

Abstract
Iodoarenes are important synthons for a wide range of organic transformations. Here we report a general strategy to prepare singly iodinated electron-rich aromatic compounds through the intermediacy of diaryliodonium salts. This process, which incorporates a phase separation that greatly simplifies product purification, is an attractive replacement for the Sandmeyer approach to iodoarenes that are otherwise difficult to access.
Keywords: aryl iodides, diaryliodonium salt, electrophilic substitution, regioselectivity, Sandmeyer reaction
The significant efforts devoted to iodoarene synthesis speak to the central roles these iodinated intermediates play in modern organic chemistry.[1] A large number of reagents have been used for electrophilic aromatic iodination,[2–6] including N-iodosuccinimide (NIS)[7–12] and related N–I compounds,[13–17] iodine monochloride (ICl),[18–20] bis(pyridine)-iodonium(I) tetrafluoroborate (Py2 IBF4),[21,22] dichloroiodates,[23,24] NaI in combination with various oxidants,[25,26] and silver[12,27–30] or mercury[31,32] salts in combination with I2. A multitude of reagents and diverse reaction conditions are required to address a central problem in iodoarene synthesis by electrophilic aromatic substitution (EAS): the electrophilicity of “I+” reagents must be matched with the overall “electron-richness” of the arene to obtain selective iodine substitution. Once on the aromatic ring, the iodine substituent is not strongly deactivating (Hammett σp=0.18, σm= 0.35, σ+ =0.14),[33] so trial and error must be used to find an iodine source which is sufficiently aggressive, but not so electrophilic that multiple electrophilic aromatic substitutions take place. This problem is summarized in Scheme 1.
Scheme 1.

Tuned electrophilicity in iodoarene synthesis.
One tried and true technique to prevent multiple iodine substitution is to generate an electron-poor aromatic intermediate which is deactivated toward further EAS, but which can be converted subsequently to an iodoarene. The classic Sandmeyer process,[34, 35] which involves nitration of an arene, reduction to the corresponding aniline, diazotization with nitrous acid, and substitution with KI, is the oldest example of the use of this approach.[36] Like the nitro group (σp=0.78, σm=0.71),[33] the aryliodonium substituent is sufficiently electron-withdrawing (σp=1.37, σm=1.35 for PhI(BF4))[37] to preclude further substitution. Moreover, it has been known for more than a century that diaryliodonium compounds[38–40] can be converted to aryl iodides efficiently using iodide salts under mild conditions.[41,42] Here we show that an operationally simple, two-step approach, which involves formation of a readily purified diaryliodonium salt intermediate and conversion of this intermediate to an iodoarene, is an effective and general process for the selective synthesis of a large variety of monoiodinated arenes. Most surprising is that this process occurs with greater reactivity and selectivity than those employing most common electrophilic iodination reagents.
The two-step iodination process is outlined in Scheme 2. The optimization of reaction conditions using aryliodination reagents 1-(diacetoxyiodo)-4-nitrobenzene 1a, and dimethyl 5-diacetoxyiodo)isophthalate 1b is summarized in Table 1. The diacetoxyiodoarene compounds 1a and 1b are readily synthesized by oxidation of the corresponding aryl iodides with m-chloroperoxybenzoic acid (m-CPBA) in acetic acid.[43] Reagents 1a and 1b are unreactive toward typical arenes, like toluene, at room temperature in CH3CN, but treatment of 1a or 1b with trimethylsilyltriflate (TMSOTf) in CH3CN increases their electrophilicity dramatically. This reagent combination gives an equilibrium distribution of IIII species (ArI(OAc)2, ArI(OAc)(OTf), and ArI(OTf)2); ligand exchange is indicated by a downfield shift in the aromatic signals in the 1H NMR spectrum upon addition of TMSOTf, and by generation of TMSOAc. (The relatively unstable aryliodonium bis(triflate) salts can also be isolated, and they show similar reactivity (see Supporting Information)). In survey experiments, 1.0 to 2.0 equivalents of 1 were combined with 1.0 to 4.0 equivalents of TMSOTf in CD3CN to generate the electrophile. This activated aryliodonium species was treated with one equivalent of toluene and the mixture was allowed to stand at room temperature until all of the toluene was converted to the diaryliodonium salt 2aa and 2ab (Table 1). Under the optimized conditions (toluene:1:TMSOTf=1:1.5:3.0), conversion of arene to the diaryliodonium salt was complete within one hour. After a second brief survey of reaction conditions, it was found that treatment with 3 equivalents of NaI and heating to 80°C for one hour was sufficient for complete conversion of the diaryliodonium salt to the corresponding aryl iodides (Table 1). The use of excess iodide serves two purposes: it drives the ion-exchange for the intermediate diaryliodonium triflate to the iodide, and it also quenches (by reduction) any excess ArIX2 reagent remaining upon completion of the reaction.
Scheme 2.

Regioselective iodination of toluene.
Table 1.
Optimization of conditions for aryliodination/iodination.a

| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| y | Reagent | X | Ratiob | t1[h] | Yield of 2aa or 2ab [%]c | Iodide source | Equivd | T [°C] | t2[h] | Yield of 3a [%]c |
| 1 | 1a | OTf | 1.0/1.5/1.5 | 1 | 66 | – | – | – | – | – |
| 2 | 1a | OTf | 1.0/1.5/1.5 | 8 | 98 | TMAI | 2.0 | 80 | 13 | 68 |
| 3 | 1a | OTf | 1.0/2.0/4.0 | 0.5 | 95 | – | – | – | – | – |
| 4 | 1a | OTf | 1.0/2.0/4.0 | 1 | >99 | TMAI | 2.0 | 120 | 1 | 46 |
| 5 | 1a | OTf | 1.0/1.5/3.0 | 1 | >99 | TMAI | 2.0 | 120 | 1 | 71 |
| 6 | 1a | OTf | 1.0/1.5/3.0 | 1 | >99 | TMAI | 2.0 | 120 | 4 | 95 |
| 7 | 1a | OTf | 1.0/1.5/3.0 | 1 | >99 | TMAI | 4.0 | 80 | 1 | > 99 |
| 8 | 1a | OTf | 1.0/1.5/3.0 | 1 | >99 | TMAI | 3.0 | 80 | 1 | > 99 |
| 9 | 1a | OTf | 1.0/1.5/3.0 | 1 | >99 | TBAI | 3.0 | 80 | 1 | > 99 |
| 10 | 1a | OTf | 1.0/1.5/3.0 | 1 | >99 | NaI | 3.0 | 80 | 1 | > 99 |
| 11 | 1b | OTf | 1.0/1.5/1.5 | 1 | 30 | – | – | – | – | – |
| 12 | 1b | OTf | 1.0/1.5/1.5 | 29 | 97 | – | – | – | – | – |
| 13 | 1b | OTf | 1.0/1.5/3.0 | 1 | >99 | NaI | 3.0 | 120 | 22 | > 99 |
| 14 | 1b | OMs | 1.0/1.5/3.0 | 1.5 | 0 | – | – | – | – | – |
| 15 | 1b | TFA | 1.0/1.5/3.0 | 1.5 | 0 | – | – | – | – | – |
Reaction conditions: 0.10 mmol scale, 1.5 mL of CD3CN, N2. For details of operations, see Supporting Information.
Toluene/1/TMSX.
Yields were determined by 1H NMR spectroscopy.
Equivalents of iodide source. TMSOTf=trimethylsilyl trifluoromethanesulfonate; TMAI=tetramethylammonium iodide; TBAI =tetrabutylammonium iodide; TMSOMs=trimethylsilyl methanesulfonate; TMSTFA= trimethylsilyl trifluoroacetate.
The exceptionally mild conditions under which ArI(OTf)2 is generated reaffirm that weakly ion-paired aryliodonium cations are quite potent, yet selective electrophiles: aryliodination of toluene occurs with quite good regioselectivity. The 90:10 para:ortho diaryliodonium salt product ratio is conserved upon conversion of these salts to 4-iodotoluene and 2-iodotoluene.[44] For comparison, the room-temperature reactivity and regioselectivity of several iodination reagents toward toluene under comparable conditions (0.1m) was examined (Table 2).
Table 2.
Comparison of toluene iodination.

| ||||
|---|---|---|---|---|
| Entry | Method | Conditions | Yield [%]a | Selectivity |
| 1 | A | NIS (1.1 equiv), In(OTf)3 (0.1 equiv), CH3CN, N2, 23°C, 3 days | 15 | 55:45 |
| 2 | B | Ag2SO4 (1.0 equiv), I2 (1.0 equiv), CH2Cl2, 23°C, 1 day | <5% | – |
| 3 | C | 0.83 equiv ICl in CH2Cl2, 23°C, N2, overnight | trace | – |
| 4 | D[45] | I2, potassium 4-iodylbenzenesulfonate, H2SO4 (5%), MeCN, 65°C | 98b | 84:16b |
Determined by 1H NMR spectroscopy using an internal standard.
The reported data (see ref. [45]).
A major advantage of this approach is that the ionic diaryliodonium salt has significantly different solubility than the reactants, permitting an intermediate purification, if desired. For example, precipitation of p-2ab (X=I) removes the last traces of the more soluble ortho-substituted diaryliodonium salt intermediate, allowing regiochemically pure 4-iodotoluene (p-3a) to be prepared easily and in good isolated yield (74%, Scheme 2).
This iodination process may be used with a variety of aryl IIII reagents. We chose reagents 1a and 1b for their disparate retention factors on normal phase silica gel TLC plates, since flash chromatography was used to isolate the iodoarene products. However, aryl IIII reagents that form aryl iodides having differential boiling points, phase behavior, or solubility could also be employed to streamline final purification of sensitive aryl iodides.
To examine the scope of this process, a number of arenes were subjected to aryliodination/iodination (Scheme 3). For most arenes, treatment with 1.5 equivalents of 1 and 3 equivalents of TMSOTf resulted in >99% conversion to the diaryliodonium intermediate within 1 h at room temperature. As expected, in no case was a multiple addition product observed. The resultant diaryliodonium salts were converted rapidly (< 1 h) to the corresponding aryl iodides by treatment with 3 equivalents of NaI in acetonitrile at 80°C. In cases where the intermediate diaryliodonium iodide (R=5-(dimethyl isophthalyl)) was sparingly soluble in acetonitrile, more aggressive heating was used.
Scheme 3.
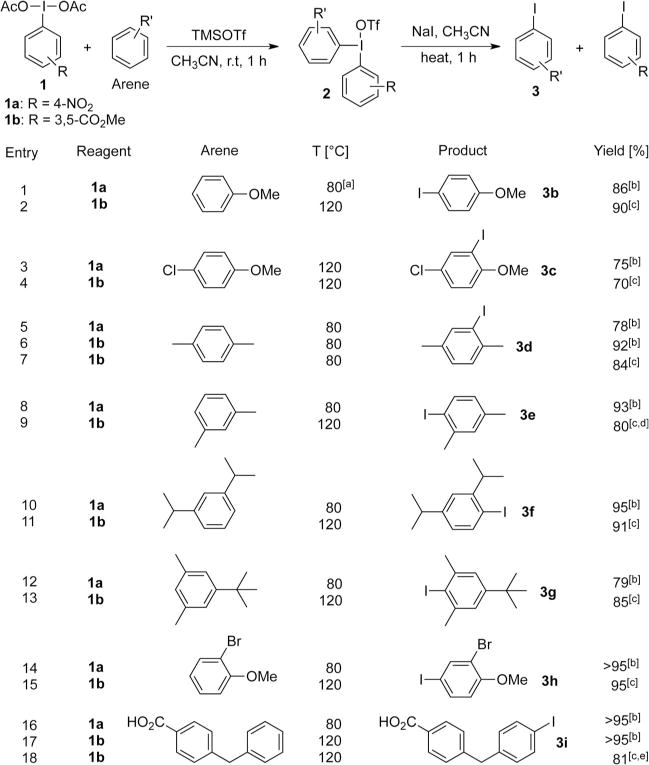
Synthetic scope of the reagents. Reaction conditions: 0.10 or 0.50 mmol scale, arene/1/TMSOTf/NaI=1:1.5:3.0:3.0, CD3CN or CH3CN, N2. For details of operations, see Supporting Information. [a] The iodination required 80°C for 2 h. [b] Yields of NMR scale were determined by 1H NMR spectroscopy using an internal integration standard. [c] Isolated yields. [d] 1.0 mmol scale. [e] 6.0 mmol scale.
These preliminary reactions, performed on simple arenes, indicated that this electrophilic iodination chemistry was quite promising for its combination of reactivity and selectivity, however, several limitations were noted. The presence of free N–H bonds and phenolic hydroxyl groups were incompatible with the aryliodination reaction, although simple protective group strategies provide a workaround to this problem. Importantly, the presence of hydrogen bonding groups (Scheme 3, entries 16–18) in the absence of a reducing functional group is tolerated. Highly reducing arenes (1,4-dimethylami-nobenzene, 1,4-dimethoxybenzene) participated in redox reactions rather than EAS, indicating that reduction of the aryliodonium salt was the principal side reaction that limits the utility of this method. For iodination of extremely electron-rich arenes, less oxidizing aryliodonium electrophiles are preferred (see below).
The attractiveness of the diaryliodonium salt synthetic approach is demonstrated by the synthesis of two compounds which are quite difficult to prepare by standard methods. Methyl 2-(3-iodo-4-isobutylphenyl)propanoate 3j has not been isolated previously, although (3-bromo-4-isobutylphenyl)propanoate has been obtained using a Sandmeyer approach.[46] Ibuprofen methyl ester (Scheme 4) is difficult to iodinate regioselectively with conventional electrophilic reagents, and the resulting mixtures of the 3-iodo and 2-iodo derivatives 2j present a separations challenge (see Supporting Information). Treatment with 1b under standard conditions yields a 90:10 mixture of the two regioisomeric diaryliodonium salts 2j, which, upon crystallization of the mixture provided one pure regioisomer 3-iodo 2j in good yield. The conversion of the regiochemically pure diaryliodonium salt to the iodoarene was followed by silica gel chromatography (EtOAc/hexane, 1:40) to remove dimethyl 5-iodoisophthalate to provide methyl 2-(3-iodo-4-isobutylphenyl)propanoate 3j in good overall yield (65%). It bears emphasis that this procedure is operationally simpler, and uses milder reagents and conditions than those required to obtain the same compound using approaches that employ a diazonium salt intermediate.
Scheme 4.

Regioselective iodination of ibuprofen methyl ester (top) and BPAD (bottom).
Perhaps the most impressive example of selective monoiodination using the aryliodonation approach is seen with bisphenol A dimethyl ether (BPAD, Scheme 4). As is expected from the symmetrical structure of BPAD, typical electrophilic iodinating reagents such as NIS/In(OTf)3, ICl, or I2/AgSO4 gave inseparable mixtures of BPAD and singly and multiply iodinated derivatives, even when a significant excess of BPAD (3 equiv) was used (see Supporting Information). In contrast, when a threefold excess of BPAD was treated with the mild and selective ArIIII reagent 1-(diacetoxyiodo)-4-methoxybenzene 1c, and trimethylsilyl trifluoroacetate (TMSTFA), a single diaryliodonium salt product 2k was obtained. The diaryliodonium salt intermediate 2k was readily separated from excess BPAD by precipitation of the salt from ethyl acetate with methyl tert-butyl ether (MTBE). The isolated diaryliodonium salt 2k was converted smoothly to the iodinated derivative in good yield. It bears emphasis that a single electrophilic aromatic substitution product is not expected from a statistical analysis that assumes that each ring of BPAD is equally susceptible to EAS. For example, treatment of BPAD with NIS gave complex mixtures of products when the reaction was run with the same stoichiometry. The addition of an ArIIII species to one ring of BPAD deactivates the second ring toward electrophilic aromatic substitution, even though the two rings are separated by a quaternary sp3-carbon atom. An analysis of the 1H NMR spectrum indicates that the aromatic signals arising from the unsubstituted ring in the diaryliodonium salt intermediate 2k move upfield, and the methyl resonance arising from the methoxy group of the unsubstituted ring moves downfield upon formation of 2k from BPAD. These chemical shift changes are in the same direction, but of larger magnitude than those observed in the direct iodination of BPAD, indicating that the increased selectivity of the aryliodination reaction most likely results from a direct, through-bond electronic effect rather than a secondary, through-space, intramolecular π–π interaction involving the electron-rich and electron-poor rings in 2k. Given the expected attenuation of such an electronic effect through the saturated bridge, the selectivity for the singly substituted product is surprising and impressive.
The aryliodoination approach to the preparation of aryl iodides retains many of the advantages of the Sandmeyer reaction, but it is operationally simpler. In cases where regioisomers are obtained, formation of polar, diaryliodonium salt intermediates that are readily purified simplifies the isolation of a single regioisomer. The solubility characteristics of the diaryliodonium salt intermediates also make it possible to obtain good yields of iodinated arenes even when the reaction is carried out at low conversion. The reactivity of the aryliodination reagents can be tuned to match a variety of electron-rich arenes, and the polarity of the aryliodoination reagents (1a, 1b, or 1c) can be varied to simplify product isolation by silica gel chromatography. This method allows one to prepare iodoarenes that normally cannot be prepared regioselectively by direct electrophilic aromatic substitution, and it also provides a means to isolate iodoarenes that would be extremely challenging to separate from their unsubstituted parent compounds. Finally, these reagents are distinguished by a unique combination of reactivity and selectivity, as they are simultaneously more aggressive, regioselective, and chemoselective than standard reagents for electrophilic aromatic iodination.
Experimental Section
Typical procedure for the synthesis of 4-iodotoluene
Under an atmosphere of dry N2, TMSOTf (1.50 mmol, 333 mg, 3.0 equiv) was added slowly to a stirred solution of dimethyl 5-(diacetoxyiodo)isophthalate 1b (0.75 mmol, 329 mg, 1.5 equiv) in dry CH3CN (1.0 mL). A solution of distilled toluene (0.50 mmol, 46 mg, 1.0 equiv) in dry CH3CN (1.0 mL) was added dropwise and the mixture was allowed to stand at 23°C for 1.0 h. The completion of the reaction was monitored by TLC. Approximately 10 min after the addition of solid sodium iodide (1.50 mmol, 225 mg, 3.0 equiv) with stirring, a yellow solid precipitated from the solution. The precipitated solid was collected by filtration, washed with acetonitrile and dried in vacuo to provide diaryliodonium iodide as a yellow solid (220 mg, 82%), m.p. 143–145°C. 1H NMR (400 MHz, [D6]DMSO): δ =8.14 (d, J=1.2 Hz, 2H), 7.73 (t, J=1.6 Hz, 1H), 7.39 (d, J=8.4 Hz, 2H), 6.50 (d, J=8.4 Hz, 2H), 3.09 (s, 3H), 3.08 (s, 3H), 1.51 ppm (s, 3H); 13C NMR (100 MHz, DMSO): δ =162.7, 141.3, 138.1, 134.1, 131.3, 131.1, 130.7, 117.8, 113.7, 51.9, 19.7 ppm; HRMS (ESI) calcd for C15H17O4INa [M–I+Na]+ 411.0069; found: 411.0078. The diaryliodonium salt (220 mg) was suspended in CH3CN (2.0 mL) and heated 120°C for 1 h. The solvent was evaporated in vacuo and the residue was purified by silica gel chromatography (100% hexane) to afford the product p-3a as a yellowish liquid (81 mg, 74% yield). 1H NMR (400 MHz, CD3CN): δ =7.58 (d, J=8.0 Hz, 2H), 6.98 (d, J=7.6 Hz, 2H), 2.27 ppm (s, 3H); 13C NMR (100 MHz, CD3CN): δ =136.3, 135.6, 129.8, 88.1, 18.6 ppm. 1H and 13C NMR data matched those previously reported.[47] In addition, the starting material 5-iododimethyl isophthalate (180 mg, 75%) for 1b was recovered from this two-step process.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health under award number R01 EB015536. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Bao Hu also thanks the National Natural Science Foundation of China (No. 21202148) for support.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201500151.
References
- 1.Waldvogel SR, Wehming KM. In: Science of Synthesis. Ramsden CA, editor. Vol. 31. Thieme; Stuttgart: 2007. p. 235. [Google Scholar]
- 2.Merkushev EB. Synthesis. 1988:923. [Google Scholar]
- 3.Kraszkiewicz L, Sosnowski M, Skulski L. Tetrahedron. 2004;60:9113. [Google Scholar]
- 4.Hanson JR. J Chem Res. 2006:277–280. [Google Scholar]
- 5.Stavber S, Jereb M, Zupan M. Synthesis. 2008:1487. [Google Scholar]
- 6.Kandepi VVKM, Narender N. Synthesis. 2012;44:15–26. [Google Scholar]
- 7.CarreÇo MC, Ruano JLG, Sanz G, Toledo MA, Urbano A. Tetrahedron Lett. 1996;37:4081. [Google Scholar]
- 8.Castanet AS, Colobert F, Broutin PE. Tetrahedron Lett. 2002;43:5047. [Google Scholar]
- 9.Olah GA, Wang Q, Sandford G, Prakash GKS. J Org Chem. 1993;58:3194. [Google Scholar]
- 10.Prakash GKS, Mathew T, Hoole D, Esteves PM, Wang Q, Rasul G, Olah GA. J Am Chem Soc. 2004;126:15770. doi: 10.1021/ja0465247. [DOI] [PubMed] [Google Scholar]
- 11.Shen H, Vollhardt KPC. Synlett. 2012:208–214. [Google Scholar]
- 12.Zhou CY, Li J, Peddibhotla S, Romo D. Org Lett. 2010;12:2104. doi: 10.1021/ol100587j. [DOI] [PubMed] [Google Scholar]
- 13.Orazi OO, Corral RA, Bertorello HE. J Org Chem. 1965;30:1101. doi: 10.1021/jo01015a036. [DOI] [PubMed] [Google Scholar]
- 14.Dolenc D. Synlett. 2000:544–546. [Google Scholar]
- 15.Ghorbani-Vaghei R. Tetrahedron Lett. 2003;44:7529. [Google Scholar]
- 16.Bailey L, Handy ST. Tetrahedron Lett. 2011;52:2413. [Google Scholar]
- 17.da Ribeiro RS, Esteves PM, de Mattos MCS. Synthesis. 2011:739–744. [Google Scholar]
- 18.Lambourne LJ, Robertson PWJ. J Chem Soc. 1947:1167. [Google Scholar]
- 19.Berliner E. J Am Chem Soc. 1956;78:3632. [Google Scholar]
- 20.Hubig SM, Jung W, Kochi JK. J Org Chem. 1994;59:6233. [Google Scholar]
- 21.Barluenga J, González JM, García-Martín MA, Campos PJ, Asensio G. J Org Chem. 1993;58:2058. [Google Scholar]
- 22.Barluenga J. Pure Appl Chem. 1999;71:431–436. [Google Scholar]
- 23.Kajigaeshi S, Kakinami T, Yamasaki H, Fujisaki S, Kondo M, Okamoto T. Chem Lett. 1987;16:2109–2112. [Google Scholar]
- 24.Kajigaeshi S, Kakinami T, Moriwaki M, Watanabe M, Fujisaki S, Okamoto T. Chem Lett. 1988;17:795–798. [Google Scholar]
- 25.Kometani T, Watt DS, Ji T, Fitz T. J Org Chem. 1985;50:5384. [Google Scholar]
- 26.Edgar KJ, Falling SN. J Org Chem. 1990;55:5287. [Google Scholar]
- 27.Sy WW, Lodge BA. Tetrahedron Lett. 1989;30:3769. [Google Scholar]
- 28.Sy WW, Lodge BA, By AW. Synth Commun. 1990;20:877–880. [Google Scholar]
- 29.Sy WW. Synth Commun. 1992;22:3215. [Google Scholar]
- 30.Al-Zoubi RM, Hall DG. Org Lett. 2010;12:2480. doi: 10.1021/ol100537x. [DOI] [PubMed] [Google Scholar]
- 31.Bachki A, Foubelo F, Yus M. Tetrahedron. 1994;50:5139. [Google Scholar]
- 32.Orito K, Hatakeyama T, Takeo M, Suginome H. Synthesis. 1995:1273–1277. [Google Scholar]
- 33.Hansch C, Leo A, Taft RW. Chem Rev. 1991;91:165. [Google Scholar]
- 34.Sandmeyer T. Ber Dtsch Chem Ges. 1884;17:2650. [Google Scholar]
- 35.Sandmeyer T. Ber Dtsch Chem Ges. 1884;17:1633. [Google Scholar]
- 36.Griess P. Justus Liebigs Ann Chem. 1866;137:39. [Google Scholar]
- 37.Ochiai M. Top Curr Chem. 2003;224:5. [Google Scholar]
- 38.Grushin VV. Chem Soc Rev. 2000;29:315. [Google Scholar]
- 39.Yusubov MS, Maskaev AV, Zhdankin VV. ARKIVOC. 2011;2011:370–409. [Google Scholar]
- 40.Merritt EA, Olofsson B. Angew Chem Int Ed. 2009;48:9052. doi: 10.1002/anie.200904689. Angew. Chem.2009, 121, 9214. [DOI] [PubMed] [Google Scholar]
- 41.Hartmann C, Meyer V. Ber Dtsch Chem Ges. 1894;27:502. [Google Scholar]
- 42.Fletcher C, Hinshelwood JMCN. J Chem Soc. 1935:596. [Google Scholar]
- 43.Iinuma M, Moriyama K, Togo H. Synlett. 2012;23:2663–2666. [Google Scholar]
- 44.Bielawski M, Olofsson B. Chem Commun. 2007:2521. doi: 10.1039/b701864a. [DOI] [PubMed] [Google Scholar]
- 45.Yusubov MS, Yusubova RY, Nemykin VN, Maskaev AV, Geraskina MR, Kirschning A, Zhdankin VV. Eur J Org Chem. 2012:5935. [Google Scholar]
- 46.Gupta K, Kaub CJ, Carey KN, Casillas EG, Selinsky BS, Loll PJ. Bioorg Med Chem Lett. 2004;14:667. doi: 10.1016/j.bmcl.2003.11.034. [DOI] [PubMed] [Google Scholar]
- 47.Wilson SR, Jacob LA. J Org Chem. 1986;51:4833. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.