age of 3 months |
GW, gestation week; BW, birth weight; NDM, neonatal diabetes mellitus; HM, homozygous; HT, heterozygous; UPDPat6, paternal uniparental disomy on Chr6q24; F/H MD, family history of monogenic diabetes; EPI, exocrine pancreas insufficiency; SU-R, sulphonylurea response; SU, sulphonylurea.
Age at NDM diagnosis.
Not tested but presumably GCK PNDM as sibling diagnosed with GCK PNDM and both parents were heterozygous carriers of GCK mutation.
Annual incidence of NDM
According to TSI, the total number of live births in the five cities (Diyarbakir, Siirt, Sirnak, Mardin and Batman) in the South-Eastern Anatolian region of Turkey between 2010 and 2013 was 387 857. In our cohort of 22 NDM patients, 13 patients (five TNDM and eight PNDM) were born during this period across these five cities. These figures suggest that the overall annual incidence of NDM (and PNDM) during this period in the South-Eastern Anatolian region of Turkey was at least one in 30 000 (one in 48 000) live births.
Mutation analysis
The underlying genetic cause for NDM was identified in 20/21 (95%) patients by undergoing a mutation anlysis. Chromosome 6q24 abnormalities, heterozygous ABCC8 and homozygous INS mutations were identified in three, one and one patients respectively (Fig. 1). Two patients with chromosome 6q24 methylation abnormalities had a homozygous mutation in ZFP57 (p.Q322RfsX13 and p.R228C) and one patient had paternal uniparental isodisomy (UPD) of chromosome 6. The p.Q322RfsX13 (c.964delC) frameshift mutation is a single-base deletion which results in the introduction of a premature termination codon. The p.R228C (c.682C>T) mutation is a missense mutation which is predicted to change the conformation of a cys-his zinc finger 3.
Figure 1.
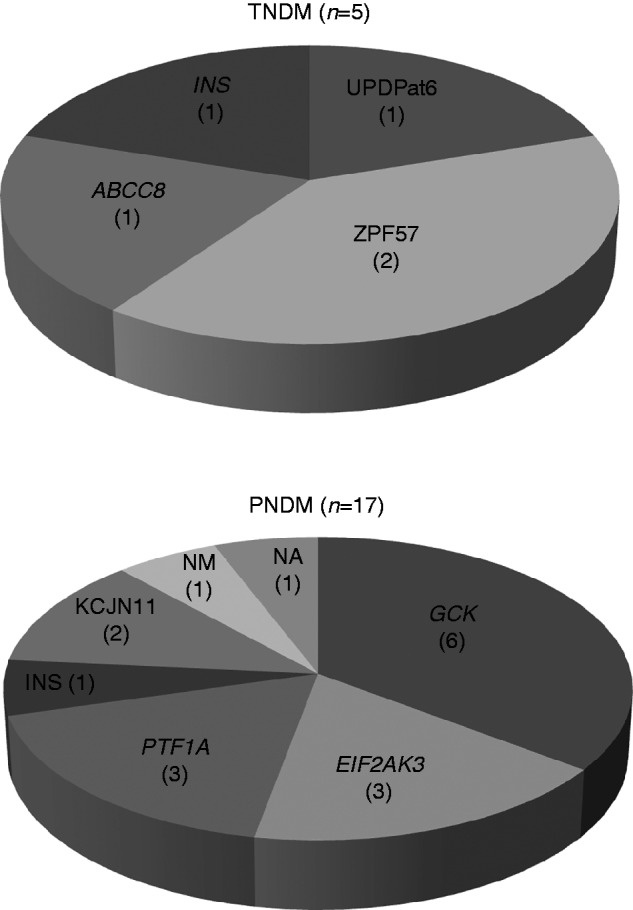
Distribution of mutations detected in patients with TNDM and PNDM (NA, not available; NM, no mutation).
The genetic cause of diabetes was identified in 15 of the 17 PNDM patients. Nine different mutations in five genes (GCK, EIF2AK3, PTF1A, KCNJ11 and INS) were identified (Fig. 1). Two different homozygous GCK mutations (p.K169R (c.506A>G) and p.C220R (c.658T>C)) were identified in four patients, two of whom were siblings. A further two NDM patients who died in early infancy of unrelated illnesses, siblings of patient 2, are likely to have had GCK NDM (although not formally tested) as both parents were heterozygous for the p.K169R GCK mutation. Both mutations were novel at the time of diagnosis, and affect residues which are highly conserved across species. A different mutation at the same amino acid residue has been reported previously (15).
Three different homozygous EIF2AK3 mutations were identified in three patients, one of which, p.W521X (c.1562G>A), has been reported previously (8, 16). The remaining two mutations were novel (c.1884-1G>C and p.Q333X (c.997C>T)).
Two different homozygous mutations (chr10:g.23508437 and chr10:g.23508365) in the distal enhancer region of PTF1A (∼25 kb downstream from PTF1A) were identified in three patients. Both mutations have been reported previously in patients with pancreatic hypoplasia (13).
A previously reported heterozygous KCNJ11 mutation (p.R201H, c.602G>A) was identified in two unrelated patients (17). One patient had a previously reported homozygous INS promoter mutation (c.-331C>A) which is predicted to disrupt a transcriptional regulatory site resulting in decreased insulin transcription (18).
No mutation was identified in two patients, one patient had been tested by targeted next-generation sequencing (7). There was inadequate DNA available for testing for the remaining patient.
Clinical details and genotype–phenotype relation
All six patients with a homozygous GCK mutation were born to consanguineous families and the median age of presentation with diabetes was 5 days (range 2–28 days). One patient had been misdiagnosed as type 1 diabetes mellitus and confirmation of GCK NDM was done at a later age rather than at initial presentation. Two siblings of this patient were diagnosed with NDM, but died in early infancy. It is presumed that they also had the identical homozygous GCK mutation. All these patients are currently on a relatively high dose of s.c. insulin (1.5 U/kg per day). In a trial to switch s.c. insulin therapy to sulphonylurea, none of these patients responded. Coincidentally, one patient also had thalassemia major and required regular blood transfusions.
The three patients with Wolcott–Rallison syndrome (WRS), due to homozygous EIF2AK3 mutations, were from consanguineous families and they presented with diabetes at a median age of 85 days (range 66–96 days). Over a mean follow-up period of 2.5 years, episodic elevation of liver enzymes and skeletal abnormalities were noticed in two patients. Imaging revealed pancreatic hypoplasia in one patient who died at the age of 3 years due to acute liver failure (Table 2).
The three patients with mutations in distal enhancer region of PTF1A presented with diabetes at a median age of 3 weeks (range 1–10 weeks). In addition to NDM, all three patients displayed symptoms of pancreatic exocrine insufficiency and pancreatic hypoplasia on imaging.
The two patients with KCNJ11 mutations presented with diabetes between 1 and 3 months of age. One of these patients had additional neurological features including epilepsy and developmental delay, which developed after acute presentation with diabetic ketoacidosis. This patient was misdiagnosed as type 1 diabetes mellitus and the confirmation of KCNJ11 NDM was done at the age of 6.2 years. An attempt was made to switch both patients to an oral sulphonylurea therapy after the molecular genetics diagnosis. Both patients responded well to glibenclamide (0.5 and 1.5 mg/kg per day respectively) and insulin therapy was successfully weaned.
The patient with an ABCC8 mutation presented at 18 days of age and the diabetes remitted at the age of 7 months. She is now 2.4 years of age and has a normal blood glucose profile. Post remission, her HbA1c improved from 7.1% (54 mmol/mol) to 5.2% (33 mmol/mol).
Both patients with the homozygous INS promoter mutation presented with NDM within the first week of life. The mean birth weight was 1650 g at 36 weeks gestational age. One patient remitted at the age of 65 days, whereas the second patient, 13 months at the time of writing, was on insulin therapy.
All three patients with NDM due to chromosome 6q methylation abnormalities presented within first week of life. The mean birth weight was 2440 g at 40 weeks gestational age. All three patients remitted before 5 months of age. One patient with TNDM due to paternal UPD died due to unrelated illness. The remaining two patients with a ZFP57 mutation, aged 1 and 3 years, are still in remission. Apart from macroglossia in one patient, no other additional features were observed.
For the two patients in whom the underlying genetic cause could not be established, there is a strong family history of diabetes mellitus in at least three generations for one patient (proband, father, brother and grandfather). None of the other diabetic members from his family had been diagnosed during the neonatal period. Mutation analysis of all 20 known NDM genes did not identify a mutation in this patient. In the second patient, who died at the age of 2.5 years, apart from NDM there were additional features of thiamine-responsive megaloblastic anaemia, pancreatic exocrine insufficiency and neurodevelopmental delay. No improvement in insulin requirement was noticed with thiamine replacement. DNA was not available from this patient.
Discussion
We describe the genetic aetiology and clinical manifestations of NDM patients from a single, large paediatric endocrine centre from South-Eastern Anatolian region of Turkey. To the best of our knowledge, our centre's cohort of 22 NDM patients is the largest single centre cohort, 20 of whom have received a genetic diagnosis.
In our study, mutations in GCK, EIF2AK3 and a distal enhancer region of PTF1A were the commonest cause of NDM, accounting for 30, 15 and 15% of genetically confirmed cases respectively. Although homozygous inactivating mutations in GCK are a rare cause of PNDM, isolated cases of GCK PNDM are frequently reported from consanguineous pedigrees (19, 20, 21). However, a recent study from Saudi Arabia, where there is a high degree of consanguinity, did not identify a single case of GCK PNDM (22). Another study looked at the genetic aetiology of Arab and British cohorts tested in Exeter Peninsula Medical School Genetics Laboratory between years 2006 and 2011 and found GCK PNDM in only five out of 88 Arabic patients (5.7%) and one out of 77 British patients (1.3%) tested (23). To the best of our knowledge, there have not been any previous large case-series from Turkey reporting the genetic cause of NDM. From our results, it seems that GCK PNDM is the commonest cause of NDM in South-Eastern Anatolian region of Turkey and accounts for more than one-quarter of NDM cases. This is the highest proportion of GCK PNDM reported to date. Our patients with homozygous GCK mutations did not respond to the trial of sulphonylurea therapy. We also noticed higher requirement of insulin (1.5–2 U/kg per day) to maintain normoglycaemia in GCK PNDM as compared with other NDM patients.
The incidence of TNDM caused by mutation of ZFP57 was also high in our cohort. A recent study of a global cohort (n=163) of TNDM reported 12 patients (7.5%) with mutation in ZFP57 (24), of which ten had homozygous mutations from consanguineous pedigrees and only two had heterozygous mutations derived from unrelated parents (Mackay 2014 unpublished data). In the cohort presented here, two out of the three TNDM cases with chromosome 6q24 methylation abnormalities, had ZFP57 mutations. It is apparent that the prevalence of ZFP57 mutations correlated with that of consanguinity; this should be borne in mind when TNDM is suspected because the recurrence risk in affected pedigrees is 25%.
WRS (EIF2AK3 mutations) has been reported as the most common cause of PNDM in consanguineous families (8). Pancreatic agenesis/hypoplasia, acute episodes of liver dysfunction and skeletal dysplasia during follow-up are major clinical characteristics of WRS that have significant influence on prognosis (8, 16, 24). Episodes of liver dysfunction, ranging from mild elevation of serum transaminases to severe hepatic failure, can develop at any time from diagnosis (8, 16, 24). In our series with a high percentage of consanguineous families, WRS was the second most common cause after G CK PNDM. In our cohort, one patient with a normal pancreas on ultrasound imaging developed a self-limiting mild elevation of transaminases, alanine transaminase and aspartate transaminase at presentation. The second patient with normal pancreatic magnetic resonance imaging did not develop any liver dysfunction. However, a skeletal dysplasia was noticed at the age of 1.5 year, which worsened during follow-up. The third patient had pancreatic hypoplasia on ultrasound imaging, developed hepatic failure and skeletal dysplasia at the age of 1 year and died at the age of 3 years due to the second episode of hepatic failure.
In our series, three patients from consanguineous pedigrees with clinical manifestations of PNDM and pancreatic exocrine insufficiency and pancreatic hypoplasia/agenesis were found to have mutations in the recently identified distal enhancer region of PTF1A (13). Our results suggest that mutations in this regulatory region of PTF1A are a relatively common cause of PNDM in consanguineous families. No additional clinical features were present in these patients, consistent with the reported clinical phenotype of isolated pancreatic agenesis with distal enhancer PTF1A mutations (13).
Although mutations in the KCNJ11 and ABCC8 genes are the commonest cause of PNDM in European and Japanese populations, in our study they accounted for 12.5% cases only (9, 25, 26, 27). Similar findings were reported in the case-series from Saudi Arabia with 17 PNDM patients, in whom no KCNJ11 or ABCC8 mutations were identified (22). Both of our patients carrying the p.R201H KCNJ11 mutation could be switched to oral sulphonylurea with distinctly different doses of glibenclamide. It is well recognised that not all KCNJ11 mutations respond to oral sulphonylurea therapy (28). Although it seemed that one patient had DEND phenotype, on careful assessment developmental delay and epilepsy in this particular patient are likely to be due to neurological insult sustained during acute prolonged period of diabetic ketoacidosis coma. No cases of DEND have been reported in association with p.R201H KCNJ11 mutation in the literature.
Our results suggest that the estimated incidence of NDM in South-Eastern Anatolian region of Turkey was higher than those of western European countries with low rates of consanguineous marriage, but similar to the incidence reported from the Arabic countries with high rates of consanguineous marriage (9, 10, 11, 22, 29). The high rate of consanguinity in South-Eastern Anatolian region of Turkey and in our cohort (89%) was consistent with these results (12). However, since South-Eastern Anatolian region has a higher rate of consanguineous marriage compared with the other regions of Turkey, this estimated incidence may not reflect the true incidence of NDM for the whole Turkish population (12). To determine the true incidence rate of NDM in Turkey, multicentre and larger nationwide studies are required.
Conclusions
We present the largest cohort of NDM from a single paediatric endocrine centre. Mutations in GCK, EIF2A K3 and the distal enhancer region of PTF1A were the commonest causes of NDM in our cohort. With the utilisation of candidate gene sequencing and targeted next-generation sequencing, the underlying genetic cause could be established in 95% of our NDM patients who had undergone a mutation analysis. The high incidence and predominance of non-KATP channel mutations are likely to reflect the increased rate of consanguinity within our cohort. Special care to the rare genetic causes in the molecular genetic analysis of NDM is clearly required in patients from consanguineous pedigrees.
Footnotes
(H Demirbilek and V B Arya contributed equally to this work)
K Hussain is now at Genetics and Epigenetics in Health and Disease, Genetics and Genomic Medicine Programme, UCL Institute of Child Health, Great Ormond Street Hospital for Children, 30 Guilford Street, London WC1N 1EH, UK
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
The genetic testing was funded by the Wellcome Trust (Senior Investigator Award to Profs S Ellard and A T Hattersley), and by Diabetes UK (Project funding to Dr D J Mackay). H Demirbilek was funded by European Society for Paediatric Endocrinology (ESPE) and The Scientific and Technological Research Council of Turkey (TUBITAK) for his 1 year clinical fellowship at University College London (UCL), Institute of Child Health, Great Ormond Street Hospital for Children, NHS Trust, Department of Paediatric Endocrinology.
References
- Rubio-Cabezas O, Ellard S. Diabetes mellitus in neonates and infants: genetic heterogeneity, clinical approach to diagnosis, and therapeutic options. Hormone Research in Paediatrics. 2013;80:137–146. doi: 10.1159/000354219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak M, Shield J. Neonatal and very-early-onset diabetes mellitus. Seminars in Neonatology. 2004;9:59–65. doi: 10.1016/S1084-2756(03)00064-2. [DOI] [PubMed] [Google Scholar]
- Temple IK, Shield JP. 6q24 transient neonatal diabetes. Reviews in Endocrine & Metabolic Disorders. 2010;11:199–204. doi: 10.1007/s11154-010-9150-4. [DOI] [PubMed] [Google Scholar]
- Mackay DJ, Temple IK. Transient neonatal diabetes mellitus type 1. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics. 2010;154c:335–342. doi: 10.1002/ajmg.c.30272. [DOI] [PubMed] [Google Scholar]
- Mackay DJ, Callaway JL, Marks SM, White HE, Acerini CL, Boonen SE, Dayanikli P, Firth HV, Goodship JA, Haemers AP, et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nature Genetics. 2008;40:949–951. doi: 10.1038/ng.187. [DOI] [PubMed] [Google Scholar]
- Flanagan SE, Patch AM, Mackay DJ, Edghill EL, Gloyn AL, Robinson D, Shield JP, Temple K, Ellard S, Hattersley AT. Mutations in ATP-sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood. Diabetes. 2007;56:1930–1937. doi: 10.2337/db07-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellard S, Lango Allen H, De Franco E, Flanagan SE, Hysenaj G, Colclough K, Houghton JA, Shepherd M, Hattersley AT, Weedon MN, et al. Improved genetic testing for monogenic diabetes using targeted next-generation sequencing. Diabetologia. 2013;56:1958–1963. doi: 10.1007/s00125-013-2962-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Cabezas O, Patch AM, Minton JA, Flanagan SE, Edghill EL, Hussain K, Balafrej A, Deeb A, Buchanan CR, Jefferson IG, et al. Wolcott–Rallison syndrome is the most common genetic cause of permanent neonatal diabetes in consanguineous families. Journal of Clinical Endocrinology and Metabolism. 2009;94:4162–4170. doi: 10.1210/jc.2009-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanik J, Gasperikova D, Paskova M, Barak L, Javorkova J, Jancova E, Ciljakova M, Hlava P, Michalek J, Flanagan SE, et al. Prevalence of permanent neonatal diabetes in Slovakia and successful replacement of insulin with sulfonylurea therapy in KCNJ11 and ABCC8 mutation carriers. Journal of Clinical Endocrinology and Metabolism. 2007;92:1276–1282. doi: 10.1210/jc.2006-2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slingerland AS, Shields BM, Flanagan SE, Bruining GJ, Noordam K, Gach A, Mlynarski W, Malecki MT, Hattersley AT, Ellard S. Referral rates for diagnostic testing support an incidence of permanent neonatal diabetes in three European countries of at least 1 in 260,000 live births. Diabetologia. 2009;52:1683–1685. doi: 10.1007/s00125-009-1416-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iafusco D, Massa O, Pasquino B, Colombo C, Iughetti L, Bizzarri C, Mammi C, Lo Presti D, Suprani T, Schiaffini R, et al. Minimal incidence of neonatal/infancy onset diabetes in Italy is 1:90,000 live births. Acta Diabetologica. 2012;49:405–408. doi: 10.1007/s00592-011-0331-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuncbilek E, Ulusoy M. Consanguinity in Turkey in 1988. Turkis Journal of Population Studies. 1988;11:35–46. [PubMed] [Google Scholar]
- Weedon MN, Cebola I, Patch AM, Flanagan SE, De Franco E, Caswell R, Rodriguez-Segui SA, Shaw-Smith C, Cho CH, Lango Allen H, et al. Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nature Genetics. 2014;46:61–64. doi: 10.1038/ng.2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay DJ, Temple IK, Shield JP, Robinson DO. Bisulphite sequencing of the transient neonatal diabetes mellitus DMR facilitates a novel diagnostic test but reveals no methylation anomalies in patients of unknown aetiology. Human Genetics. 2005;116:255–261. doi: 10.1007/s00439-004-1236-1. [DOI] [PubMed] [Google Scholar]
- Massa O, Meschi F, Cuesta-Munoz A, Caumo A, Cerutti F, Toni S, Cherubini V, Guazzarotti L, Sulli N, Matschinsky FM, et al. High prevalence of glucokinase mutations in Italian children with MODY. Influence on glucose tolerance, first-phase insulin response, insulin sensitivity and BMI. Diabetologia. 2001;44:898–905. doi: 10.1007/s001250100530. [DOI] [PubMed] [Google Scholar]
- Senee V, Vattem KM, Delepine M, Rainbow LA, Haton C, Lecoq A, Shaw NJ, Robert JJ, Rooman R, Diatloff-Zito C, et al. Wolcott–Rallison syndrome: clinical, genetic, and functional study of EIF2AK3 mutations and suggestion of genetic heterogeneity. Diabetes. 2004;53:1876–1883. doi: 10.2337/diabetes.53.7.1876. [DOI] [PubMed] [Google Scholar]
- Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, Howard N, Srinivasan S, Silva JM, Molnes J, et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. New England Journal of Medicine. 2004;350:1838–1849. doi: 10.1056/NEJMoa032922. [DOI] [PubMed] [Google Scholar]
- Garin I, Edghill EL, Akerman I, Rubio-Cabezas O, Rica I, Locke JM, Maestro MA, Alshaikh A, Bundak R, del Castillo G, et al. Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. PNAS. 2010;107:3105–3110. doi: 10.1073/pnas.0910533107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloyn AL, Ellard S, Shield JP, Temple IK, Mackay DJ, Polak M, Barrett T, Hattersley AT. Complete glucokinase deficiency is not a common cause of permanent neonatal diabetes. Diabetologia. 2002;45:290. doi: 10.1007/s00125-001-0746-9. [DOI] [PubMed] [Google Scholar]
- Bennett K, James C, Mutair A, Al-Shaikh H, Sinani A, Hussain K. Four novel cases of permanent neonatal diabetes mellitus caused by homozygous mutations in the glucokinase gene. Pediatric Diabetes. 2011;12:192–196. doi: 10.1111/j.1399-5448.2010.00683.x. [DOI] [PubMed] [Google Scholar]
- Njolstad PR, Sovik O, Cuesta-Munoz A, Bjorkhaug L, Massa O, Barbetti F, Undlien DE, Shiota C, Magnuson MA, Molven A, et al. Neonatal diabetes mellitus due to complete glucokinase deficiency. New England Journal of Medicine. 2001;344:1588–1592. doi: 10.1056/NEJM200105243442104. [DOI] [PubMed] [Google Scholar]
- Habeb AM, Al-Magamsi MS, Eid IM, Ali MI, Hattersley AT, Hussain K, Ellard S. Incidence, genetics, and clinical phenotype of permanent neonatal diabetes mellitus in northwest Saudi Arabia. Pediatric Diabetes. 2012;13:499–505. doi: 10.1111/j.1399-5448.2011.00828.x. [DOI] [PubMed] [Google Scholar]
- Habeb AM, Flanagan SE, Deeb A, Al-Alwan I, Alawneh H, Balafrej AA, Mutair A, Hattersley AT, Hussain K, Ellard S. Permanent neonatal diabetes: different aetiology in Arabs compared to Europeans. Archives of Disease in Childhood. 2012;97:721–723. doi: 10.1136/archdischild-2012-301744. [DOI] [PubMed] [Google Scholar]
- Ozbek MN, Senee V, Aydemir S, Kotan LD, Mungan NO, Yuksel B, Julier C, Topaloglu AK. Wolcott–Rallison syndrome due to the same mutation (W522X) in EIF2AK3 in two unrelated families and review of the literature. Pediatric Diabetes. 2010;11:279–285. doi: 10.1111/j.1399-5448.2009.00591.x. [DOI] [PubMed] [Google Scholar]
- Flanagan SE, Edghill EL, Gloyn AL, Ellard S, Hattersley AT. Mutations in KCNJ11, which encodes Kir6.2, are a common cause of diabetes diagnosed in the first 6 months of life, with the phenotype determined by genotype. Diabetologia. 2006;49:1190–1197. doi: 10.1007/s00125-006-0246-z. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Makita Y, Mukai T, Matsuo K, Ueda O, Fujieda K. Molecular basis of neonatal diabetes in Japanese patients. Journal of Clinical Endocrinology and Metabolism. 2007;92:3979–3985. doi: 10.1210/jc.2007-0486. [DOI] [PubMed] [Google Scholar]
- Edghill EL, Flanagan SE, Patch AM, Boustred C, Parrish A, Shields B, Shepherd MH, Hussain K, Kapoor RR, Malecki M, et al. Insulin mutation screening in 1,044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes. 2008;57:1034–1042. doi: 10.2337/db07-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson ER, Flechtner I, Njolstad PR, Malecki MT, Flanagan SE, Larkin B, Ashcroft FM, Klimes I, Codner E, Iotova V, et al. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. New England Journal of Medicine. 2006;355:467–477. doi: 10.1056/NEJMoa061759. [DOI] [PubMed] [Google Scholar]
- Bappal B, Raghupathy P, de Silva V, Khusaiby SM. Permanent neonatal diabetes mellitus: clinical presentation and epidemiology in Oman. Archives of Disease in Childhood. Fetal and Neonatal Edition. 1999;80:F209–F212. doi: 10.1136/fn.80.3.F209. [DOI] [PMC free article] [PubMed] [Google Scholar]