

Abstract
Drugs that can protect against organ damage are urgently needed, especially for diseases such as sepsis and brain stroke. We have discovered that terazosin (TZ), a widely marketed alpha1-adrenergic receptor agonist, alleviated organ damage and improved survival in rodent models of stroke and sepsis. Through combined studies of enzymology and X-ray crystallography, we have discovered that TZ binds to a novel target, phosphoglycerate kinase 1 (Pgk1) and activates its enzymatic activity, probably through 1,3-diamino-6,7-dimethoxyisoquinoline's ability to promote ATP release from Pgk1. Mechanistically, the ATP generated from Pgk1 may enhance the chaperone activity of Hsp90, an ATPase known to associate with Pgk1. Upon activation, Hsp90 promotes multi-stress resistance. Our studies have demonstrated that TZ has a novel protein target, Pgk1, and has revealed its corresponding biological effect. As a clinical drug, TZ may be quickly translated into treatment of devastating diseases including stroke and sepsis.
Introduction
Symptoms of a wide variety of human diseases, such as neurodegenerative diseases, autoimmune diseases, heart failure, stroke and sepsis, are caused by the overwhelming activation of cell death and tissue damage. Sepsis, for example, is initiated by bacterial infection, which triggers massive apoptosis in immune systems and the failure of multiple organ functions 1. However, the clinical treatment of sepsis has extensively relied on antimicrobial treatment and supportive care, which have limited efficacy due to complications such as drug resistance, antibiotic toxicity, endotoxins, cell death and organ dysfunction 2, 3. Similarly, brain stroke, a age-related vascular disease, is another leading cause of death worldwide with massive cell death that occurs in the brain. Currently, no effective neuroprotective therapy has been developed beyond thrombolytic therapy to restore clotted blood vessels 4.
Apoptosis, which is mediated by caspases, was discovered in the 1990s 5. However, over the past 20 years no caspase inhibitor has passed clinical trials, and the discovery of novel inhibitors remains a significant challenge in biomedical research 6. In order to manipulate apoptosis, efforts have been focused on the endogenous regulatory factors, including the inhibitor of apoptosis (IAP) family proteins, Bcl-2 family proteins and chaperone proteins 7, 8. For example, the IAP family proteins are heavily pursued drug targets for cancer therapeutics 9. Hsp90, a taxonomically highly conserved chaperone, has been reported to play important roles in the regulation of cellular homeostasis and stress response 10. It has been reported that Hsp90 has many biologically important client proteins, especially kinases and hormone receptors 11, 12. Hsp90 is highly dynamic and its conformational change is regulated by its ATPase activity, which stabilizes its interactions with client proteins 10. Considering its prominent anti-apoptotic and protective effects in cancer cells, many Hsp90 inhibitors have been developed as potential anti-cancer drugs to attenuate cancer cell survival. However, no Hsp90 activator that may protect cells from stresses and cell death has been reported.
Here, we have devised a screen to search for novel anti-apoptotic drugs from a pool of bioactive small molecules 13. Once a candidate compound is identified, investigations of its targets and mode of action may enable its rapid translation to clinical applications. We found that terazosin could rescue rodent models of sepsis and stoke in vivo. Strikingly, it inhibited apoptosis in macrophages likely through activation of Pgk1 and Hsp90.
Results
TZ rescued reaper-mediated apoptosis in Drosophila
We designed our search for apoptosis inhibitors in Drosophila on the rationale that its apoptotic pathway is conserved in mammals 14 and that flies are economical for compound screening. To induce apoptosis, the expression of UAS-reaper (rpr) was driven by a heat-activated promoter, HS-Gal4 (their progeny flies were simplified as HS>rpr). Consistent with the previous report that ectopically expressed rpr can induce widespread apoptosis and organ death in Drosophila 15, we observed that approximately 75% of the progenies were dead after 24 hours upon heat shock at 37 °C for 45 minutes (Supplementary Results, Supplementary Fig. 1a and b).
To screen for apoptosis inhibitors, we first assessed global transcriptional alteration during apoptosis using Affymetrix microarrays. Comparing the HS>rpr flies to the genetic background matched control HS x y w67c23 flies (progenies of HS-Gal4 crossed with y w67c23), 2169 genes showed more than 50% altered expression levels (the genes identified from the microarrays are listed in Supplementary Data Set 1). Then, these genes were converted to homologous human genes (the apoptotic signature genes after conversion into human homologs are listed in Supplementary Data Set 2) and entered into the Cmap for compound matching 13 (The matched drugs are listed in Supplementary Data Set 3). Twenty-five compounds were selected (Supplementary Data Set 4) and fed the HS>rpr flies with concentrations indicated (Supplementary Data Set 5). From this screen, we identified terazosin (TZ), an α1-adrenergic receptor antagonist and a widely marketed anti-hypertension drug, as the only compound that significantly improved survival of the HS>rpr flies (Supplementary Fig. 1b).
TZ inhibited apoptosis in cultured mammalian cells
To examine whether TZ inhibits apoptosis in cultured mammalian cells, RAW 264.7 cells were pre-treated for 18-24 hours before induction of apoptosis by lipopolysaccharide (LPS) and interferon-γ (IFN-γ)16. After treatment for 18-24 hours, apoptosis was determined by Annexin V staining. The result showed that TZ suppressed apoptosis in these cells (Fig. 1a and Supplementary Fig. 2a), and further verified by the LDH assay (Supplementary Fig. 2b). Furthermore, to test whether TZ blocks apoptosis induced by a different stressor, hydrogen peroxide (H2O2) was applied, which induces caspase-mediated apoptosis, and this type of cell death can be blocked by zVad, a pan-caspase inhibitor (Supplementary Fig. 2c). Again, TZ exhibited a protective effect against H2O2-induced apoptosis by an LDH release assay (Fig. 1b), active caspase-3 Western blot (Fig. 1c and Supplementary Fig. 2d) and caspase activity assay (Fig. 1d). These results demonstrated that TZ inhibited caspase-mediated apoptosis triggered by diverse inducers in macrophages.
Figure 1. TZ blocked apoptosis in cultured mammalian cells.
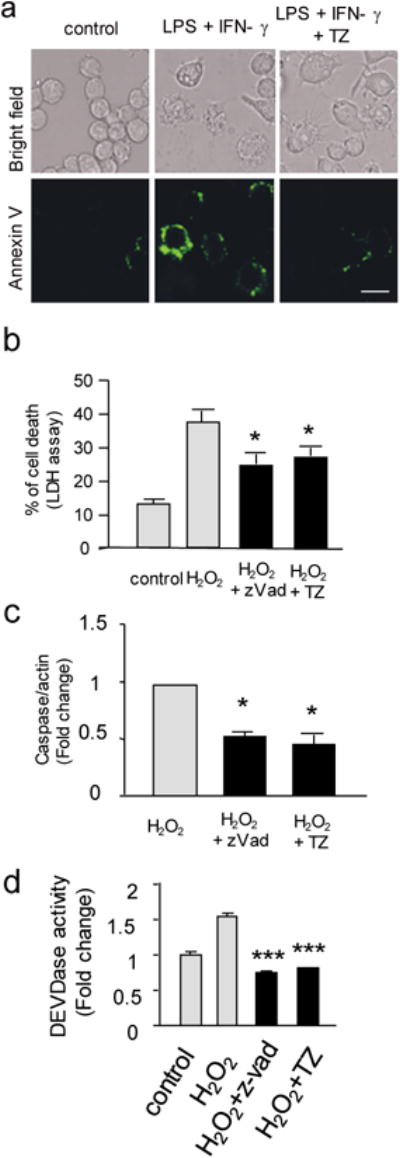
(a) Examples of bright field and fluorescent images of the RAW 264.7 cells stained with Annexin V after LPS and IFN-γ. The bar represents 25 μm. 3 independent experiments were performed. (b) The LDH assay of cell death induced by hydrogen peroxide. Th e R A W 264.7 cells treated with 0.1 % DMSO (control), H2O2 (1 mM), H2O2 plus Z-vad (100 μM) or H2O2 plus TZ (10 μM) in 0.1 % DMSO. The percentage of cell death (%) = LDHmedium/(LDHmedium + LDHcell) × 100%; Trial n = 3. All data are presented as the mean + standard error, unless otherwise indicated. The P values of a two-tailed t-test for the comparison of the two data sets comparison, the one-way ANOVA with post-hoc multiple comparison Sidak for three or more data sets comparisons, and the Kaplan-Meier survival analysis with a log rank algorithm are represented as *** for P<0.001, ** for P<0.01, and * for P<0.05, respectively, throughout all figures. (c) The statistical result of the Western blot to detect the active form of caspase 3. The same condition as (b). The relative ratio of caspase 3/β-actin from the H2O2 treatment was set as 1, and the relative ratio of other treatments were shown. Trial n = 3. (d) The effect of TZ on the activity of caspase 3/7 measured by a DEVDase activity assay. The data were normalized to the control conditions (without treatment). Trial n = 3.
TZ was an activator of phosphoglycerate kinase 1 (Pgk1)
To test whether the effect of TZ on apoptosis was mediated through the α1-adrenergic receptor, we modified TZ by replacing a nitrogen positioned on the quinazoline ring with a carbon (Fig. 2a, TZ-md), which is known to reduce its binding to the α1- adrenoceptor by approximately 1000-fold 17. Indeed, TZ-md could not block intracellular calcium elevation induced by metaraninol, an α1 receptor agonist (Supplementary Fig. 3a). However, TZ-md still displayed an anti-apoptotic effect in Raw 264.7 cells (Supplementary Fig. 3b). This result suggested that TZ might exert an anti-apoptotic effect via an unknown target.
Figure 2. Pgk1 as the target of TZ.
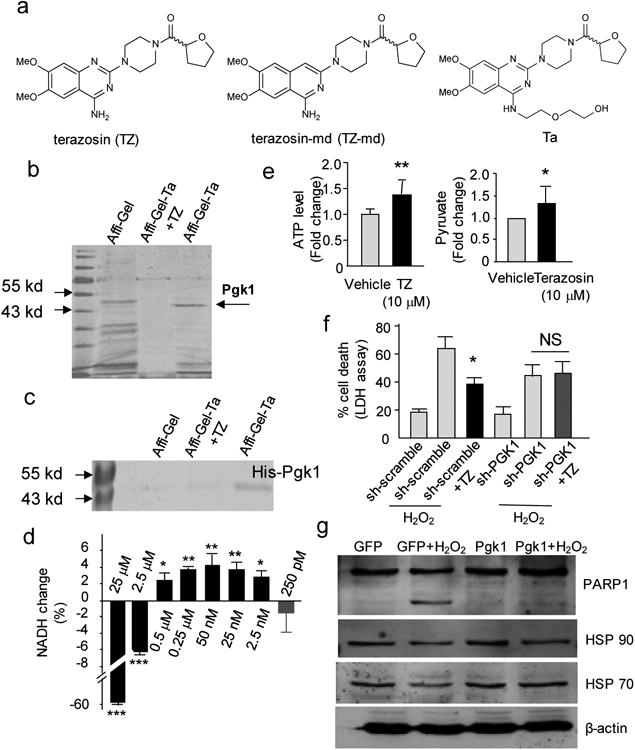
(a) The chemical structures of TZ, modified TZ (TZ-md) and the bait compound (TZ-TA). (b) The in vitro pull-down assay to identify the TZ target. Naked Affi-Gel beads andAffi-Gel-TZ-TA beads with saturated soluble competitor of TZ were used as controls. The full gel was shown on Supplementary Fig. 15. (c) The in vitro pull down of the recombinant mouse His-tagged Pgk1 protein by Affi-Gel-TZ-TA. The full gel was shown on Supplementary Fig. 15. (d) TZ effect on purified mouse Pgk1 enzymatic activity. The dosage TZ was indicated. Trial n = 4. (e) The effect of TZ on the ATP and pyruvate production in the cell lysate of RAW 264.7 cells. For the ATP level, the transient 1st minute was measured. Trial n = 5. For the pyruvate production, the steady state (the 10th minute) was measured. Trial n = 3. (f) The effect of stably expressed shRNA for Pgk1 (scrambled shRNA as the control) on cell death mediated by H2O2. The percentage of cell death was determined by the LDH assay; Trial n = 5. (g) The effect of stably expressing Pgk1 or EGFP (control) on the PARP1 cleavage upon H2O2 treatment. The full gel was shown on Supplementary Fig. 15. Trial n = 3.
To search for potential new targets, we synthesized a TZ derivative, “TZ-TA”, by conjugating a chemical handle at the 4-amino position of the quinazoline motif (Fig. 2a). “TZ-TA” had preserved the anti-apoptotic property of TZ in RAW 264.7 cells (Supplementary Fig. 3b). Then, “TZ-TA” was immobilized onto an Affi-Gel support to immobilize any binding proteins (see Supplementary Note 1). Comparing the pull-down results from the RAW 264.7 cell lysate and the lysate containing saturated TZ as a soluble competitor, we observed a distinct protein band specifically bound to the Ta-beads, with a molecular weight of ∼46 kDa (Fig. 2b). This band was extracted and identified by mass spectrometry as phosphoglycerate kinase 1 (Pgk1) (Supplementary Fig. 3c and Supplementary Note 2). Moreover, the purified His-tagged mouse Pgk1 could be pulled down by the Affi-Gel-TZ-TA beads (Fig. 2c), suggesting that TZ directly bound Pgk1.
As one of the core enzymes in glycolysis, Pgk1 had been reported to act as a suppressor of apoptosis in yeast and improve the viability in the human SH-SY5Y cell line upon amyloid stimulus 18, 19. To test the effect of TZ on Pgk1 activity, we performed an in vitro assay by providing the purified Pgk1 with substrates (ADP and 1,3-Bisphosphoglycerate) and measured the forward reaction that would accumulate NADH (see Online Method for more information). Intriguingly, at higher dosages (2.5 and 25 μM), TZ inhibited Pgk1; it activated Pgk1 at lower dosages (0.5 μM to 2.5 nM) (Fig. 2d). To further validate the TZ effect on Pgk1, we measured ATP (a product of Pgk1) and pyruvate (a downstream product of glycolysis) levels in the cell lysate. The ATP level was transiently elevated by nearly 40% in the first minute of the reaction, and the pyruvate level was stably increased approximately 30% (Fig. 2e). Together, these results suggested that TZ activated Pgk1 at low concentrations.
Furthermore, we determined the effective dosages of TZ to inhibit H2O2-induced apoptosis in macrophages. The results showed that TZ suppressed apoptosis with concentrations as low as 0.1 μM (Supplementary Fig. 4a).
To further explore the functional role of Pgk1 in modulating apoptosis, we knocked down Pgk1 in RAW 264.7 cells by stably expressing shRNA of Pgk1. The knockdown efficiency was shown (Supplementary Fig. 4b). Although knocking down Pgk1 did not affect cell survival, the protective effect of TZ against H2O2-induced apoptosis was abolished by the Pgk1 shRNA, determined by LDH release assays (Fig. 2f), and caspase 3 activation and PARP-1 cleavage by Western blots (Supplementary Fig. 4c). These results indicated that the anti-apoptosis activity of TZ required Pgk1. Consistently, stable expression of Pgk1 in the RAW 264.7 cells by lentiviral transfection reduced PARP-1 cleavage and LDH release after H2O2 treatment (Fig. 2g, and Supplementary Fig. 4d and e). Collectively, these results indicated that Pgk1 was required for the protective effect of TZ.
Crystallization of Pgk1 with TZ
To investigate how TZ interacts with Pgk1, we crystallized mouse and human Pgk1 (Supplementary Fig. 5 showed protein purification of Pgk1). Pgk1 consists of an N-terminal and C-terminal domains with roughly equal size and a similar Rossman fold. With various ligands hPgk1 has been crystallized in open 20 and closed 21 forms, respectively. The enzyme spends most of its time in a fully open and resting conformation for substrate-binding and product release with a short period of time in a closed form which is utilized for quick catalysis 22. Although mPgk2 structure has been resolved 23, there has been no report on mPgk1 structure. First, we have grown mPgk1 crystals in the presence of 3-phosphoglycerate (3PG), a substrate of Pgk1. Next, we obtained the ternary co-crystallization complexes for both the mouse and the human Pgk1-3PG-TZ, with the addition of saturated amount of TZ (10 mM). The structures were determined using molecular replacement to 2.1Å. Consistent with the 98% sequence identity between Pgk1 of these two species (Supplementary Fig. 6a), the structure of hPGK1-3PG-TZ could be superposed onto mPgk1-3PG-TZ well, indicating an identical TZ-binding mode to hPGK1 and mPGK1 (Fig. 3a and Supplementary Fig. S6b-g). The only difference between these two structures occurred within a small disordered loop region between residues Thr378 and Asp387 in mPGK1 located near the C-terminus, but structurally located at the junction between the N-terminal and C-terminal domains (Supplementary Fig. 6c).
Figure 3. Crystal structure of Pgk1 and TZ binding.
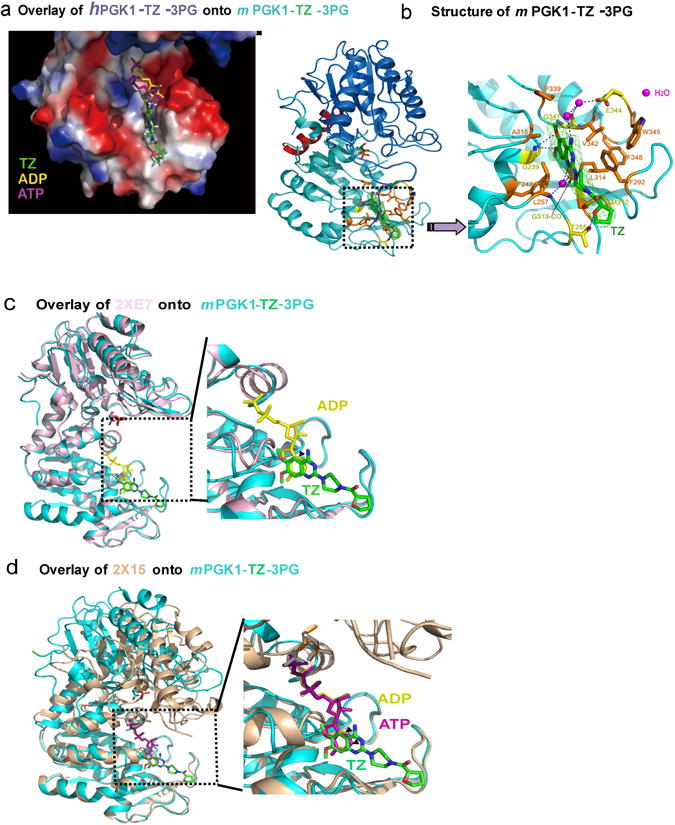
(a) The relative positions of TZ, ADP and ATP were shown. The red, white, and blue colors indicated acidic, neutral, and alkalic distributions, respectively. (b) The structure of mPgk1-3PG-TZ. The enlarged view showed a hydrophobic cleft at the C-terminal domain of Pgk1. The dashed lines indicated predicted hydrogen bonds of TZ with the amino acids from Pgk1. Interactions of potential water molecules with TZ were also shown (in pink). (c) Overlaid of the mPgk1-3PG-TZ structure onto the ADP-bound open form structure of human Pgk1. “2XE7” indicated the open form of hPgk1. (d) Overlaid of the mPgk1-3PG-TZ structure onto the ATP-bound closed form structure of human Pgk1. “2X15” indicated the closed form of hPgk1.
From the structure of mPgk1-3PG-TZ, we observed that the three hydrophobic rings of the TZ molecule were surrounded by many hydrophobic residues of the Pgk1 molecule, including the aromatic side chains of Phe242, Phe292, Phe348, Trp345 and also Leu257, Met312 and Leu314 (Fig. 3b). Strikingly, the oxygen and nitrogen atoms of TZ formed a network of hydrogen bonds directly or through water molecules to Pgk1, indicating that the drug/enzyme recognition was quite specific (Fig. 3b). We displayed the electrostatic surface of the mPgk1-3PG-TZ structure to demonstrate the relative position of ADP, ATP and TZ (Fig. 3b). We observed that the phosphate groups of ADP and ATP were oriented towards a positively charged well; while the TZ molecule extended its hydrophobic tail further away on the opposite direction toward the end of the domain (Fig. 3b). Further, we overlaid the mPgk1-3PG-TZ structure onto the ADP-bound open form of the hPgk1 (Fig. 3c) and the ATP-bound closed form of the hPgk1(Fig. 3d). These results indicated that the 6-membered benzene ring of TZ's quinazoline core sited at the position of the 6-membered pyrimidine ring of purine of ADP and ATP. Together, our results demonstrated that the TZ-binding site overlaps with the ADP and ATP binding site to the enzyme, and they were all inserted deeply into the same hydrophobic cleft at the C-terminal domain of Pgk1 (Fig. 3b).
The binding mode revealed by the Pgk1-3PG-TZ structures raises an intriguing paradox: in vitro enzymatic activity assays (Fig. 2d) indicated that TZ was stimulatory for Pgk1 at lower concentrations; however, as TZ occupies the binding site of ADP/ATP, it should be a competitive inhibitor instead of an activator. We rationalize that, based on the chemical equilibrium principle, the effective TZ dosage, for either inhibition or activation, should depend on the relative affinity and the concentration of TZ, ADP and ATP for Pgk1. The Pgk1-binding affinity (Kd) of ADP and ATP is 2.9×10-5 M and 3.3×10-4 M, respectively 24. According to our measurement by ITC (Isothermal Titration Calorimetry), TZ and Ta bind to Pgk1 with the affinity (Kd) of 2.78×10-6 M and 2.5×10-6 M, respectively (Supplementary Fig. 7a and b), which is approximately 10 and 100 stronger than ADP and ATP.
The anti-apoptotic effect of TZ might depend on Hsp90
Although up-regulation of glycolytic pathway has been reported to benefit cell survival 25, the mechanism for the anti-apoptotic activity of activated Pgk1 is unclear. We have observed that the ATP level increased upon TZ treatment in cell lysate (Fig. 2e). However, the ATP level declined to 90% of the baseline level after 10 minutes incubation with TZ (Fig. 4a). This suggests that ATP consumption is increased, likely through activation of an ATPase. Interestingly, Pgk1 has been reported to interact with Hsp90, a highly abundant chaperone protein containing ATPase activity 11, 12. To test whether Hsp90 was the ATPase activated by TZ, we examined the effect of geldanamycin (GA), a potent inhibitor of the ATPase of Hsp90. With GA treatment alone, the ATP level was not altered in the cell lysate (Fig. 4a), suggesting the basal ATPase activity of Hsp90 is very low as reported 26. However, GA reversed the ATP decline triggered by TZ (Fig. 4a). To verify the interaction between Pgk1 and Hsp90, we overexpressed HA-tagged mPgk1 (Pgk1-HA) in RAW 264.7 cells. We found that IP with an anti-HA antibody, Hsp90 could be pulled down (Fig. 4b). Consistently, IP with an anti-Hsp90 antibody, endogenous p23 (a known binding partner of Hsp90) 27 could be pulled down, as well as Pgk1 (Supplementary Fig. 8a). These results suggest a possible interaction between Pgk1 and Hsp90. Moreover, we expect more proteins associating with Hsp90 upon activating the ATPase of Hsp90 by TZ 27. Indeed, the anti-Hsp90 antibody could pull down more proteins upon addition of TZ than without TZ (Supplementary Fig. 8b), suggesting TZ may activate Hsp90.
Figure 4. The anti-apoptosis effect of TZ was dependent on Hsp90.
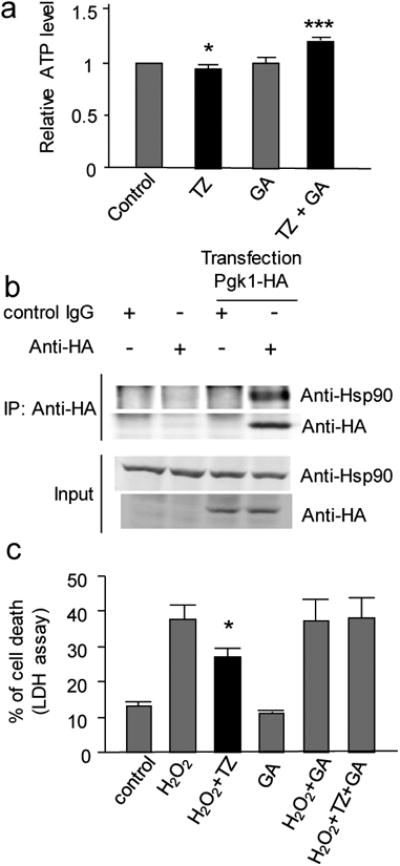
(a) The effect of TZ and GA on the ATP levels in the cell lysate of RAW 264.7 cells. The steady state (after the 10th minute) ATP level in the cell lysate was measured after 0.1% DMSO (control), TZ (10 μM), GA alone (100 μM) or TZ plus GA (100 μM) were added. Tail n = 3. (b) The co-immunoprecipitation of Pgk1-HA and Hsp90. Pgk1-HA was transfected into RAW 264.7 cells. An anti-HA antibody was used for a protein pull-down from the cell lysate. The full gel was shown on Supplementary Fig. 15. (c) Effect of TZ against H2O2-induced cell death in the presence of GA (10 nM). The LDH release assay was shown. Trial n = 3.
Next, we asked whether the function of Hsp90 was required for TZ to block apoptosis. Quantified by the LDH release and caspase activity assays, we found that the anti-apoptotic effect of TZ was completely abolished in the presence of GA (10 nM), while GA alone did not affect cell survival (Fig. 4c and Supplementary Fig. 8c). As a control, the anti-apoptotic effect of zVad was unaffected by 10 nM GA (Supplementary Fig. 8c). We further verified this result by PARP1 cleavage (Supplementary Fig. 8d). Interestingly, GA treatment could increase Hsp70, as reported by others 28 (Supplementary Fig. 8d). This result further suggested the protective effect was mediated by Hsp90, but not Hsp70. Together, these data suggested that TZ may require the ATPase activity of Hsp90 to block apoptosis.
TZ protected flies and rodents from multiple stresses
To examine effect of TZ in vivo, we first studied several stress conditions, including hypoxia, oxidative stress and apoptosis in Drosophila, based on the rationale that Hsp90 activation induced by TZ may promote multi-stress resistance. To induce hypoxia, fly larvae were immersed in Drosophila HL3 saline for two hours, then their lethality was determined six hours later after re-oxygenation. The result showed that TZ reduced lethality in a dose-dependent manner (Supplementary Fig. 9a). For oxidative stress, the lethality induced by feeding the flies with 20 mM of paraquat was determined. The results demonstrated that TZ (10 μM) was protective against paraquat (Fig. 5a). Moreover, the expression of Pgk1 RNAi or Hsp83 (the Drosophila homolog of mammalian Hsp90) RNAi which effectively knocked down the targeted transcripts (Supplementary Fig. 9b) abolished the TZ effect (Fig. 5a), suggesting the TZ effect was dependent on Pgk1 and Hsp83. In contrast, overexpression of Pgk1 was sufficient to rescue reaper-mediated apoptosis in the fly eye (Fig. 5b). Together, these results suggest that TZ protects flies from multiple stresses, and this effect depends on the function of Pgk1 and Hsp83.
Figure 5. TZ showed multiple stress resistance in flies and rodents.
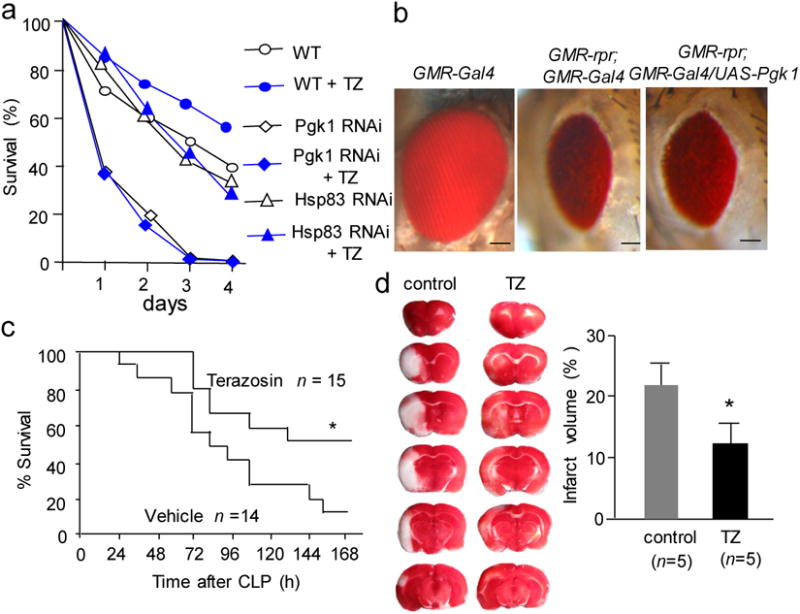
(a) The TZ effect on adult flies under oxidative stress. Survival rates of different genotypes of flies feeding on 20 mM paraquat were shown with or without feeding 10 μM TZ. Trial n = 3, each trial tested 60 flies. (b) The effect of Pgk1 overexpression on apoptosis in Drosophila. GMR-rpr represents a transgene with the GMR promoter directly upstream of the reaper cDNA, which causes eye-specific apoptosis in flies. The bars represent a length of 50 μm. (c) The CLP model of sepsis. TZ (0.08 mg/kg was injected s.c. at 1.5 and 24 hours after CLP), or saline (vehicle) was injected as a control. The Kaplan-Merier survival analysis was performed, and the number of mice tested was shown. (d) Effect of TZ on MCAO in rats. The micrograph showed the representative TTC staining from a sectioned whole brain. The quantification data was shown on the bar graph.
Next, we assessed the effect of TZ in mammals and studied two mouse models of sepsis. For the LPS model of sepsis, we used the immunocompetent BALB/C mice, due to their high sensitivity to septic stress 29. We found that intraperitoneal (i.p.) injection of TZ (0.4 mg/kg) at 1.5 hours after LPS infusion significantly improved the mouse survival (Supplementary Fig. 9c). Similarly, subcutaneous (s.c.) injection of TZ (0.08 mg/kg) at 1.5 hours and 24 hours after the surgery showed protection against the cecal ligation and puncture (CLP) model of sepsis, which is considered as a better model to reflect the complex nature of human sepsis 30 (Fig. 5c). Strikingly, TZ still exhibited protective effects when administered at 6 hours post-surgery (Supplementary Fig. 9d). For the pharmacodynamic marker, we measured the serum lactate level, because serum acidosis has been considered as a biomarker of sepsis correlating with lethality in patients 31. We observed that TZ was able to reduce the serum lactate level in the CLP mice (Supplementary Fig. 9e). This was consistent with the rescue of septic survival by TZ.
To determine the drug effect on apoptosis, we observed that DNA fragmentation, a marker of apoptosis, occurred in the thymus in both septic models (Supplementary Fig. 10a), which is consistent with previous reports 6. Importantly, TZ significantly reduced DNA fragmentation in these septic models (Supplementary Fig. 10a). We further confirmed this observation by staining the thymus with TUNEL (dUTP Nick End Labeling) (Supplementary Fig. 10b). Collectively, these results have demonstrated that TZ was protective against mouse models of sepsis.
As for the TZ dose at the site of action, we measured TZ concentrations in the blood serum at various time points of after i.p injection of TZ (0.4 mg/kg) in mice. The result showed that its concentration reduced from 200 nM to 1 nM in 2 hours (Supplementary Fig. 11a). Meanwhile, we also determined TZ concentrations in tissues, including thymus, heart, spleen, liver and kidney at 20 minutes, 1 hour, 2 hours and 6 hours after TZ injection. The result showed that concentrations of TZ declined with time and it became undetectable after 6 hours (Supplementary Fig. 11b). Within this time, we found that the pyruvate level was increased in thymus, where prominent apoptosis occurred in sepsis (Supplementary Fig. 11c).
Based on our data, gain of function Pgk1 may phenocopy TZ effect, therefore, we infected mice with Pgk1 or the control EGFP lentivirus 32. The transfection efficiency of lenti-EGFP was low but detectable (Supplementary Fig. 12a and b). After the CLP treatment, we observed that mice expressing Pgk1 showed significant reduction of lethality (Supplementary Fig. 12c) and apoptosis in thymus (Supplementary Fig. 12d). This result further confirmed the functional role of Pgk1 in sepsis.
One caveat for the in vivo effect of TZ is that it may have inhibited the sympathetic nervous system 33, and resulted in the reduced cytokine release during sepsis 34. We found that administration of TZ or overexpression of Pgk1 could not alter the elevated levels of cytokines including IL-6, IL-10, HMGB1 and TNF-α at multiple time points after CLP surgery (Supplementary Fig. 13a and b). In addition, the TZ dosage we used (0.4 mg/kg on LPS and 0.08 mg/kg on CLP) was much lower than the 10 mg/kg dosage that can affect blood pressure in mice 35. We also measured the blood pressure change after i.p injection of 0.4 mg/kg TZ, and observed no blood pressure reduction (Supplementary Fig. 14a). These results suggested that the anti-septic effect of TZ was not mediated through inhibition of the sympathetic nervous system.
Finally, we tested the effect of TZ on the unilateral middle cerebral artery occlusion (MCAO) model in rat. The coronal sectioned brain slices were stained with triphenyl-tetrazolium chloride (TTC), which can be oxidized by dehydrogenase from intact mitochondria to yield formazan, a red color product 36. Therefore, the defective brain area is not stained by TTC and displays a white color. We found that i.p. injection of TZ (0.08 mg/kg) at 30 minutes before occlusion had no protective effect on the infarct volume compared with the control rats (Supplementary Fig. 14b). Then, we reduced the TZ dosage to 0.03 mg/kg, and observed significant protection in MCAO (Fig. 5d). This result suggested that TZ was protective in neural cells during stroke, and it required lower dosage than treatment for sepsis.
Discussion
Our studies have demonstrated that TZ inhibits apoptosis in fruit flies, mammalian cells, as well as in models of mouse sepsis and rat brain stroke. In contrast to its well-known role in antagonizing the α1- adrenergic receptor, our results demonstrated that TZ reconciles its effects through the activation of a novel target Pgk1, defined by the fact that the anti-apoptotic effect of TZ was abolished by the loss of Pgk1. Consistent with this protective role, Pgk1 deficiency in humans causes muscle stiffness, hemolytic anemia, and mental retardation 37, whereas the overexpression of Pgk1 renders a multi-drug resistant phenotype in human ovarian cancer cells or the progression of hepatic carcinoma 38, 39.
Regarding the mechanism of TZ activation of Pgk1, our co-crystal structure data from both the mouse and the human Pgk1 with TZ suggested that the 1,3-diamino-6,7-dimethoxyisoquinoline structural motif fits into the pocket of ADP/ATP binding site. Because ATP release is likely to be a key rate limiting step for the enzymatic reaction 40, TZ may kinetically facilitate the reaction through accelerating ATP release. However, the binding affinity of TZ to Pgk1 was approximately 10 fold higher than the substrate ADP, therefore the concentration of TZ needs to be sufficiently low in order for ADP to compete off TZ for the next catalytic cycle.
Our mechanistic studies suggested that activated Pgk1 might promote the ATPase activity of Hsp90. Under basal conditions, Hsp90 is known to possess a very low ATPase activity 41, and activation of its ATPase can enhance its interaction with client proteins 8. Hsp90 is a well-known inhibitor of caspase-mediated apoptosis through diverse mechanisms. For instance, Hsp90 may interact with Apaf-1 to prevent the formation of the apoptosome 42 or prevent Bid cleavage 43. Activated Hsp90 may also promote pro-survival pathways by increasing NF-κB activity or by maintaining Akt kinase activity 44, 45. Consistent with our hypothesis, Hsp90 has been shown to protect mice from LPS-induced sepsis 46. Although our results suggest that TZ may enhance the ATPase activity of Hsp90, this observation is indirect, which warrants further investigation.
Different from of our hypothesis, some studies have suggested that the administration of some Hsp90 inhibitors such as GA and 17-AAG were protective against sepsis 47, 48. However, these inhibitors were also known to activate the function of HSF-1 through inactivation of Hsp90 to release HSF-1 28, 49. The effect of TZ on Hsp90 activation warrants further investigation, especially during sepsis and stroke.
The discovery of TZ as a novel protective agent and Pgk1 as a novel target has great potential to be implemented in many medical conditions to protect cells from damages. The fact that TZ is an approved drug with well-established pharmacokinetic and safety profiles in humans may accelerate its potential development and repurposing as a novel clinical drug.
Online Methods
Fly stocks and compound screen
UAS-rpr and HS-Gal4 flies were obtained from Bloomington Stock Center. The F1 progenies (HS>rpr) from the cross of these two lines were raised at 18 °C for the compound screen. The Pgk1 transgenic fly was generated using a pUAST vector. For the compound screen, chemicals (purchased from Sigma and Santa Cruz, the purity of each chemical was shown in Supplementary data set 4) were dissolved in 5% sucrose at the concentrations listed in the Cmap. The compound solution (150 μL) was added to a vial containing three layers of filter papers. Twenty adult HS>rpr flies aged 1-3 days were then placed into each vial for 24 hours. Expression of rpr was subsequently induced by placing the vials in a 37 °C incubator for 45 min. These flies were then placed at 25 °C for another 24 hours before calculation of the survival rate.
Antibody information
Caspase 3 (9662, Cell Signaling); PARP1 (9532, Cell Signaling); HSP70 (4872, Cell Signaling); HSP90 for IP (ab1429, Abcam); β-actin (P30002, Abmart); HA (H6908, sigma); Hsp90 for WB (4874, Cell Signaling); Pgk1 for IP (sc-17943, Santa Cruz); P23 (sc-376725, Santa Cruz); GFP (BE2001, Easybio); Pgk1 for WB (sc-48342, Santa Cruz); HSF-1 (12972, Cell Signaling); IgG (invitrogen).
Microarray processing and data analysis
Ten vials of HS>rpr flies and progenies of HS-Gal4 and yw67c23 (a genetic-background-matched control line) aged 1-3 days were collected and frozen in liquid nitrogen before heat shock (as time 0), and after heat shock at 1, 2, 3, 4, 5, 6, 7, 8 and 12 hours. Total RNA was prepared by RNeasy mini kit (Qiagen). Drosophila 2.0 DNA microarrays from Affymetrix were used (Affymetrix, Inc). Data analysis was performed using the Arrayassist 5.0 (Affymetrix, Inc). The algorithm of “RMA” was used to analyze the data. The fold changes of transcripts were listed in Supplementary Table 1.
Exploration of candidate compounds by Cmap
We converted up-regulated and down-regulated genes from the Drosophila microarray to mammalian homologous genes based on an online database (www.affymetrix.com) (Supplementary Table 2). The signature files were then generated based on the protocol provided by Cmap and the list of predicted compounds with the best positive or negative correlations was generated (Supplementary Table 3). The 25 tested compounds were listed Supplementary Table 4, and their concentrations used in the microarrays were listed in Supplementary Table 5.
Cell culture and apoptosis induction
The RAW 264.7 cells and 293T cells were cultured in the DMEM medium (Gibco) supplemented with 10% FBS (Gibco), streptomycin (0.1 mg/mL) and penicillin (0.06 mg/mL) (Gibco). To induce apoptosis, RAW 264.7 cells at 40% confluence were incubated in DMEM (10% FBS) containing 2 μg/mL lipopolysaccharide (2 μg/mL, LPS; Escherichia coli O111:B4, Sigma-Aldrich, >500,000 EU/mg) and murine IFN-γ in 0.1 % DMSO (50 U/mL, Peprotech, >98%), or LPS and IFN-γ plus TZ in 0.1 % DMSO (10 μM) for 18 to 30 hours. For H2O2 treatment, the final concentration of H2O2 was 1 mM. For geldanamycin (GA, purchased from Biovision, >99%) treatment, the concentration was 10 nM. Z-Val-Ala-DL-Asp (OMe)-fluoromethylketone (Z-vad, Bachem) was used at the final concentration of 100 μ;M.
Annexin V staining and analysis in the RAW 264.7 cells
After incubating with LPS and IFN-γ, the cells were stained with anti-Annexin V antibody (Invitrogen) for 15 min. The cells were then washed once with Annexin-binding buffer and imaged with a confocal microscope (TCS SP5, Leica). The percentage of apoptotic cells are calculated as the number of annexin V positive cells to the number of total cells.
LDH assay
The LDH assay (Promega) was performed according to the manufacture's instructions with minor modifications. Cells were plated in 96-well plates and treated as indicated for 24 hours. LDH levels in both medium and cells were measured. The absorbance at 490 nm was obtained from SpectraMax M2 Microplate Reader (Molecular Devices). The cytotoxicity is calculated as LDHmedium/(LDHmedium + LDHcell).
Western blotting and Co-immunoprecipitation
Proteins were extracted with the RIPA buffer (50 mM Tris-HCl, pH 7.8, 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate and 1 mM EDTA), and then added with protease inhibitors (Roche). After disrupting on ice for 30 min, the cell lysates were centrifuged at 13,000 g and boiled with SDS sample buffer at 95° C for 5 min.
For the co-IP experiment, Pgk1-HA was transfected in the RAW264.7 cells for 36 hours. The cells were lysed in the lysis buffer (20 mM Tris-HCl, pH=8.0, 130 mM NaCl, 10 mM KCl, 1.5 mM MgCl2, 10% glycerol, 0.25% Triton X-100 and protease inhibitors). After centrifugation at 13,000 g for 10 min at 4° C, the supernatant was incubated with HA antibody or control IgG at 4° C overnight. The protein A/G-agarose beads were added to precipitate the complexes. After washing with lysis buffer for six times, the agarose beads were boiled in a 2×SDS loading buffer. For endogenous co-IP, the same sample preparation protocol was followed.
Activity assay of caspase 3/7
Cell lysates were analyzed with a caspase-GloR 3/7 Assay kit (Promega).
Target identification of TZ with affinity chromatography
RAW 264.7 cells were sonicated in a modified RIPA buffer (50 mM Tris-HCl, pH 7.8, 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate and 1 mM EDTA) supplemented with a protease inhibitor cocktail (Roche). For the control experiment, 50 μL of naked Affi-Gel 102 (Bio-Rad) beads were added to 1 mL of 4 mg/mL cytosolic extracts. For the soluble competitor experiment, 10 mg of TZ was incubated with 1 mL of cell lysates for 1 hour before combined with 50 μL of TZ-TA immobilized Affi-Gel (Affi-Gel-TZ-TA). For the bait beads experiment, 50 μL of Affi-Gel-TZ-TA was directly incubated with 1 mL of cell lysates. The incubation lasted overnight at 4° C. The beads were then washed with the RIPA buffer three times. Bound proteins were eluted by boiling the beads in 0.1% SDS and were analyzed on a 12% SDS-PAGE gel and visualized by Coomassie brilliant blue staining.
Mass spectrometry analysis
The bands on the protein gels were excised and digested using the standard tryptic digestion method (from ABRF). The peptides from digestion were analyzed on an LTQ-Orbitrap mass spectrometer (Thermo Scientific). The analytical reverse phase column was 100 μm (ID) × 8 cm (length) with a pulled tip, packed with 3 μm, 125 Å Aqua C18 resin (Phenomenex). Gradients from 5% to 80% of ACN were used. The MS2 data was acquired in data-dependent mode, as one full scan followed by eight CID MS2 scans for the top eight peaks. The MS2 spectra were searched against a NCBI-mouse database using Prolucid, and filtered with DTASelect2.
Measurement of intracellular calcium level
The intracellular Ca2+ concentration ([Ca2+]i) was measured using the Fura-2 staining kit (Invitrogen). Cells were loaded with the Fura 2-AM (2 μM) for 30 min at 37° C in the dark, and then washed three times in standard external solution (141 mM NaCl, 2.5 mM KCl, 2.4 mM CaCl2, 1.3 mM MgCl2, 1.25 mM NaH2PO4, 10 mM HEPES, and 11 mM glucose). Imaging was performed on an inverted Olympus microscope, and the Fura-2 was excited at 340 and 380 nm. The emission was then collected with a 480-540 nm bandpass filter. The 340/380 nm ratio was calculated and normalized to the baseline ratio at the beginning of the experiment. The Ca2+ signals are plotted as relative Fura-2 intensity, which denotes the 340/380 nm ratio (background intensity was subtracted). Metaraminol (Sigma, ≥98%) was used at 200 μM. TZ and TZ-md were used at 4 μg/mL.
Activity assay of Pgk1
To induce Pgk1 expression in vitro, the mouse Pgk1 cDNA was cloned into a pEASY™-E2 vector containing a His-tag (TransGen Biotech). Protein expression was induced with 0.5 mM IPTG in BL21 (DE3) chemically competent cells (TransGen Biotech). After the cells were lysed in an extraction buffer (20 mM sodium phosphate, 500 mM NaCl, 5 mM imidazole, 5% glycerol, protease inhibitor cocktail, pH 7.9), the supernatant was purified with nickel beads (AdarBiotech). The elution buffer (20 mM sodium phosphate, 500 mM NaCl, 80 mM imidazole, protease inhibitor cocktail, pH 7.9) was used to elute the His-tagged Pgk1.
To measure the Pgk1 activity, a saturated amount of the substrates (1.6 mM GAP, 1 mM (β-NAD, 1 mM ADP, 20 ng/μL GAPDH) was mixed with the purified recombinant mouse Pgk1-His protein (2 μg/mL) and the reaction was conducted in a buffer (20 mM Tris, 100 mM NaCl, 0.1 mM MgSO4,10 mM Na2HPO4, 2 mM DTT at pH 8.6). Because the forward reaction of Pgk1 can lead to reduction of ADP level, and this will promote GAPDH to produce more NADH. NADH can be detected at the absorbance wavelength of 339 nm. Therefore, the levels of relative NADH changes to the baseline can indicate Pgk1 activity.
Affinity assay of TZ and Pgk1
To examine whether TZ and Pgk1 bind directly, naked Affi-Gel, Affi-Gel-TZ-TA with competitor TZ or Affi-Gel-TZ-TA alone was incubated with His-tagged Pgk1 overnight at 4° C. The proteins on the beads were eluted and analyzed on a 15% SDS-PAGE.
Cloning expression and purification
Recombinant mPGK1 was produced in BL21 Escherichia coli cells harboring the mPGK1-PET28a expression plasmid with a TEV cleavage sequence inserted. Protein expression was induced with 0.5 mM IPTG overnight at 16° C. After the cells were lysed in the extraction buffer (20 mM sodium phosphate, 500 mM NaCl, 5 mM imidazole, 5% glycerol, pH 7.9), the supernatant was loaded onto a nickel affinity column (GE Healthcare) and washed with the buffer containing 10 mM imidazole. The bound protein to the column was eluted using 100 mM imidazole in the same buffer. The elution was then mixed with TEV enzyme and dialyzed in the buffer containing 20 mM sodium phosphate, 500 mM NaCl, pH 7.9. After dialysis, the nickel affinity column was used to remove the peptide with the His-tag and the TEV sequence. The mPGK1 without the His-tag was concentrated and further purified by size-exclusion chromatography (GE Healthcare), and final fractions containing pure mPGK1 in the buffer containing 20 mM Tris-HCl pH 7.0, 100 mM NaCl, 2 mM DTT were pooled and concentrated to 20–30 mg/ml.
Crystallization
The mPGK1-3PG crystals were crystallized at room temperature using the hanging drop vapor diffusion method from 25-30% PEG 4000 0.2 M sodium acetate trihydrate and 0.1 M Tris-HCl, pH 8.0. The drop contained 1μl of the protein solution mixed with 2 μl of the reservoir solution. The protein solution (10 mg/ml) consisting of Tris-HCl pH 7.0 contained 25 mM MgCl2, 50 mM PG, 10 mM DTT and 0.02% NaN3.
To obtain the PGK1-3PG-TZ complex, both co-crystallization and soaking methods were used. The mPGK1-3PG-TZ co-crystals were grown from the same crystallization condition described previously. The protein solution contained the same components as mPGK1-3PG solution, with the addition of 10 mM TZ. Meanwhile, hPGK1-3PG-TZ co-crystals were obtained from 25-30% PEG 4000, 0.1 M Tris-HCl, pH 8.0. To ensure complex formation, mPGK1-3PG crystals were soaked in the buffer containing 30% PEG 4000 0.1M Tris-HCl, pH 8.0, 25 mM MgCl2, 50 mM PG and 10 mM TZ.
Cryoprotection
Crystallization buffer contained 30% PEG 4,000, which was sufficient for cryoprotection in both human and mouse PGK. Therefore, no additional cryoprotectant was needed to freeze the crystals. Individual crystals were mounted in a cryoloop (Hampton), followed by flash cooling in liquid nitrogen for data collection.
Data collection and refinement of crystal structures
Diffraction data of mPGK1-3PG-TZ was collected from cryo-cooled crystals to a resolution of 2.1 Å (see Table S2) at the KEK Photon Factory Synchrotron located in Japan, using the BL17A b eamline using the Q270 X-ray diffractometer. Diffraction data of hPGK1-3PG-TZ was collected at the Shanghai Synchrotron Radiation Facility, BL17U1 beamline, with a resolution of 2.1 Å (see Structure Statistic Table). Data were processed with the program HKL2000, and molecular replacement was carried out using MolRep in the CCP4 Program Suite. The search model for the mPGK1-3PG-TZ structure is 3C39 in the PDB, while the PDB 2XE6 was used to determine hPGK1-3PG-TZ structure. Refinement was performed in PHENIX with rigid body refinement, and alternating cycles of TLS, positional, and individual B-factor refinement. The resulting model was manually inspected and modified with the program COOT.
ITC
The raw heat were measured over a series of injections of TZ or TZ-TA into mPGK1 at 25 °C by MicroCal™ iTC200 (GE Healthcare).
RT-qPCR
The method for qRT-PCR followed the “relative study” standard protocol from Applied Biosystem (7500 real time PCR system, ABI, Inc). Actin was used as a reference of the total RNA quantity.
Lentivirus package
The coding sequences of EGFP and mouse Pgk1 were amplified by PCR and separately cloned into the pLentiCMVMCSSVBsd vector. Lenti-EGFP and Lenti-PGK-1 viruses were produced by cotransfection with the VSV-G/pMD2G and pCMV-dR8.74 plasmids into the packaging cell line HEK 293T by Lipofectamine™ 2000 (Invitrogen). The shRNA lentiviruses were packaged in a similar fashion.
Infection of cells and mice with lentivirus
RAW264.7 cells were incubated with the virus and the polybrene (8 μg/mL) overnight. Starting from the second day, stable cell lines were selected in a growth medium supplemented with 5-10 μg/mL blasticidin for a week. To infect a mouse, 2 × 107 Infection Units (IU) of virus were diluted in 200 μL saline and injected (s.c.) twice at day 1 and 4 before CLP. One week later, the mice were subjected to CLP.
Detection of ATP and pyruvate in cell lysate and mouse tissue
The ATP level was detected using the kit from Promega. The cell lysates from RAW 264.7 cells were prepared as described previously. After adding DMSO or TZ (10 μM) into the cell lysates, the total ATP in the reaction system was detected immediately or 10 min after the reaction system was established. For the GA treatment, it was added directly into the reaction system and the final concentration was 100 μM. Pyruvate measurement was followed a protocol from a kit (EnzyChrom™ pyruvate Assay Kit, BioAssay Systems).
Drosophila hypoxia assay
The third instar larvae were immersed in a 1.5 ml tube filled with HL3 saline for 2 hours. Then, the saline was removed to allow re-oxygenation of the larvae. After 6 hours recovery, their survival rate was determined.
Drosophila oxidative stress assay
The wild type, UAS-Pgk1RNAi and UAS-Hsp83RNAi flies were crossed with the Actin5c-Gal4 promoter line. Their progeny male flies were collected at an age of 1-3 days-old. The 20 flies then were put into a testing vial, which contains 450 μL of solutions (20 mM paraquat in 5% of sucrose) on 3 layers of filter papers. Their lethality was recorded each day for 4 days.
Mouse models of sepsis
Male BALB/C mice from the Vital River (Beijing, China) were housed in the Animal Center of Peking University. The animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) of the Peking University (Approve number: LSC-LiuL-1 and LSC-LiuL-2). For the LPS model of sepsis, each mouse was injected with LPS (13.5 mg/kg, intraperitoneal injection, i.p.) once. TZ (0.4 mg/kg) or saline was then injected (i.p.) 1.5 hours after LPS infusion. Lethality was recorded every 12 hours for 7 days.
For the CLP model of sepsis, mice were anesthetized with Avertin (240 mg/kg, i.p.). Then, a 1.5-2.0 cm long abdominal midline incision was made and the cecum was exposed. After ligation at 5 mm from the cecal tip, the cecum was punctured once with a 22-gauge needle and about 1 mm of stool was squeezed out. TZ (0.08 mg/kg) or saline was injected after 1.5 hours and 24 hours post-surgery. Lethality was recorded every 12 hours for 7 days.
Rat permanent MCAO model
We used adult male Sprague-Dawley rats (250–270g, Vital River, Beijing). The animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) of the Peking University. Briefly, a suture was inserted through the left external carotid artery of the anesthetized rats. A laser Doppler blood flow monitor was used to ensure successful occlusion (>80% drop from pre-stroke baseline). Then, TZ or saline (control) was injected intraperitoneally at a concentration of 0.08 mg/kg and 0.03 mg/kg respectively, 30 minutes before the occlusion. The infarct volume was determined 4 hours later by TTC staining (1%, at 37° C for 15 minutes).
DNA fragmentation in mouse thymus
The thymi were collected from each mouse, 6-48 hours after CLP. The genomic DNA of thymus was then extracted with a genomic DNA purification kit (Biofuture). DNA fragmentation was assessed on a 1% agarose gel.
TUNEL assay
The assay was performed based on the manufacture's protocol (Millipore).
Cytokine detection
Blood samples were collected from mice 6 to 48 hours after LPS infusion or CLP surgery. The samples were then incubated at room temperature for 45 min., and serum was obtained by centrifugation at 1,900 g for 15 min. The serum samples were then analyzed by TNF-α, IL-6 and IL-10 ELISA kits (Boster) and mouse HMGB-1 ELISA kit (SUNBIO).
Pharmacokinetics of TZ
After i.p. injection of TZ, blood or tissue samples collected at various time points. The tissues were grinded and sonicated in RIPA buffer (50 mM Tris-HCl, pH 7.8, 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate and 1 mM EDTA). For standard curve of TZ, 30 μL of TZ (200 nM) was added to 100 μL of blood sample and mixed with 40 μL of 1 M NaOH. Then, 200 μL of acetic ether was added and mixed for 2 minutes. After centrifuge at 2000g for 3 minutes, the supernatant was harvested; and 200 μL of acetic ether was added again. Then, combine the extracts (600 μL of acetic ether total). Then, the samples were analyzed by a WATERS UPLC system (Phenomenex Kinetex) coupled with an AB API4000Q mass spectrometer. Mass signals were collected under positive mode.
Statistical analyses
A one-way ANOVA tests were used for group comparison (Fig. 1b, 1c, 1d, 2d, 2f, 4a, 4c; and Supplementary Fig. 1b, 2a, 2b, 3a, 3b, 4a, 4e, 8c, 9a, 9e, 10b, 11a, 11b, 11c, 12d, 13a, 13b an 14a). The Student-t tests were used to compare two data sets (Fig. 2e and 5d; and Supplementary Fig. 9b). The Kaplan-Meier tests were used under the Log Rank algorithm for survival analysis (Fig. 5c, and Supplementary Fig. 9c, 9d and 12c).
Database
The crystal structure data has been deposited at RCBS PDB (http://www.rcsb.org), submission ID: 4O33 and 4O3F.
Supplementary Material
Acknowledgments
This work was supported by the Chinese Ministry of Science and Technology (2013CB530700), the National Natural Science Foundation of China (NSFC31171324) and the Peking-Tsinghua Center for Life Sciences to Lei Liu. By the Ministry of Education of China and NIH grant HL48675 and funds from the Peking-Tsinghua Center for Life Sciences to Jia-huai Wang. We also acknowledge the Ministry of Science and Technology Basic Research Program (2011CB809100) the National Natural Science Foundation of China (21272016), and the Doctoral Fund of Ministry of Education of China (20120001110083) to Xiaoyu Li. We thank Prof. Luhua Lai (PKU) for her generous support in the ITC experiments.
Footnotes
Author contributions: XC, CZ, TW and LL designed the experiments. XC, CZ and TW performed the biological experiments. XL, LF and JW performed the crystal structure study. YD and MD performed the mass spectrometry analysis. YL, CC and XL performed the chemical synthesis. XL designed the chemical study. XC, CZ, JW, XL and LL wrote the manuscript.
Competing financial interests: The authors declare competing financial interests for novel use of terazosin and its related chemicals to target Pgk1 and their related disease treatments.
Additional information: Supplementary information is available in the online version of the paper.
References
- 1.Annane D, et al. Corticosteroids in the treatment of severe sepsis and septic shock in adults: a systematic review. JAMA. 2009;301:2362–2375. doi: 10.1001/jama.2009.815. [DOI] [PubMed] [Google Scholar]
- 2.Kumar A. Optimizing antimicrobial therapy in sepsis and septic shock. Crit Care Clin. 2009;25:733–751. viii. doi: 10.1016/j.ccc.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 3.Ulloa L, Brunner M, Ramos L, Deitch EA. Scientific and clinical challenges in sepsis. Curr Pharm Des. 2009;15:1918–1935. doi: 10.2174/138161209788453248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yuan J. Neuroprotective strategies targeting apoptotic and necrotic cell death for stroke. Apoptosis. 2009;14:469–477. doi: 10.1007/s10495-008-0304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuan JY, Horvitz HR. The Caenorhabditis elegans genes ced-3 and ced-4 act cell autonomously to cause programmed cell death. Dev Biol. 1990;138:33–41. doi: 10.1016/0012-1606(90)90174-h. [DOI] [PubMed] [Google Scholar]
- 6.Lang JD, Matute-Bello G. Lymphocytes, apoptosis and sepsis: making the jump from mice to humans. Crit Care. 2009;13:109. doi: 10.1186/cc7144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jana S, Paliwal J. Apoptosis: potential therapeutic targets for new drug discovery. Curr Med Chem. 2007;14:2369–2379. doi: 10.2174/092986707781745569. [DOI] [PubMed] [Google Scholar]
- 8.Lanneau D, de Thonel A, Maurel S, Didelot C, Garrido C. Apoptosis versus cell differentiation: role of heat shock proteins HSP90, HSP70 and HSP27. Prion. 2007;1:53–60. doi: 10.4161/pri.1.1.4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ndubaku C, Cohen F, Varfolomeev E, Vucic D. Targeting inhibitor of apoptosis proteins for therapeutic intervention. Future Med Chem. 2009;1:1509–1525. doi: 10.4155/fmc.09.116. [DOI] [PubMed] [Google Scholar]
- 10.Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11:515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 11.Falsone SF, Gesslbauer B, Tirk F, Piccinini AM, Kungl AJ. A proteomic snapshot of the human heat shock protein 90 interactome. FEBS letters. 2005;579:6350–6354. doi: 10.1016/j.febslet.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 12.Zhao R, et al. Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the hsp90 chaperone. Cell. 2005;120:715–727. doi: 10.1016/j.cell.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 13.Lamb J, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 14.Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742–758. doi: 10.1016/j.cell.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White K, Tahaoglu E, Steller H. Cell killing by the Drosophila gene reaper. Science. 1996;271:805–807. doi: 10.1126/science.271.5250.805. [DOI] [PubMed] [Google Scholar]
- 16.Albina JE, Cui S, Mateo RB, Reichner JS. Nitric oxide-mediated apoptosis in murine peritoneal macrophages. J Immunol. 1993;150:5080–5085. [PubMed] [Google Scholar]
- 17.Bordner J, Campbell SF, Palmer MJ, Tute MS. 1,3-Diamino-6,7-dimethoxyisoquinoline derivatives as potential alpha 1-adrenoceptor antagonists. J Med Chem. 1988;31:1036–1039. doi: 10.1021/jm00400a026. [DOI] [PubMed] [Google Scholar]
- 18.Han J, Miyamae Y, Shigemori H, Isoda H. Neuroprotective effect of 3,5di-O-caffeoylquinic acid on SH-SY5Y cells senescence-accelerated-prone mice8 through the up-regulation of phosphoglycerate kinase-1. Neuroscience. 2010;169:1039–1045. doi: 10.1016/j.neuroscience.2010.05.049. [DOI] [PubMed] [Google Scholar]
- 19.Mazzoni C, Torella M, Petrera A, Palermo V, Falcone C. PGK1, the gene encoding the glycolitic enzyme phosphoglycerate kinase, acts as a multicopy suppressor of apoptotic phenotypes in S. cerevisiae. Yeast. 2009;26:31–37. doi: 10.1002/yea.1647. [DOI] [PubMed] [Google Scholar]
- 20.Banks RD, et al. Sequence, structure and activity of phosphoglycerate kinase: a possible hinge-bending enzyme. Nature. 1979;279:773–777. doi: 10.1038/279773a0. [DOI] [PubMed] [Google Scholar]
- 21.Bernstein BE, Michels PA, Hol WG. Synergistic effects of substrate-induced conformational changes in phosphoglycerate kinase activation. Nature. 1997;385:275–278. doi: 10.1038/385275a0. [DOI] [PubMed] [Google Scholar]
- 22.Zerrad L, et al. A spring-loaded release mechanism regulates domain movement and catalysis in phosphoglycerate kinase. J Biol Chem. 2011;286:14040–14048. doi: 10.1074/jbc.M110.206813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sawyer GM, Monzingo AF, Poteet EC, O'Brien DA, Robertus JD. X-ray analysis of phosphoglycerate kinase 2, a sperm-specific isoform from Mus musculus. Proteins. 2008;71:1134–1144. doi: 10.1002/prot.21801. [DOI] [PubMed] [Google Scholar]
- 24.Flachner B, et al. Substrate-assisted movement of the catalytic Lys 215 during domain closure: site-directed mutagenesis studies of human 3-phosphoglycerate kinase. Biochemistry. 2005;44:16853–16865. doi: 10.1021/bi051726g. [DOI] [PubMed] [Google Scholar]
- 25.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jackson SE. Hsp90: structure and function. Topics in current chemistry. 2013;328:155–240. doi: 10.1007/128_2012_356. [DOI] [PubMed] [Google Scholar]
- 27.Kamal A, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 28.Guo F, et al. Abrogation of heat shock protein 70 induction as a strategy to increase antileukemia activity of heat shock protein 90 inhibitor 17-allylamino-demethoxy geldanamycin. Cancer research. 2005;65:10536–10544. doi: 10.1158/0008-5472.CAN-05-1799. [DOI] [PubMed] [Google Scholar]
- 29.Remick DG, Newcomb DE, Bolgos GL, Call DR. Comparison of the mortality and inflammatory response of two models of sepsis: lipopolysaccharide vs. cecal ligation and puncture. Shock. 2000;13:110–116. doi: 10.1097/00024382-200013020-00004. [DOI] [PubMed] [Google Scholar]
- 30.Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4:31–36. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maciel AT, Noritomi DT, Park M. Metabolic acidosis in sepsis. Endocrine, metabolic & immune disorders drug targets. 2010;10:252–257. doi: 10.2174/187153010791936900. [DOI] [PubMed] [Google Scholar]
- 32.Naldini L, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 33.Akduman B, Crawford ED. Terazosin, doxazosin, and prazosin: current clinical experience. Urology. 2001;58:49–54. doi: 10.1016/s0090-4295(01)01302-4. [DOI] [PubMed] [Google Scholar]
- 34.Miksa M, Wu R, Zhou M, Wang P. Sympathetic excitotoxicity in sepsis: pro-inflammatory priming of macrophages by norepinephrine. Front Biosci. 2005;10:2217–2229. doi: 10.2741/1691. [DOI] [PubMed] [Google Scholar]
- 35.Lee DL, Webb RC, Brands MW. Sympathetic and angiotensin-dependent hypertension during cage-switch stress in mice. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1394–1398. doi: 10.1152/ajpregu.00306.2004. [DOI] [PubMed] [Google Scholar]
- 36.Rupadevi M, Parasuraman S, Raveendran R. Protocol for middle cerebral artery occlusion by an intraluminal suture method. J Pharmacol Pharmacother. 2011;2:36–39. doi: 10.4103/0976-500X.77113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shirakawa K, Takahashi Y, Miyajima H. Intronic mutation in the PGK1 gene may cause recurrent myoglobinuria by aberrant splicing. Neurology. 2006;66:925–927. doi: 10.1212/01.wnl.0000203500.63884.39. [DOI] [PubMed] [Google Scholar]
- 38.Ai J, et al. FLNA and PGK1 are two potential markers for progression in hepatocellular carcinoma. Cell Physiol Biochem. 2011;27:207–216. doi: 10.1159/000327946. [DOI] [PubMed] [Google Scholar]
- 39.Duan Z, et al. Overexpression of human phosphoglycerate kinase 1 (PGK1) induces a multidrug resistance phenotype. Anticancer Res. 2002;22:1933–1941. [PubMed] [Google Scholar]
- 40.Geerlof A, Schmidt PP, Travers F, Barman T. Cryoenzymic studies on yeast 3-phosphoglycerate kinase. Attempt to obtain the kinetics of the hinge-bending motion. Biochemistry. 1997;36:5538–5545. doi: 10.1021/bi962842+. [DOI] [PubMed] [Google Scholar]
- 41.Jackson SE. Hsp90: structure and function. Top Curr Chem. 2012;328:155–240. doi: 10.1007/128_2012_356. [DOI] [PubMed] [Google Scholar]
- 42.Pandey P, et al. Negative regulation of cytochrome c-mediated oligomerization of Apaf-1 and activation of procaspase-9 by heat shock protein 90. EMBO J. 2000;19:4310–4322. doi: 10.1093/emboj/19.16.4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao C, Wang E. Heat shock protein 90 suppresses tumor necrosis factor alpha induced apoptosis by preventing the cleavage of Bid in NIH3T3 fibroblasts. Cell Signal. 2004;16:313–321. doi: 10.1016/j.cellsig.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 44.Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. Proc Natl Acad Sci U S A. 2000;97:10832–10837. doi: 10.1073/pnas.170276797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lewis J, et al. Disruption of hsp90 function results in degradation of the death domain kinase, receptor-interacting protein (RIP), and blockage of tumor necrosis factor-induced nuclear factor-kappaB activation. J Biol Chem. 2000;275:10519–10526. doi: 10.1074/jbc.275.14.10519. [DOI] [PubMed] [Google Scholar]
- 46.Li X, et al. The role of the Hsp90/Akt pathway in myocardial calpain-induced caspase-3 activation and apoptosis during sepsis. BMC Cardiovasc Disord. 2013;13:8. doi: 10.1186/1471-2261-13-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chatterjee A, et al. Heat shock protein 90 inhibitors prolong survival, attenuate inflammation, and reduce lung injury in murine sepsis. Am J Respir Crit Care Med. 2007;176:667–675. doi: 10.1164/rccm.200702-291OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chatterjee A, et al. Heat shock protein 90 inhibitors attenuate LPS-induced endothelial hyperpermeability. Am J Physiol Lung Cell Mol Physiol. 2008;294:L755–763. doi: 10.1152/ajplung.00350.2007. [DOI] [PubMed] [Google Scholar]
- 49.Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998;94:471–480. doi: 10.1016/s0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.