

Abstract
mRNA alternative polyadenylation (APA) has been increasingly recognized as a widespread and evolutionarily conserved mechanism for eukaryotic gene regulation. Here we describe a method called poly(A) site sequencing that can not only map RNA polyadenylation sites on a transcriptome level but also provide quantitative information on the relative abundance of polyadenylated RNAs. This method has been successfully used for both global APA analysis and digital gene expression profiling.
Keywords: Alternative polyadenylation, mRNA 3′ processing, Genomics, High-throughput sequencing, PAS-seq
1 Introduction
In eukaryotes, a large portion (30–70 %) of genes have been estimated to produce mRNAs bearing multiple alternative 3′ ends, a phenomenon known as alternative polyadenylation (APA) [1, 2]. To globally characterize APA, early experimental studies used various microarray platforms [3–5]. In these studies, APA changes are monitored by calculating the ratio between the average signal intensities of the probes targeting the extended regions found only in the longer APA isoforms and those of the probes for the common regions. Although these microarray-based methods can be used to detect APA changes, there are several serious limitations. For example, microarrays cannot be used to map polyadenylation sites (PASs), and the quantification is challenging, especially for genes with more than two APA isoforms.
In the past several years, many high-throughput sequencing (HTS)-based methods have been introduced for global characterization of mRNA polyadenylation [1], which can be generally classified into three distinct types. The first type, called direct RNA sequencing (DRS) [6], is based on the Helicos single-molecule sequencing platform. As polyadenylated RNAs are directly captured and sequenced by synthesis without library construction, DRS is believed to be more quantitative. However, when compared to other more commonly used HTS systems such as the Illumina platform, the disadvantages of DRS include lower read counts, shorter read length, higher error rate, and lack of multiplexing capacity [7]. The second method, called 3P-seq, utilizes a series of enzymatic steps designed to map the true 3′ ends of polyadenylated RNAs [8]. However, 3P-seq is labor intensive, and experimental bias may be introduced at various steps. The third type and most commonly used HTS method, including poly(A) site sequencing (PAS-seq), is based on oligo(dT)-primed reverse transcription [9–11]. The advantages of this method include its simplicity and quantitative performance. One limitation is the possibility of oligo(dT) primers hybridizing to internal A-rich RNA sequences, thereby identifying false-positive PASs. Computational methods can be applied to identify and remove the majority of these sites. In addition, various modifications have been introduced to this basic method to reduce internal priming and facilitate library construction and sequencing. For example, PAS-seq takes advantage of the SMART reverse transcription system in library construction [12]. Using this method, reverse transcription and linker addition on both ends are accomplished in one step, thereby significantly simplifying library construction. Additionally, HTS is carried out on the Illumina platform using a custom sequencing primer, which allows sequencing to start at the poly(A) junction and avoid the problematic A stretch at the beginning of the reads. Below we describe the detailed protocol for PAS-seq and offer technical advice on optimization and troubleshooting.
2 Materials
2.1 Solutions
Ammonium acetate (10 M).
100 % Ethanol.
1×TBE buffer (89 mM Tris base, 89 mM boric acid, 2 mM EDTA).
2.2 Enzymes, Reagents, Equipment
Trizol (Life Technologies).
Dynal Beads (dT)25 (Life Technologies).
10× RNA Fragmentation Buffer (10× Fragmentation Reagent, Life Technologies).
10× Stop Buffer (10× Stop Solution, Life Technologies).
6× DNA Loading Dye (Thermo Scientific).
Glycogen (Life Technologies).
RNAseOUT (Life Technologies).
Superscript III reverse transcriptase (Life Technologies).
QIAquick PCR purification kit (Qiagen).
Phusion DNA polymerase (New England Biolabs).
25 bp DNA ladder (Promega).
QIAquick Gel Extraction kit (Qiagen).
NanoDrop 1000 (Thermo Scientific).
PCR Thermal Cycler (Eppendorf).
2.3 Primer Sequences
-
HITS-5′:
CGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCTr (GGG).
-
HITS-3′:
ACACTCTTTCCCTACACGACGCTCTTCCGATCTTTTTTTTTTTTTTTTTTTTVN (V:A/C/G; N:A/T/C/G).
-
PE 1.0:
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT.
-
PE 2.0:
CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT.
-
PAS-seq:
ACACTCTTTCCCTACACGACGCTCTTCCGATCTTTTTTTTTTTTTTTTTTTT.
3 Methods
3.1 Poly(A+) RNA Purification
Purify total RNAs from cells/tissues using Trizol or other reagent as per manufacturer’s instructions.
Purify poly(A+) RNAs from total RNAs using Dynal Beads (dT)25 as per manufacturer’s instructions.
3.2 Poly(A+) RNA Fragmentation
-
Prepare the following mixture:
9 μl poly(A+) RNA (0.5–1 μg).
1 μl 10× RNA fragmentation buffer.
Incubate at 70 °C for 10 min (minutes).
Add 1 μl STOP buffer (10×), and leave on ice for 2 min.
-
Add:
190 μl H2O.
50 μl ammonium acetate (10 M).
750 μl 100 % ethanol.
0.5 μl glycogen (20 μg/μl).
Incubate on dry ice for 10 min, and spin at top speed in a microfuge for 15 min at 4 °C.
Rinse the pellet with 70 % ethanol, air-dry the pellet, and resuspend it in 22 μl of H2O.
3.3 Reverse Transcription (RT)
-
Prepare the following mixture:
22 μl RNA.
2 μl HITS-3′ (12 μM).
2 μl dNTP mix (10 mM).
Incubate at 65 °C for 5 min and then on ice for 5 min.
-
Add:
8 μl 5× First-strand reverse transcriptase buffer.
2 μl DTT (0.1 M).
2 μl HITS-5′ (10 μM).
2 μl RNaseOUT (40 U/μl).
2 μl Superscript III reverse transcriptase (200 U/μl).
Incubate at 50 °C for 30 min, then 42 °C for 30 min, and 90 °C for 5 min, and cool down to room temperature.
3.4 First-Round PCR mplification of cDNAs
Add 60 μl of H2O to the 40 μl RT reaction, and isolate cDNAs using the QIAquick PCR purification kit. Elute cDNA from the column with 40 μl of H2O.
-
Prepare the following PCR mixture (50 μl total volume):
36.4 μl cDNA.
10 μl 5× High-fidelity phusion polymerase buffer.
1 μl PE-1.0 primer (12 μM).
1 μl PE-2.0 primer (12 μM).
1 μl dNTP mix (10 mM).
0.5 μl High-fidelity phusion polymerase (2 U/μl).
-
Run the following PCR program:
98 °C 30 s.
98 °C 10 s.
60 °C 30 s.
72 °C 30 s.
Go to step 2 twice.
72 °C 5 min.
4 °C HOLD.
3.5 Size Selection of cDNAs
Add 10 μl of 6× DNA loading dye to the PCR reaction, and run the above PCR reaction on a 2 % agarose gel.
Cut out 200–300 bp band.
Extract DNA using QIAquick Gel Extraction kit, and elute DNA from the columns with 40 μl H2O.
3.6 Second-Round PCR Amplification of cDNA Fragments
-
Prepare the following PCR mixture (50 μl total volume):
36.5 μl cDNA (eluate from the previous step).
10 μl 5× High-fidelity Phusion polymerase buffer.
1 μl PE-1 primer (12 μM).
1 μl PE-2 primer (12 μM).
1 μl dNTP mix (10 mM).
0.5 μl High-fidelity phusion polymerase (2 U/μl).
-
Run PCR program:
98 °C 30 s.
98 °C 10 s.
65 °C 30 s.
72 °C 30 s.
Go to step 2 14 times.
72 °C 5 min.
4 °C HOLD.
Purify DNA using QIAquick PCR purification kit.
Run 10 % of the PCR product on a 2 % agarose gel to examine the size. A typical gel picture of the library is shown in Fig. 1.
Measure the DNA concentration using a Nanodrop spectrophotometer, and adjust the concentration according to the requirement of your sequencing facility.
Submit the library for sequencing on an Illumina Genome Analyzer or Hi-Seq using a custom sequencing primer (PAS-seq).
Fig. 1.
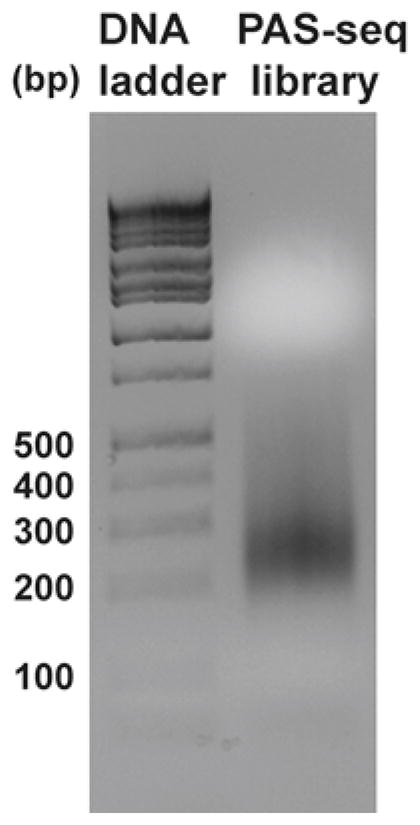
PAS-seq library. 10 % of the second-round PCR reaction (Subheading 3.6) is resolved on a 2 % agarose gel to check the size of DNA fragments
3.7 Bioinformatic Analysis
The PAS-seq raw reads are first trimmed by removing Ts from the beginning of the reads.
The remaining sequences are mapped to the proper reference genome using Bowtie with the setting (-n 2 -m 1 -s 1) (up to two nucleotide mismatches and one unique match to the reference genome allowed) [13].
To address the internal poly(A) priming issue, mapped reads that have six or more consecutive As or seven As in total within the ten nucleotides downstream of mapped poly(A) junction are removed.
PASs that are found within 40 nt of one another are likely due to heterogeneity of the cleavage reaction and are thus pooled together, and the weighed average position of all the PASs are designated as the final PAS. This process is repeated iteratively until there is no more PAS found within 40 nt.
For further bioinformatics analysis, please refer to references 9–11, 14.
4 Trouble-Shooting Tips
It is important to purify the cDNAs after the RT step and remove unused RT primers. Otherwise the RT primers may be used in the PCR amplification, and produce DNAs with incomplete linker sequences.
The purpose for the first short PCR step is to convert cDNAs into double-stranded DNAs. Gel purification helps to make sure that the DNAs are relatively homogenous in size, therefore minimizing the possible amplification bias.
Be extra careful with each DNA purification step. Maximize the recovery by eluting twice.
The low-cycle-number PCR is important for quantification. If the starting RNA amount is low, PCR cycle number may be increased to obtain the necessary DNA amount. However this may negatively influence the quantification performance.
If sequencing using the custom primer is not possible at your sequencing facility, one can change the orientation of the library by redesigning the primer sequences such as sequencing reads start at the 5′ end of the corresponding RNA sequence and read into the poly(A) tail. For this approach, we usually gel purify 175–225 bp DNA fragments and perform single-end 100 nt sequencing.
PAS-seq can be used for digital gene expression profiling (DEG) analysis. RNA-seq, one of the most commonly used methods for DEG analysis, relies on the average read counts over the entire genes for quantification. As each mRNA has only one 3′ end, PAS-seq and related methods should be at least as accurate as RNA-seq for DEG analysis [14].
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grant R01 GM090056 and American Cancer Society Grant RSG-12-186.
References
- 1.Shi Y. Alternative polyadenylation: new insights from global analyses. RNA. 2012;18:2105–2117. doi: 10.1261/rna.035899.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Di Giammartino DC, Nishida K, Manley JL. Mechanisms and consequences of alternative polyadenylation. Mol Cell. 2011;43:853–866. doi: 10.1016/j.molcel.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sandberg R, Neilson JR, Sarma A, et al. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science. 2008;320:1643–1647. doi: 10.1126/science.1155390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flavell SW, Kim TK, Gray JM, et al. Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron. 2008;60:1022–1038. doi: 10.1016/j.neuron.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ji Z, Lee JY, Pan Z, et al. Progressive lengthening of 3′ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc Natl Acad Sci U S A. 2009;106:7028–7033. doi: 10.1073/pnas.0900028106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ozsolak F, Kapranov P, Foissac S, et al. Comprehensive polyadenylation site maps in yeast and human reveal pervasive alternative polyadenylation. Cell. 2010;143:1018–1029. doi: 10.1016/j.cell.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 2011;12:87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jan CH, Friedman RC, Ruby JG, et al. Formation, regulation and evolution of Caenorhabditis elegans 3≥UTRs. Nature. 2010;469:97–101. doi: 10.1038/nature09616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shepard PJ, Choi E, Lu J, et al. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA. 2011;17:761. doi: 10.1261/rna.2581711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fox-Walsh K, Davis-Turak J, Zhou Y, et al. A multiplex RNA-seq strategy to profile poly(A+) RNA: application to analysis of transcription response and 3≥ end formation. Genomics. 2011;98:266–271. doi: 10.1016/j.ygeno.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fu Y, Sun Y, Li Y, et al. Differential genome-wide profiling of tandem 3≥ UTRs among human breast cancer and normal cells by high-throughput sequencing. Genome Res. 2011;21:741–747. doi: 10.1101/gr.115295.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu YY, Machleder EM, Chenchik A, et al. Reverse transcriptase template switching: a SMART approach for full-length cDNA library construction. Biotechniques. 2001;30:892–897. doi: 10.2144/01304pf02. [DOI] [PubMed] [Google Scholar]
- 13.Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Derti A, Garrett-Engele P, Macisaac KD, et al. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012;22:1173–1183. doi: 10.1101/gr.132563.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.