

Abstract
Angiogenesis is regulated by hyperglycemic conditions, which can induce cellular stress responses, reactive oxygen species (ROS), and anti-oxidant defenses that modulate intracellular signaling to prevent oxidative damage. The RUNX2 DNA-binding transcription factor is activated by a glucose-mediated intracellular pathway, plays an important role in endothelial cell (EC) function and angiogenesis, and is a target of oxidative stress. RUNX2 DNA-binding and EC differentiation in response to glucose were conserved in ECs from different tissues and inhibited by hyperglycemia, which stimulated ROS production through the aldose reductase glucose-utilization pathway. Furthermore, the redox status of cysteine and methionine residues regulated RUNX2 DNA-binding and reversal of oxidative inhibition was consistent with an endogenous Methionine sulfoxide reductase-A (MsrA) activity. Low molecular weight MsrA substrates and sulfoxide scavengers were potent inhibitors of RUNX2 DNA binding in the absence of oxidative stress, but acted as antioxidants to increase DNA binding in the presence of oxidants. MsrA was associated with RUNX2:DNA complexes, as measured by a sensitive, quantitative DNA-binding ELISA. The related RUNX2 protein family member, RUNX1, which contains an identical DNA-binding domain, was a catalytic substrate of recombinant MsrA. These findings define novel redox pathways involving aldose reductase and MsrA that regulate RUNX2 transcription factor activity and biological function in ECs. Targeting of these pathways could result in more effective strategies to alleviate the vascular dysfunction associated with diabetes or cancer.
Keywords: Angiogenesis, Endothelial cell differentiation, Transcriptional factors, Oxidative stress, Antioxidant enzymes
Introduction
Angiogenesis contributes nutrients and energy for tumor growth and is an essential component of wound healing (Potente et al., 2011). We, and others, have shown that rodents exhibit deficits in angiogenesis with age that account for the reduced growth of tumors and poor wound healing (Pili et al., 1994; Sato, 2003). One of the hypotheses proposed to explain these reduced angiogenic responses with aging is the elevation of ROS as a consequence of impaired antioxidant defenses (Lass et al., 1998). Angiogenesis is regulated by the coordinated expression of many cytokines, receptors, and transcription factors (DNA binding or associated factors) that control the expression of genes important in vascular endothelial cell (EC) migration, invasion, proliferation, and survival (Cao, 2014). One of these transcription factors, the Runt domain family member RUNX2, has been shown by our laboratory (Sun et al., 2001) and others (Namba et al., 2000) to regulate EC migration, invasion, differentiation, proliferation, and survival. RUNX2 promotes angiogenesis through expression of angiogenic cytokines, matrix molecules, and metalloproteinase target genes (Bronckers et al., 2005; Qiao et al., 2006; Qiao et al., 2004; Sun et al., 2001; Sun et al., 2004). RUNX2 knockout mice exhibit defects in placental vessels (Komori et al., 1997), skin and hair follicle development (Glotzer et al., 2008), and fail to express VEGF in hypertrophic chondrocytes, resulting in lack of blood vessel infiltration and no bone formation (Zelzer et al., 2001). Adult RUNX2(+/−) heterozygote mice, but not young mice exhibit poor wound healing after bone marrow ablation (Juttner and Perry, 2007; Tsuji et al., 2004).
Oxidative stress in mammalian cells reflects a balance between the production of reactive oxygen species (ROS) and the activation of antioxidant pathways (Jones, 2008), which regulate angiogenesis (Brautigam et al., 2013; Sohal and Orr, 1998). ROS contribute to immune responses, act as essential physiological signaling molecules that regulate basic cell biology (Hamanaka and Chandel, 2010), and promote oxidative damage with age (Stadtman et al., 2005). Sources of ROS are numerous and include ligand/receptor activation (Sundaresan et al., 1996) and enzymatic generation (Leto and Geiszt, 2006) while ROS targets include all components of the cell including proteins, lipids, and nucleic acids (Kohen and Nyska, 2002; Wood et al., 2003). Cellular oxidant sensors and antioxidant defenses consist of DNA damage response enzymes (ribonucleotide reductases), superoxide dismutase and catalase, Keap1/Nrf1-induced activation of antioxidant genes (Kohen and Nyska, 2002), the thioredoxin/thioredoxin reductase complex, Ref1 response systems (Gius, 2004), and the protein repair enzymes Methionine sulfoxide reductases (MsrA; MsrB) (Oien and Moskovitz, 2008). The Msr enzymes not only act directly on oxidized methionine (MetO) residues to restore transcriptional activity of proteins such as p53 (Hanson et al., 2005; Nomura et al., 2009) but can also reduce overall cellular ROS by neutralizing H2O2 in the presence of sulfoxide antioxidant scavenger substrates (Weissbach et al., 2005). These enzymes play important roles in anti-oxidant defenses and oxidative stress-related neurological malfunction (Fomenko et al., 2009; Moskovitz et al., 2001; Moskovitz et al., 1995; Oien et al., 2008; Oien et al., 2010; Ortiz et al., 2011), controlling transcriptional activation (Agbas and Moskovitz, 2009), and targeting proteins for proteolytic degradation (Oien et al., 2009b; Stadtman et al., 2003). Therefore, identification of new target proteins that might be regulated by Msr antioxidant activity will be important in understanding how cellular ROS control cell signaling, transcriptional activation, enzymatic function, and aging.
RUNX2 expression is downregulated in hyperglycemic (HG) diabetic mice, which may account for the observed angiogenic dysfunction in these mice (Fowlkes et al., 2008; Lu et al., 2003). Recent findings from our laboratory revealed that RUNX2 is regulated by nutrients in the microenvironment and its DNA-binding activity is especially sensitive to glycemic status (D’Souza et al., 2009). HG inhibited, while anti-oxidants increased, RUNX2 activity and wound healing (D’Souza et al., 2009). Glucose activated RUNX2 DNA binding by increasing phosphorylation through the cdk pathway, which is less active in HG (Pierce et al., 2012). RUNX2 DNA binding may be sensitive to oxidation because of the presence of two conserved cysteine (Cys) residues in the Runt DNA binding domain that are necessary for correct protein folding and interaction with DNA (Akamatsu et al., 1997; Kurokawa et al., 1996). DNA binding is also controlled by the presence of a conserved Met residue in the Runt domain that mediates heterodimer formation with the RUNX2 binding partner, Cbfβ, which enhances DNA binding ten-fold relative to the RUNX2 monomer (Tahirov et al., 2001). However, the possible role of HG stress in regulating the redox status of Met residues in RUNX2:Cbfβ interactions has not been studied.
To test the hypothesis that HG activation of the AR pathway in ECs could lead to elevated oxidative stress and negative regulation of RUNX2, we examined EC redox status in HG and how this affects RUNX2 DNA binding. We found that HG regulated RUNX2 activation through the AR glucose utilization pathway by elevating redox stress. Maintenance of RUNX2 activity was mediated by the Met-specific MsrA system, which can restore global redox balance of Cys residues after oxidative stress and regulates the redox status of critical Met residue(s) in the DNA-binding Runt domain of RUNX2. The results from this study provide new evidence that RUNX2 activity is regulated by glycemic status and oxidative stress. This may encourage new approaches to modulate the redox status of ECs to inhibit tumor angiogenesis or alleviate the vascular dysfunction associated with diabetes (Cao et al., 2011).
Methods
Reagents
2,2-Bis(3-allyl-4-hydroxyphenyl) hexafluoropropane was prepared by dissolving hexafluorobisphenol A and K2CO3 in acetone (200 ml) and heating to reflux. After 30 min, allyl bromide was added drop wise and the resulting mixture was stirred at reflux for an additional 7 h and concentrated under reduced pressure. The remaining residue was taken up in ethyl acetate, washed with water and brine, dried over MgSO4, and concentrated under reduced pressure. BCl3 in dichloromethane was added drop wise to a solution of the crude 2,2-bis(4-allyloxyphenyl)hexafluoropropane in dry dichloromethane (100 ml) at −78 °C. The resulting mixture was allowed to reach room temperature and stirred for another 3 h. The reaction mixture was quenched with water (100 ml), extracted with chloroform, washed with brine, dried over MgSO4, and concentrated under reduced pressure. The crude product was purified by flash chromatography to generate 2,2-bis(3-allyl-4-hydroxyphenyl) hexafluoropropane as a yellow oil. For DNA-binding assays, sulindac sulfoxide or sulfide was diluted from a 100 mM stock in 100% ethanol. Control treatment with ethanol alone (<0.1%) did not affect DNA binding.
Cell culture
Human bone marrow microvascular ECs (HBME) were obtained from Dr. Ken Pienta (University of Michigan); human microvascular adipose-derived ECs (HAdEC) were prepared by Dr. John McLenithan (NORC core facility at the University of Maryland); human microvascular dermal ECs (HMEC1) were obtained from the Centers for Disease Control and Prevention, Atlanta, GA; human retroviral telomerase (HRVT) ECs were prepared in our laboratory by infecting primary human umbilical vein endothelial cells (Huvec from Cell Systems, Kirkland, WA) with a retroviral vector expressing the human telomerase gene (hTert) from Dr. R. Weinberg (Counter et al., 1998); rat pancreatic β-cells (RINr) were a generous gift from Dr. Jun Hayashi (A&G Pharmaceuticals, Columbia, MD). All cells, except HRVT ECs, were cultured in DMEM + 10%FBS (complete medium) (Qiao et al., 2006); HRVT ECs were cultured in endothelial basal media (EBM2) plus growth supplements and 2% FBS (Lonza, Gaithersburg, MD). In some experiments, cells were cultured in glucose and serum-free, phenol red-free, DMEM supplemented with pyruvate, NaHCO3 (starvation medium). Cells were routinely tested for Ac-LDL uptake (fluorescence microscopy) and CD31 expression (FACS analysis) to confirm endothelial origin.
Measurement of redox status
For ROS measurements, cells were harvested, and replated for 24 h to 48 h in defined medium containing 5 mM or 25 mM glucose prior to preparation of nuclear extracts for use in Western blots. The aldose reductase (AR) inhibitors alrestatin (Tocris Cookson, Inc. Ellisville, MO) or ranirestat (AS-3201; Eisai Limited, United Kingdom) and the ROS quencher N-acetyl Cys (NAC; Sigma-Aldrich, St. Louis, MO), were used as antioxidants. Cellular ROS was measured using the CDCF-DA reagent (Enzo Life Sciences, New York, NY; #52103). Briefly, cells were cultured in black 96-well tissue culture plates for 2 days and starved for 16 h prior to glucose or oxidant treatments. After 20 min, the plate was washed with PBS and CDCF-DA (2 μM) was added for 20 min at 37 °C. Fluorescence was measured using 485 nm/528 nm excitation/emission wavelengths. Redox status was determined by measuring the levels of NADP+, NADPH, and reduced glutathione (GSH). The NADP/NADPH Quantification Kit (Cell Technology, Inc., Mountain View, CA) was used to measure the ratio of NADP/NADPH without interference from NAD/NADH. A standard curve using NADPH was generated in a cycling reaction with reaction product detected at 450 nm. The fluorescent thiol detection kit (Cell Technology, Inc. Mountain View, CA) was used to measure GSH levels. Monitoring fluorescence emission at 515–530 nm with excitation at 488 nm generated a GSH standard curve.
Antibodies, Western blots, and IP protocol
Western blot protocols were described previously (Vitolo et al., 2007). Protein concentration was determined with the Bio-Rad Protein Assay. Specific proteins were detected by enhanced chemiluminescence (Immobilon Western, Millipore, Inc., Billerica, MA). Co-immunoprecipitation assays used the RUNX2-specific antibody (MBL D130-3, Woburn, MA) to isolate RUNX2 followed by Western blotting with the Cbfβ-specific antibody (Aviva Systems Biology, San Diego, CA). MsrA antibody (ab-16803) was from Abcam (Cambridge, MA) and anti-MetO antibody was used as described previously (Oien et al., 2009a).
EC tube formation and wound healing in response to oxidants
ECs were isolated from subconfluent growing cultures and replated on 96-well plates containing 50 μl of matrigel (BD Biosciences) per well in the presence or absence of the indicated oxidants or antioxidants. Tube formation was documented for 16 h with a Nikon TMS microscope equipped with a digital camera. Images were quantified for tube formation by scanning (n = 3–4 fields per treatment) using the generic gray gamma 2.2-profiler in the Apple ColorSync Utility (version 4.6.2) program. Mean ± SD (representing arbitrary units (AU) of cellular area) and p-values were calculated from Excel spreadsheets using Student’s t-Test. In some cases, images were quantified for tube formation by counting the number of nodes per well, with nodes defined as the intersection of at least 3 tubular structures and were expressed as mean number of nodes ± SD.
DNA-binding electrophoretic mobility shift assay (EMSA)
Electrophoretic mobility shift assays (EMSA) were used to measure RUNX2 DNA-binding activity as described previously (Qiao et al., 2006). Nuclear proteins (5 mg) isolated by the low salt/high salt method were diluted in binding buffer and incubated with a double-stranded (32P)-labeled oligonucleotide derived from the human osteocalcin promoter (−141 to −165) containing the human RUNX2 consensus binding sequence (shown in bold): 5′-CGTATTAACCACAATACTCG-3′. Specific binding complexes were resolved on DNA retardation gels (Invitrogen Corporation, Mountain View, CA).
DNA-binding enzyme-linked immunosorbent assay (D-ELISA)
DNA binding was quantified with a specific ELISA format method (Renard et al., 2001) as modified for RUNX2-specific detection (Underwood et al., 2012). Avidin-coated 96-well plates were fixed with sodium carbonate for 2 h at 24 °C. After rinsing 3× with wash buffer, a 3′-biotin labeled oligonucleotide containing 3 osteocalcin RUNX2-binding sites was added for 2 h. Wash buffer was used after each of the subsequent steps of the assay. Nuclear extracts containing RUNX2 protein were exposed to the plate with rocking for 16 h at 4 °C. Primary antibody (for RUNX2) was added for 1 h at 24 °C and a secondary antibody-HRP was allowed to react with the primary antibody for 30 min at 24 °C. RUNX2 antibody (MBL, 1:5000) or MsrA Ab (Abcam, 1:1000) or non-specific IgG (1:1000) was used. TMB substrate (50 μl) was allowed to react with the HRP in the dark. Once color development was confirmed (10–20 min), 50 μl of stop solution was added to each well. A BioTrak II plate reader spectrophotometer (Amersham Biosciences/GE Healthcare, Piscataway, NJ) was used to obtain absorbance readings (A450 nm). For continuous (kinetic) monitoring of each reaction, automated absorbance readings were determined each minute at 635 nm with equivalent results using a BIO-TEK Synergy HT (Biotek Industries, Winooski, VT) and analyzed with Gen 5 Data Analysis Software.
Recombinant RUNX1 and MsrA
Human recombinant RUNX1 (rRunx1; NM_001001890) was obtained from Origene, Inc. (Rockville, Md). This protein contains a DNA-binding Runt domain that is 96% identical to the RUNX2 DNA-binding domain. It was produced by transient transfection of the TrueORF clone, RC223854, which contains a C-terminal Myc/DDK tag in HEK293T cells. Cells were lysed 48 h after transfection in modified RIPA buffer (25 mM Tris-HCl pH7.6, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 1× proteinase inhibitor cocktail mix (Sigma), 1 mM PMSF and 1 mM Na3VO4), and recombinant protein was purified through anti-DDK affinity columns. Protein concentration was measured by BCA kit. RUNX1 protein was stored in 10% glycerol, 100 mM glycine, 25 mM Tris-HCl, and pH 7.3 at −80C. Recombinant human MsrA was obtained from Sigma-Aldrich (M8698; St. Louis, MO), was produced in Escherichia coli, and was stored in buffered aqueous solution at >8 U/mg protein where 1 Unit of recombinant protein is defined as the oxidation of 1 mmol of NADPH per minute at 37 °C at pH 7.6. For D-ELISA measurements, 0.24 μg of MsrA was used for each treatment (1.92 mU). For analysis of rRunx1 Met oxidation, 0.1 μg of MsrA was used for each treatment (0.8 mU). Western blots were used to detect Met-sulfoxide (MetO) with anti-MetO antibody (Oien et al., 2009a). Gels were stripped and reprobed with anti-MsrA antibody (Abcam ab16803).
Statistical analysis
Results for culture assays were calculated from at least three replicate samples and expressed as the mean (±SD) per data point. Western blot quantitation of scanned gels (n = 3 per lane) used the generic gray gamma 2.2 profiler in the Apple ColorSync Utility (version 4.6.2) program. Results from in vivo experiments were calculated from 4–6 data points (matrigel angiogenesis assays). To determine statistical significance, comparison of measurements relative to control samples used Student’s t-test. For comparison of multiple measurements, Tukey’s post-hoc adjustment for 2-by-2 comparisons following ANOVA was used for data analysis. p-values < 0.05 were considered significant.
Results
HG and oxidants inhibit RUNX2 DNA-binding and EC differentiation
Hyperglycemia (HG) elevates oxidative stress through the aldose reductase (AR) alternative glucose utilization pathway, which inhibits RUNX2 activity in bone marrow EC (HBME) without altering the levels of total RUNX2 protein (D’Souza et al., 2009). To determine whether this response was conserved in other cells, primary adipose (HAdEC), dermal (HMEC1), and umbilical vein (HRVT) ECs, as well as the classic glucose-responsive cell, the pancreatic β-cell (RINr) were treated with glucose after starvation to lower basal levels of RUNX2 DNA binding. Treatment with 5 mM glucose (euglycemic conditions) increased DNA binding. However, hyperglycemia (HG; 25 mM glucose) was not as effective at stimulating DNA binding in these cells (Fig. 1A). The antioxidant NAC restored RUNX2 DNA binding in HG (HBME, HAdEC, HRVT) to levels observed in 5 mM glucose, suggesting that the RUNX2 response to glucose and the anti-oxidant effect were conserved in several ECs (Fig. 1A). Several pro-oxidants are well-known inhibitors of protein tyrosine phosphatases through inactivation of a Cys residue in the enzyme active site (Miki and Funato, 2012). These oxidants (VAN and PAO) were as effective as H2O2 or curcumin at inhibiting RUNX2 DNA binding in 5 mM glucose (Fig. 1B; lanes 1–7). The anti-angiogenic agents honokiol (Fig. 1B; lane 8) and hexafluoro-(HF)-honokiol (Fig. 1B; lanes 10–13) were also potent inhibitors of RUNX2 DNA binding. This inhibition may be mediated by the AR pathway since the AR inhibitor alrestatin restored RUNX2 DNA binding in the presence of honokiol (Fig. 1B; lane 9), consistent with our previous report showing that the AR pathway mediates RUNX2 inhibition in HG (D’Souza et al., 2009).
Fig. 1.

Regulation of RUNX2 DNA binding by HG and oxidative stress. (A) Human bone marrow microvascular ECs (HBME), human adipose ECs (HAdEC), human microvascular dermal ECs (HMEC1), telomerized human umbilical vein ECs (HRVT), or rat pancreatic β-cells (RIN-r) were cultured in starvation media for 16 h and treated with 0, 5, 12.5, or 25 mM glucose for 4 h or with 10 mM of the anti-oxidant N-acetyl Cys (NAC). Nuclear extracts were prepared and RUNX2 DNA binding activity measured by EMSA with a RUNX2-specific oligonucleotide. Assays were repeated three times. Relative band intensity is shown for a representative set of results, normalized to total protein in each lane. (B) Glucose and serum-starved ECs were treated with 5 mM or 25 mM glucose or with 5 mM glucose and the oxidants curcumin, H2O2 honokiol, HF-honokiol or the phosphatase inhibitors VAN or PAO before isolation of nuclear extracts. Relative band intensities are indicated. (C) Oxidants inhibit EC tube formation. ECs were treated with 5 mM glucose or glucose + oxidants (H2O2, diquat, honokiol, hexafluoro-honokiol, IBMX) or phosphatase inhibitors (PAO, orthovanadate), for 6 h on matrigel. Essentially similar results were obtained in three other experiments.
RUNX2 regulates EC tube formation on extracellular matrix, a measure of differentiation during angiogenesis (Sun et al., 2001). Since HG and oxidants inhibited RUNX2 DNA binding (Fig. 1A, Fig. 1B), we tested whether oxidants inhibited EC tube formation. Several oxidants potently and completely inhibited EC tube formation in 5 mM glucose (Fig. 1C). To determine whether HG regulated EC differentiation, tube formation using nutrient-starved ECs on matrigel was performed. The addition of 5 mM glucose promoted tube formation, but not 25 mM glucose (Fig. 2A). Improved EC differentiation (5× increase) was observed in HG if cells were treated with the AR inhibitor ranirestat (Fig. 2A) consistent with improved EC wound healing using the related inhibitor, alrestatin, in HG (D’Souza et al., 2009). Further, treatment of cells with nM doses of ranirestat also prevented HG-induced inhibition of RUNX2 DNA-binding (Fig. 2B).
Fig. 2.
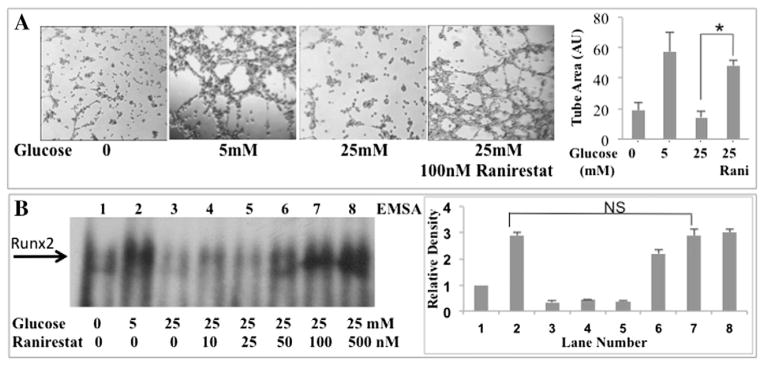
RUNX2-mediated EC differentiation is regulated through aldose reductase in HG. (A) The aldose reductase inhibitor ranirestat (100 nM) was added to ECs cultured on matrigel and tube formation was measured in starved cells (0 mM glucose) or cells treated with 5 mM or 25 mM glucose after 16 h. Bars represent mean area occupied by tubes; n = 4; *p < 0.01 (ANOVA). (B) ECs were starved for 16 h and treated for 4 h with the indicated doses of glucose. Nuclear extracts were examined for RUNX2 DNA binding after treatment of cells with 25 mM glucose and the AR-selective inhibitor ranirestat (0–500 nM). Relative band intensity normalized to total protein is shown for a representative assay (repeated three times). Ranirestat (100 nM) prevented inhibition of DNA binding in HG (NS, not significant, p > 0.05; lane 2 vs lane 7).
The AR pathway alters redox status in ECs exposed to HG
Activation of the AR pathway by HG can lead to elevated oxidative stress and the depletion of NADPH, a glutathione reductase cofactor used to generate reduced glutathione (GSH) (Brownlee, 2001). AR inhibitors can restore RUNX2 DNA binding in HG (D’Souza et al., 2009) and have been used to reduce flux through the AR pathway (Srivastava et al., 2005). To determine if HG-associated ROS was responsible for inhibition of RUNX2 activity and EC differentiation, we measured cellular ROS levels in ECs exposed to HG. The levels of total ROS were measured using the fluorescent indicator CDCF-DA, which detects both superoxide anion ( ) and hydrogen peroxide (H2O2). Cells under glucose starvation exhibited 2-fold greater ROS levels than cells that were treated with euglycemic (5 mM) levels of glucose for 4 h (Fig. 3A). However, treatment with 25 mM glucose (HG) did not lower ROS levels. Cells treated with the oxidants H2O2 or diquat in 5 mM glucose also exhibited elevated ROS while cells treated with the osmotic control sugar, mannitol, did not. Inhibition of AR with two different AR inhibitors (alrestatin or ranirestat) did not lower ROS levels in cells cultured under HG conditions. Two measures of oxidative stress in cells are the levels of reduced glutathione (GSH) and the ratio of oxidized NADP+ to reduced NADPH. Cells under oxidative stress generally exhibit high NADP:NADPH ratios (Brownlee, 2001). To determine if elevated ROS stress was dependent on the AR pathway, alrestatin (D’Souza et al., 2009) and ranirestat (Fig. 2) were used. Enzymatic assays to measure the levels of GSH, NADPH, and NADP+, confirmed that cells treated with 5 mM glucose exhibited low levels of oxidative stress (lower NADP+:NADPH) relative to starved or HG-treated (25 mM glucose) cells (Fig. 3B). Treatment with HG (25 mM glucose) did not lower the NADP+:NADPH ratio but inhibition of AR with alrestatin or ranirestat (Fig. 3B) reduced the NADP+:NADPH ratio 5 to 10-fold, suggesting that AR contributed to increased oxidative stress in HG. Starvation or glucose treatment after starvation did not have a significant impact on cellular redox status as measured by reduced glutathione (GSH) levels (Fig. 3C). However, inhibitors of AR in 25 mM glucose increased the levels of GSH by 1.5 to 2-fold.
Fig. 3.

HG regulates ROS production and redox status in ECs. (A) ECs were cultured in triplicate in the absence of glucose and serum for 16 h before treatment with 0, 5, or 25 mM glucose for 4 h. Total ROS levels were measured after incubation with CDCF-DA. In some cases, cells treated with 5 mM glucose were also treated with H2O2, diquat, or mannitol or cells were treated with AR inhibitors (alrestatin, 10 μM; ranirestat, 1 μM) in the presence of 25 mM glucose. NS = not significant. (B) NADP:NADPH ratios (triplicate determination) were measured in response to glucose and AR inhibitors (alrestatin, 10 μM; ranirestat, 1 μM) (*p < 0.05, Student’s t-test; **p < 0.05, ANOVA). (C) GSH concentrations (μM) in response to glucose or glucose + AR inhibitors were measured after treatment as in panel B (*p < 0.05, ANOVA).
Role of cysteine and methionine residues in RUNX2 DNA binding
Cysteine (Cys) and Methionine (Met) residues in proteins that are sensitive to oxidative stress regulate transcription factor DNA binding (Brash and Havre, 2002; Hanson et al., 2005). The DNA-binding Runt domain of RUNX2 contains two oxidation-sensitive Cys residues that mediate proper folding for Arg+/DNA interactions and DNA binding (Akamatsu et al., 1997; Kurokawa et al., 1996). A critical Met residue within the Runt domain also mediates association with its essential cofactor Cbfβ, which increases DNA binding 10-fold relative to Runt alone (Tahirov et al., 2001). Using a quantitative DNA-binding assay (D-ELISA) specific for RUNX2 (Underwood et al., 2012; Underwood et al., 2013), we showed that RUNX2 with a Cys mutation in the Runt domain (Cys118Ser) did not bind DNA (Underwood et al., 2012). Furthermore, a GFP-tagged Runt domain vector mutated at position Met-143 of the Runt domain (which inhibits Cbfβ binding) exhibited reduced DNA binding relative to wild type Runt domain (Underwood et al., 2012). The Cys-selective oxidant diamide (Akamatsu et al., 1997) dose-dependently inhibited endogenous RUNX2 DNA binding, which was completely reversed by addition of the reducing agent DTT at 24 °C (Fig. 4A). Treatment of nuclear extracts with H2O2, which oxidizes both Cys and Met residues (Winterbourn and Hampton, 2008), inhibited RUNX2 activity as well. This inhibition was not reversible when nuclear extracts were incubated at 24 °C for 30 min, but was reversible after incubation at 37 °C for 30 min (Fig. 4A). To further characterize this response, redox reactions were carried out for various periods of time at 37 °C after oxidation at 24 °C. EC nuclear extracts prepared without DTT exhibited an initial lower level of DNA binding relative to RUNX2 prepared with DTT (Fig. 4B). Without DTT, DNA binding declined with incubation at 37 °C. Nuclear extracts prepared with DTT in the extraction buffer maintained stable RUNX2 DNA binding when incubated at 37 °C. As expected, RUNX2 DNA binding was almost completely abolished by H2O2 but addition of DTT to the H2O2 treated nuclear extracts resulted in a rapid increase in RUNX2 DNA binding, reaching an activity equal to unoxidized samples within 20 min at 37 °C (Fig. 4C), consistent with an antioxidant and possibly enzymatic response.
Fig. 4.
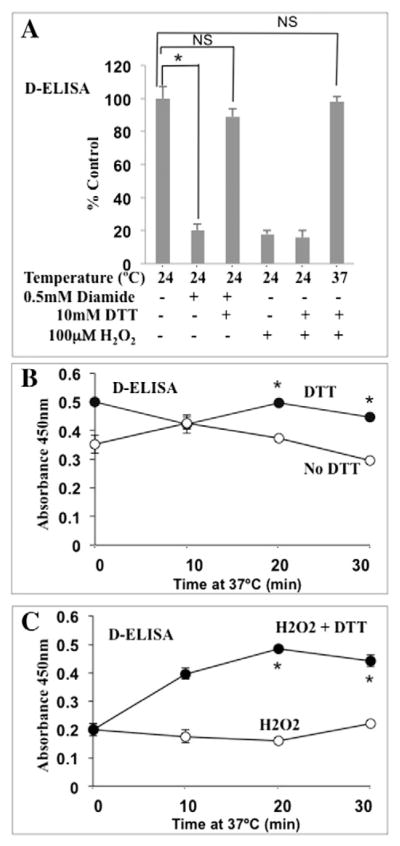
Redox regulation of RUNX2 DNA binding. (A) EC nuclear extracts were treated with diamide (15 min) to specifically oxidize Cys residues followed by reduction with DTT (15 min) at 24 °C. Diamide oxidation of Cys residues inhibited RUNX2 activity, which was reversible by DTT treatment at 24 °C (for untreated vs diamide, *p < 0.0015; for untreated vs diamide + DTT; NS, p < 0.06 m, not significant). H2O2 treatment (100 μM) inhibited DNA binding (p < 0.005), which was reversible only at 37 °C for 30 min (NS, p < 0.12; not significant relative to untreated). RUNX2-specific DNA binding was measured quantitatively by D-ELISA. (B) Comparison of RUNX2 DNA binding (triplicate determinations) in nuclear extracts incubated at 37 °C for 30 min. DTT (30 mM) prevented loss of RUNX2 activity. *p < 0.05 relative to no DTT. (C) Nuclear extracts treated with 100 μM H2O2 showed low DNA binding. Addition of DTT (30 mM) after H2O2 treatment restored DNA-binding activity within 20 min at 37 °C. *p < 0.05 relative to H2O2 alone.
Redox regulation of RUNX2 by Met-sulfoxide reductase (MsrA) substrates
Oxidized Cys residues in proteins can be reduced with endogenous (GSH) or exogenous (DTT) reducing agents. However, reduction of oxidized Met (sulfoxides) requires the Methionine sulfoxide reductase (Msr) system consisting of two genes encoding MsrA and MsrB enzymes, which can reduce the S-form or the R-form of the sulfoxide stereoisomer, respectively (Moskovitz et al., 2000; Moskovitz et al., 2002). To determine if endogenous MsrA was present in the nuclear extracts from ECs and if it was associated with RUNX2 in a DNA-binding complex, nuclear extracts were incubated with DNA oligonucleotide specific for RUNX2 and reacted with MsrA antibody. DNA binding was measured spectrophotometrically using the D-ELISA quantitative method (Fig. 5A, MsrA antibody). These nuclear extracts also contained RUNX2 (Fig. 5A, RUNX2 antibody). Nonspecific IgG was the negative control. MsrA (but not MsrB) enzymatic activity is sensitive to exogenous sulfoxide substrates, such as dimethyl sulfoxide (DMSO) or sulindac — a sulfoxide compound (Weissbach et al., 2005). DMSO inhibited RUNX2 DNA binding in a dose-dependent manner with half-maximal inhibition at 0.1% DMSO (not shown). DMSO also inhibited (by 50%) the ability of DTT to restore RUNX2 activity after H2O2 treatment (Fig. 5B). MsrA converts sulindac sulfoxide to sulindac sulfide, which is an ROS scavenger that can be oxidized to sulindac, leading to depletion of free radicals in the presence of MsrA (Weissbach et al., 2005). Treatment of RUNX2 nuclear extracts with sulindac in the absence of H2O2 (active RUNX2) resulted in a dose-dependent inhibition of RUNX2 DNA binding with significant inhibition between 1 and 100 μM (Fig. 5C, upper curves −H2O2). Sulindac sulfide was not an effective inhibitor, except at higher concentrations (500 μM). In the presence of H2O2 (low RUNX2 activity), addition of sulindac sulfoxide (10nM) restored about 50% while addition of sulindac sulfide restored about 40% of the original RUNX2 DNA-binding activity, consistent with H2O2 quenching (Fig. 5C, lower curves +H2O2).
Fig. 5.
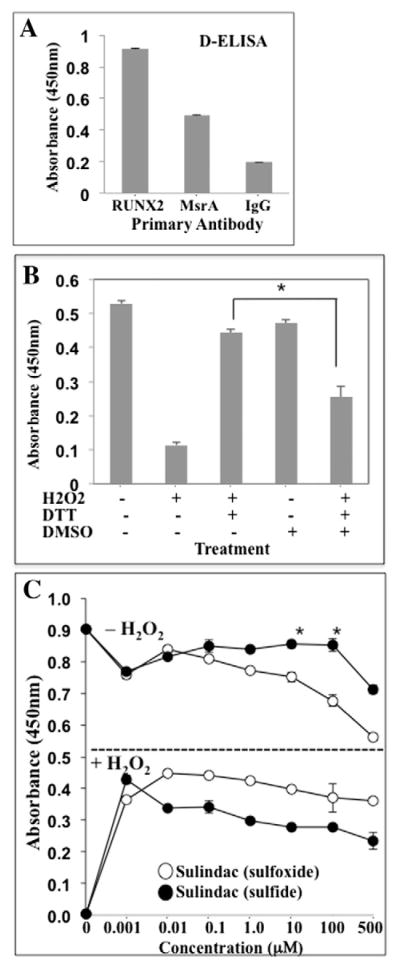
Sulfoxide regulation of RUNX2 DNA binding activity. (A) MsrA is associated with RUNX2:DNA binding complexes. DNA binding (D-ELISA) was used to quantify RUNX2 and MsrA binding to oligonucleotide containing RUNX2-binding sites. Secondary antibody-HRP reaction monitored at A450 nm as described for D-ELISA. Absorbance from secondary antibody alone was subtracted from each value. All stock antibody concentrations were 1 mg/ml. Mean ± SD calculated from triplicate samples. (B) Nuclear extracts from HBME cells were treated with H2O2 (100 μM), followed by DTT (30 mM) in the presence or absence of DMSO (0.1%). H2O2 treatment was for 30 min at RT. DMSO and DTT were added at 37 °C and incubated in a test tube for 20 min before adding to the assay plates. D-ELISA was used to quantify DNA binding. DMSO inhibited restoration of RUNX2 activity by DTT after H2O2 treatment (*p < 0.05). (C) Nuclear extracts were treated with or without H2O2 (100 μM) in the presence or absence of the indicated concentrations of sulindac (sulfoxide) or the reduced sulindac sulfide (1 nM to 500 μM). Sulindac sulfoxide inhibited RUNX2 DNA binding in the absence of DTT or H2O2 (top curves) (*p < 0.05 relative to sulindac sulfide). In the absence of DTT, sulindac sulfoxide or sulindac sulfide activated RUNX2 DNA binding after H2O2 treatment (lower curves) (p < 0.05 relative to untreated). All data points were in triplicate.
Redox regulation of RUNX2 and RUNX1 by MsrA
As a further test of MsrA-regulated RUNX2 DNA binding, EC nuclear extracts were treated with recombinant MsrA after oxidation and DNA binding was continuously monitored in a kinetic reaction (absorbance, 635 nm) as described (Underwood et al., 2012). Treatment of RUNX2 nuclear extracts with 1 μM H2O2 inhibited RUNX2 DNA binding by 50% relative to untreated samples (Fig. 6A). Addition of DTT (30 mM) and recombinant MsrA restored RUNX2 DNA binding to levels 24% higher than pretreatment (after 1 μM H2O2). Treatment with DTT and MsrA (without prior H2O2 treatment) also increased DNA binding by 24% (Fig. 6A). These samples were incubated at 37 °C, which is optimal for the MsrA reaction. Incubation of nuclear extracts at 37 °C with DTT after H2O2 treatment also restored RUNX2 activity, without the need for recombinant MsrA (Fig. 6A), consistent with the presence of MsrA in the RUNX2:DNA complexes (Fig. 5A). To detect endogenous MsrA, nuclear extracts were isolated from live cells that had been incubated with the oxidants H2O2 or honokiol and MsrA was immune-purified with an MsrA-specific antibody. Recombinant MsrA was used as a positive control (Fig. 6B, lane 1). MsrA protein (26 kDa) was present in nuclear extracts from untreated cells (Fig. 6B, lane 2). Reduced levels of MsrA were evident in nuclear extracts from honokiol-treated cells (Fig. 6B, lane 3) but not from H2O2-treated cells (Fig. 6B, lane 4) relative to untreated controls. The amount of RUNX2 protein associated with MsrA was lower by 6-fold in H2O2-treated cells (Fig. 6B, lane 4). Mutation of a specific Met residue in the Runt-homology DNA binding domain (RHD) inhibits interactions of RUNX2 with its binding partner Cbfβ and reduces DNA binding (Tahirov et al., 2001). ECs were starved and treated with 5 mM glucose or 5 mM glucose + H2O2 before isolation of nuclear extracts and immunoprecipitation with anti-RUNX2 antibody. The levels of Cbfβ associated with RUNX2 increased 2.4-fold in cells treated with 5 mM glucose, relative to untreated cells, while H2O2 prevented the glucose-mediated increase in associated Cbfβ (Fig. 6C).
Fig. 6.

MsrA-regulated oxidation of RUNX methionine residues. (A) EC nuclear extracts were treated with the indicated compounds or with recombinant MsrA. Each treatment was performed in triplicate and RUNX2 DNA binding was monitored kinetically after the addition of HRP substrate (absorbance 635 nm) in a 96-well plate. Shown are results after 32 min (significant differences were noted between 10 and 32 min of reaction). p-Values relative to untreated (t-test): 1 μM H2O2 (p < 0.005); 1 μM H2O2 + 30 mM DTT (p > 0.05; NS); 30 mM DTT + MsrA or 1 μM H2O2 + 30 mM DTT + MsrA (p < 0.05). rMsrA increased RUNX2 DNA binding in the presence of H2O2 (*p < 0.05, ANOVA). (B) MsrA association with RUNX2 by co-immunoprecipitation (IP): representative gel from three determinations. ECs were treated in vivo with honokiol (10 μM) or H2O2 (100 μM). Nuclear extracts were isolated, immunoprecipitated with MsrA-specific antibody and immunoblotted with RUNX2 or MsrA-specific antibody. Recombinant MsrA control, lane 1; untreated cells, lane 2; cells + honokiol, lane 3; cells + H2O2, lane 4. Relative density of RUNX2 (normalized to MsrA) in each lane is indicated as fold changes. (C) Live cells were starved for 16 h (0 mM glucose) and treated with glucose (5 mM) or glucose + H2O2 (100 μM). RUNX2 antibody was used for immuneprecipitation of RUNX2-associated Cbfβ cofactor. Relative density of Cbfβ (normalized to Runx2) in each lane is indicated as fold changes. (D) RUNX1 (a surrogate for RUNX2) is an MsrA substrate. Recombinant proteins rRUNX1 or rMsrA were incubated individually or together at 24 °C or 37 °C for 30 min and resolved on SDS-PAGE. Western blot with specific antibody (Ab) detects Met-sulfoxide (MetO) or MsrA. Experiment was repeated with essentially similar results. Indicated are rRunx1 (49 kDa), rRunx1 dimers (98 kDa), and rMsrA (26 kDa).
RUNX1 contains a DNA-binding Runt domain (and conserved Met residue that regulates Cbfβ binding) that is 96% identical to the RUNX2 Runt domain at the amino acid level (Blyth et al., 2005). Since recombinant RUNX2 was not available, recombinant RUNX1 (rRUNX1) was used as a surrogate to determine whether Met residues in RUNX1 could be directly oxidized to Met sulfoxide (MetO) by H2O2. rRUNX1 at 24 °C, exists as a 49 kDa monomer and a 98 kDa dimer (Fig. 6D; lane 2) while rMsrA resolved at 26 kDa (Fig. 6D; lane 3) when probed with anti-MetO antibody. Incubation of rRUNX1 with rMsrA/DTT at 24 °C resulted in the expected oxidized rRUNX1 and rMsrA species (Fig. 6D; lane 4). However, incubation of rRUNX1 with rMsrA/DTT at 37 °C resulted in reduced MetO antibody reactivity for monomeric or dimeric rRUNX1 and for rMsrA itself (Fig. 6D; lane 5). When H2O2 was included in the incubation mixture with rRUNX1 and rMsrA/DTT at 37 °C, reduction of rRUNX1 was not observed (Fig. 6D; lane 6). These results suggest that MsrA can associate with RUNX2 in EC nuclear extracts and that RUNX1 can function as an MsrA substrate.
Discussion
HG conditions contribute to vascular dysfunction, cardiovascular disease and stroke, and are associated with diabetes (Aronson, 2008; Cao, 2013; Kim et al., 2006). HG can also modulate EC redox status (Brownlee, 2001) and many cells, including ECs, adapt to oxidative stress by inducing an antioxidant response that provides the cells with an added survival advantage (Hamanaka and Chandel, 2010). Modulation of cellular ROS balance in ECs could, therefore, either “normalize” dysfunctional vessels or destabilize existing vessels to inhibit angiogenesis. Characterization of redox pathways that regulate the RUNX2 transcription factor is important in understanding vascular dysfunction associated with aging, diabetes, and cancer. Glucose levels and post-translational phosphorylation regulate RUNX2 DNA binding (Pierce et al., 2012). We have now found that euglycemic levels of glucose activated RUNX2 DNA binding and that ECs exposed to HG exhibited increased ROS and oxidative stress, which resulted in the inhibition of RUNX2 DNA-binding activity. HG increased ROS production and inhibited DNA binding through an AR alternative glucose utilization pathway. In contrast, MsrA, an enzyme that repairs protein Met residues and mediates cellular anti-oxidant defenses (Moskovitz, 2005; Stadtman et al., 2005), restored RUNX2 DNA binding under oxidative stress conditions.
HG inhibited RUNX2 DNA binding in a variety of human ECs, including immortal (HBME; HMEC1) and primary (HRVT; HAdEC) cells (Fig. 1) while the general anti-oxidant N-acetyl-cysteine (NAC) alleviated this repression. Interestingly, in HAdECs, NAC decreased basal DNA binding in the absence of glucose, which suggests that some oxidation events may lead to activation of RUNX2 DNA binding (D’Souza et al., 2009; Leto and Geiszt, 2006). HG can elevate cellular ROS in ECs and may have global effects on redox balance. We have shown that inhibition of AR with alrestatin or selective knockdown with siRNA targeting AR prevented HG-induced inhibition of RUNX2 activity and deficiencies in EC wound healing (D’Souza et al., 2009). Inhibition of AR with two different compounds (alrestatin; ranirestat) lowered cellular oxidative stress in HG and increased RUNX2 DNA binding (Figs. 2 and 3). Although AR inhibition did not appear to decrease the overall levels of ROS in 25 mM glucose, the anti-oxidant response (NADP:NADPH and GSH) was increased with AR inhibitor treatment (Fig. 3). These results suggest that HG may exert its inhibitory effects on RUNX2 and angiogenesis by lowering the overall anti-oxidant response in these cells. Therefore, blockade of glucose utilization by the AR pathway could reduce oxidative stress and alleviate the EC dysfunction associated with diseases such as diabetes, which are characterized by HG and hyperinsulinemia (Kim et al., 2006).
RUNX2 is a nuclear protein in proliferating ECs (Qiao et al., 2006) and glucose activation does not depend on new mRNA or protein synthesis (D’Souza et al., 2009). RUNX2 DNA binding was very sensitive to low doses of diamide (a Cys-selective reagent) or H2O2 (Fig. 4), which oxidizes both Cys and Met residues in proteins (Winterbourn and Hampton, 2008). Diamide treatment of the RUNX2 DNA-binding domain inhibited DNA binding activity (Akamatsu et al., 1997; Kurokawa et al., 1996). This inhibition was partially or fully reversed by treatment with the reducing agents DTT, GSH, Trx, or Ref1. We observed similar results with wild type (full-length) RUNX2 DNA binding using DTT as a reducing agent, which reversed diamide-induced oxidation and restored RUNX2 activity. H2O2-induced oxidation of RUNX2 DNA binding was not reversed by treatment with DTT at any dose when samples were incubated at 24 °C (Fig. 4). Since Met residues are oxidized by H2O2 and are not reduced by DTT alone (Oien and Moskovitz, 2008), we incubated nuclear extracts with recombinant MsrA at 37 °C. MsrA was able to restore RUNX2 activity above control levels in the presence of DTT. If MsrA were acting as a general antioxidant to neutralize H2O2, then all Cys residues would be in a reduced form and all RUNX2 activity would be restored. Nuclear extracts prepared without DTT in the extraction buffer exhibited lower RUNX2 DNA-binding activity than did extracts containing DTT, suggesting that either some RUNX2 protein was already oxidized or that MsrA was in an oxidized state. Therefore, restoration of RUNX2 activity after DTT addition in the absence of recombinant MsrA is consistent with an endogenous MsrA activity. MsrA protein was found associated with RUNX2 either in the presence of DNA (Fig. 5A) or in the absence of DNA using immunoprecipitation (Fig. 6B). Although DTT can be used as a reducing agent to regenerate active MsrA in this system, it is likely that nuclear extracts contain other components of the redox system to maintain homeostasis. For example, endogenous reducing agents such as NADPH are cofactors for thioredoxin reductase (TrxR) to regenerate thioredoxin (Trx) and activate Msr enzymes (Gius, 2004; Moskovitz et al., 2001; Stadtman et al., 2005; Weissbach et al., 2005).
Sulfoxides are low molecular weight antioxidants (LMWA) that act as scavengers of ROS (Kohen and Nyska, 2002). DMSO inhibited RUNX2 DNA binding and prevented restoration of H2O2-inhibited activity after DTT treatment (Fig. 5). Sulindac sulfoxide is an MsrA substrate that acts as a catalytic antioxidant: each cycle of sulindac reduction to sulindac sulfide, followed by oxidation of sulindac sulfide by ROS, is expected to neutralize one equivalent of ROS. Met oxidation results in the formation of equimolar quantities of two stereoisomerically unique Met sulfoxides: Met-S-sulfoxide and Met-R-sulfoxide, which are substrates of MsrA and the genetically unrelated MsrB repair enzyme, respectively (Moskovitz et al., 2000; Moskovitz et al., 2002). MsrA, which exhibits tumor suppressor activity for breast (De Luca et al., 2010) and liver (Lei et al., 2007) cancers, may function as a repair system for oxidized proteins and as a ROS scavenger in a cyclic reaction involving Met oxidation/reduction (Weissbach et al., 2005). Sulindac (sulfoxide or sulfide) is a specific substrate for MsrA but not MsrB (Weissbach et al., 2005). Since sulindac inhibited only half of RUNX2 activity and half of the activity was restored by sulindac after oxidation (Fig. 5C), the results are consistent with an MsrA-regulated RUNX2 activity where sulindac can neutralize H2O2 and restore RUNX2 activity. Noteworthy is that higher concentrations of sulindac or sulindac sulfide showed a distinct inhibition of RUNX2 DNA binding in H2O2 treated samples (Fig. 5C), consistent with inhibition of MsrA itself or other anti-oxidants that regenerate MsrA. The catalytic antioxidant ability of sulindac is limited because it is not a substrate for MsrB. However, 100% of the RUNX2 activity was restored by DTT treatment at 37 °C after oxidation with H2O2 (Fig. 4). Therefore, nuclear extracts may contain both MsrA and MsrB activity. In future, it will be important to determine whether MsrB is also present in EC nuclear extracts and whether it regulates RUNX2 activity.
The RUNX2 DNA-binding domain contains a critical Met residue, which is essential for binding of the RUNX2 cofactor Cbfβ and DNA-binding (Tahirov et al., 2001). The homologous Met in RUNX1 (Met106) is exposed on a planar, hydrophobic surface (Tahirov et al., 2001). Addition of oxygen may convert the normally flexible nonpolar side chain to a more rigid and polar species with a hydrophobicity index similar to that of lysine. Oxidized Met (MetO) has been detected in native proteins in vivo and is a modification that affects protein function (Oien and Moskovitz, 2008). Incubation of recombinant RUNX1 and MsrA in vitro resulted in reduction of MetO residues as detected with anti-MetO antibody (Fig. 6). Oxidative stress reduced the association of the RUNX2 binding partner, Cbfβ, with RUNX2. We showed that Cbfβ is present in the RUNX2:DNA binding complex (Underwood et al., 2012). Therefore, MsrA most likely is associated with a complex containing DNA, RUNX2, and its cofactor Cbfβ, consistent with a redox-regulated DNA-binding complex. There are many examples of other transcription factors whose DNA interaction and transcriptional activities are redox regulated — such as p53 and Hif1-alpha (Hanson et al., 2005; Qutub and Popel, 2008). The need for regulation of redox status in the nucleus may arise from changes in oxidative conditions, maintenance of active transcriptional complexes, or may reflect rapid modulation of signaling events to control transcriptional activity (Hamanaka and Chandel, 2010). Therefore, RUNX2 may be a novel MsrA substrate with a redox-regulatable and functional Met residue. However, MsrA may regulate RUNX2 activity globally by altering ROS balance to reverse Cys residue oxidation, which also regulates RUNX2 DNA binding.
Conclusions
Oxidative stress plays a role in age-associated pathologies and shortening of lifespan in rodents and other species (Lass et al., 1998; Stadtman et al., 2005). Compromised antioxidant defenses may account for reduced angiogenesis, vascular deficits, and neuronal dysfunction associated with aging (Moskovitz, 2005). The RUNX2 transcription factor regulates angiogenesis by activating specific target genes that control EC invasion, migration, and survival (Pierce et al., 2012; Qiao et al., 2006; Sun et al., 2001; Sun et al., 2004). Our results show that (1) HG and oxidative stress inhibited RUNX2 DNA binding and EC differentiation, (2) the aldose reductase pathway mediated inhibition of RUNX2 and EC differentiation in HG, (3) substrates of Methionine sulfoxide reductase-A regulated RUNX2 DNA binding, and (4) Methionine sulfoxide reductase-A and RUNX proteins interacted to regulate oxidative damage. Further study of the novel redox pathways regulating this important transcription factor could lead to strategies to control EC redox balance, improve vessel normalization or inhibit angiogenesis (Cao and Langer, 2010).
Acknowledgments
We thank Rod Junor (Pain Clinical Section Head & Ranirestat IPTL; Eisai Product Creation Systems; Eisai Limited; United Kingdom) for providing ranirestat. We also thank Michael Quon, Anil Jaiswal, and Mark Williams for critical discussions in the preparation of the manuscript. The support of the Biology Core of the Mid-Atlantic Nutrition Obesity Research Center (NORC), Alan Shuldiner, Director, is gratefully appreciated. This study was supported in part by a pilot grant from the Mid-Atlantic Nutrition Obesity Research Center (NORC)–NIH Grant P30 DK072488 (A. Shuldiner, P.I.) to A.P., NIH RO1 CA108846 (A.P.), AHA Grant-in-Aid GRNT2130014 (A.P.), and by the University of Maryland Cancer Restitution Funds (CRF) provided to A.P. by the Marlene & Stewart Greenebaum Cancer Center at the University of Maryland. This paper is subject to the NIH Public Access Policy.
Abbreviations
- AR
aldose reductase
- D-ELISA
DNA-based enzyme-linked immunosorbent assay
- DTT
dithiothreitol
- EMSA
electrophoretic mobility shift assay
- HAdEC
human microvascular adipose-derived ECs (HAdEC)
- HBME
human bone marrow microvascular ECs
- HG
hyperglycemia
- HMEC1
human microvascular dermal ECs (HMEC1)
- HRVT
Huvec retroviral telomerase
- MetO
methionine sulfoxide
- MsrA
Methionine sulfoxide reductase A
- NAC
N-acetyl cysteine
- PAO
phenyl arsine oxide
- RIN-r
rat pancreatic β-cells
- ROS
reactive oxygen species
- RUNX2
human Runt-related transcription factor
- VAN
vanadate
Footnotes
Authorship and disclosures
M.T.M., K.F.U., B.C., A.P., and C.N. performed the experiments; A.P. conceived and designed the experiments; A.P., M.T.M., and K.F.U. analyzed data; M.T.M. and A.P. interpreted experimental results; M.T.M., K.F.U., B.C., and A.P. prepared figures; M.T.M. and A.P. drafted the manuscript; J.A., R.A.M., A.P., J.C.M., and J.M. provided scientific input and reagents, edited the manuscript, and approved the final version of the manuscript. The authors declare no conflicts of interest, financial or otherwise.
References
- Agbas A, Moskovitz J. The role of methionine oxidation/reduction in the regulation of immune response. Curr Signal Transduct Ther. 2009;4:46–50. doi: 10.2174/157436209787048748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akamatsu Y, et al. A simple screening for mutant DNA binding proteins: application to murine transcription factor PEBP2alpha subunit, a founding member of the Runt domain protein family. Gene. 1997;185:111–117. doi: 10.1016/s0378-1119(96)00644-0. [DOI] [PubMed] [Google Scholar]
- Aronson D. Hyperglycemia and the pathobiology of diabetic complications. Adv Cardiol. 2008;45:1–16. doi: 10.1159/000115118. [DOI] [PubMed] [Google Scholar]
- Blyth K, et al. The RUNX genes: gain or loss of function in cancer. Nat Rev Cancer. 2005;5:376–387. doi: 10.1038/nrc1607. [DOI] [PubMed] [Google Scholar]
- Brash DE, Havre PA. New careers for antioxidants. Proc Natl Acad Sci U S A. 2002;99:13969–13971. doi: 10.1073/pnas.232574399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brautigam L, et al. Glutaredoxin regulates vascular development by reversible glutathionylation of sirtuin 1. Proc Natl Acad Sci U S A. 2013;110:20057–20062. doi: 10.1073/pnas.1313753110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronckers AL, et al. Expression of Runx2/Cbfa1/Pebp2alphaA during angiogenesis in postnatal rodent and fetal human orofacial tissues. J Bone Miner Res. 2005;20:428–437. doi: 10.1359/JBMR.041118. [DOI] [PubMed] [Google Scholar]
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- Cao Y. Angiogenesis and vascular functions in modulation of obesity, adipose metabolism, and insulin sensitivity. Cell Metab. 2013;18:478–489. doi: 10.1016/j.cmet.2013.08.008. [DOI] [PubMed] [Google Scholar]
- Cao Y. VEGF-targeted cancer therapeutics-paradoxical effects in endocrine organs. Nat Rev Endocrinol. 2014;10:530–539. doi: 10.1038/nrendo.2014.114. [DOI] [PubMed] [Google Scholar]
- Cao Y, Langer R. Optimizing the delivery of cancer drugs that block angiogenesis. Sci Transl Med. 2010;2:15 ps3. doi: 10.1126/scitranslmed.3000399. [DOI] [PubMed] [Google Scholar]
- Cao Y, et al. Forty-year journey of angiogenesis translational research. Sci Transl Med. 2011;3:114rv3. doi: 10.1126/scitranslmed.3003149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counter CM, et al. Telomerase activity is restored in human cells by ectopic expression of hTERT (hEST2), the catalytic subunit of telomerase. Oncogene. 1998;16:1217–1222. doi: 10.1038/sj.onc.1201882. [DOI] [PubMed] [Google Scholar]
- De Luca A, et al. Methionine sulfoxide reductase A down-regulation in human breast cancer cells results in a more aggressive phenotype. Proc Natl Acad Sci U S A. 2010;107:18628–18633. doi: 10.1073/pnas.1010171107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza DR, et al. Hyperglycemia regulates RUNX2 activation and cellular wound healing through the aldose reductase polyol pathway. J Biol Chem. 2009;284:17947–17955. doi: 10.1074/jbc.M109.002378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fomenko DE, et al. MsrB1 (methionine-R-sulfoxide reductase 1) knock-out mice: roles of MsrB1 in redox regulation and identification of a novel selenoprotein form. J Biol Chem. 2009;284:5986–5993. doi: 10.1074/jbc.M805770200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowlkes JL, et al. Runt-related transcription factor 2 (RUNX2) and RUNX2-related osteogenic genes are down-regulated throughout osteogenesis in type 1 diabetes mellitus. Endocrinology. 2008;149:1697–1704. doi: 10.1210/en.2007-1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gius D. Redox-sensitive signaling factors and antioxidants: how tumor cells respond to ionizing radiation. J Nutr. 2004;134:3213S–3214S. doi: 10.1093/jn/134.11.3213S. [DOI] [PubMed] [Google Scholar]
- Glotzer DJ, et al. Impaired skin and hair follicle development in Runx2 deficient mice. Dev Biol. 2008;315:459–473. doi: 10.1016/j.ydbio.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010;35:505–513. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson S, et al. Redox factor 1 (Ref-1) enhances specific DNA binding of p53 by promoting p53 tetramerization. Oncogene. 2005;24:1641–1647. doi: 10.1038/sj.onc.1208351. [DOI] [PubMed] [Google Scholar]
- Jones DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol. 2008;295:C849–C868. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juttner KV, Perry MJ. High-dose estrogen-induced osteogenesis is decreased in aged RUNX2(+/−) mice. Bone. 2007;41:25–32. doi: 10.1016/j.bone.2007.03.012. [DOI] [PubMed] [Google Scholar]
- Kim JA, et al. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–1904. doi: 10.1161/CIRCULATIONAHA.105.563213. [DOI] [PubMed] [Google Scholar]
- Kohen R, Nyska A. Oxidation of biological systems: oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol Pathol. 2002;30:620–650. doi: 10.1080/01926230290166724. [DOI] [PubMed] [Google Scholar]
- Komori T, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts [see comments] Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- Kurokawa M, et al. A conserved cysteine residue in the runt homology domain of AML1 is required for the DNA binding ability and the transforming activity on fibroblasts. J Biol Chem. 1996;271:16870–16876. doi: 10.1074/jbc.271.28.16870. [DOI] [PubMed] [Google Scholar]
- Lass A, et al. Caloric restriction prevents age-associated accrual of oxidative damage to mouse skeletal muscle mitochondria. Free Radic Biol Med. 1998;25:1089–1097. doi: 10.1016/s0891-5849(98)00144-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei KF, et al. Identification of MSRA gene on chromosome 8p as a candidate metastasis suppressor for human hepatitis B virus-positive hepatocellular carcinoma. BMC Cancer. 2007;7:172. doi: 10.1186/1471-2407-7-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leto TL, Geiszt M. Role of Nox family NADPH oxidases in host defense. Antioxid Redox Signal. 2006;8:1549–1561. doi: 10.1089/ars.2006.8.1549. [DOI] [PubMed] [Google Scholar]
- Lu H, et al. Diabetes interferes with the bone formation by affecting the expression of transcription factors that regulate osteoblast differentiation. Endocrinology. 2003;144:346–352. doi: 10.1210/en.2002-220072. [DOI] [PubMed] [Google Scholar]
- Miki H, Funato Y. Regulation of intracellular signalling through cysteine oxidation by reactive oxygen species. J Biochem. 2012;151:255–261. doi: 10.1093/jb/mvs006. [DOI] [PubMed] [Google Scholar]
- Moskovitz J. Methionine sulfoxide reductases: ubiquitous enzymes involved in antioxidant defense, protein regulation, and prevention of aging-associated diseases. Biochim Biophys Acta. 2005;1703:213–219. doi: 10.1016/j.bbapap.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Moskovitz J, et al. Escherichia coli peptide methionine sulfoxide reductase gene: regulation of expression and role in protecting against oxidative damage. J Bacteriol. 1995;177:502–507. doi: 10.1128/jb.177.3.502-507.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskovitz J, et al. Identification and characterization of a putative active site for peptide methionine sulfoxide reductase (MsrA) and its substrate stereospecificity. J Biol Chem. 2000;275:14167–14172. doi: 10.1074/jbc.275.19.14167. [DOI] [PubMed] [Google Scholar]
- Moskovitz J, et al. Methionine sulfoxide reductase (MsrA) is a regulator of antioxidant defense and lifespan in mammals. Proc Natl Acad Sci U S A. 2001;98:12920–12925. doi: 10.1073/pnas.231472998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskovitz J, et al. Purification and characterization of methionine sulfoxide reductases from mouse and Staphylococcus aureus and their substrate stereospecificity. Biochem Biophys Res Commun. 2002;290:62–65. doi: 10.1006/bbrc.2001.6171. [DOI] [PubMed] [Google Scholar]
- Namba K, et al. Indispensable role of the transcription factor PEBP2/CBF in angiogenic activity of a murine endothelial cell MSS31. Oncogene. 2000;19:106–114. doi: 10.1038/sj.onc.1203257. [DOI] [PubMed] [Google Scholar]
- Nomura T, et al. Oxidation of methionine residue at hydrophobic core destabilizes p53 tetrameric structure. Biopolymers. 2009;91:78–84. doi: 10.1002/bip.21084. [DOI] [PubMed] [Google Scholar]
- Oien DB, Moskovitz J. Substrates of the methionine sulfoxide reductase system and their physiological relevance. Curr Top Dev Biol. 2008;80:93–133. doi: 10.1016/S0070-2153(07)80003-2. [DOI] [PubMed] [Google Scholar]
- Oien DB, et al. MsrA knockout mouse exhibits abnormal behavior and brain dopamine levels. Free Radic Biol Med. 2008;45:193–200. doi: 10.1016/j.freeradbiomed.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oien DB, et al. Detection of oxidized methionine in selected proteins, cellular extracts and blood serums by novel anti-methionine sulfoxide antibodies. Arch Biochem Biophys. 2009a;485:35–40. doi: 10.1016/j.abb.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oien DB, et al. Clearance and phosphorylation of alpha-synuclein are inhibited in methionine sulfoxide reductase a null yeast cells. J Mol Neurosci. 2009b;39:323–332. doi: 10.1007/s12031-009-9274-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oien DB, et al. Caloric restriction alleviates abnormal locomotor activity and dopamine levels in the brain of the methionine sulfoxide reductase A knockout mouse. Neurosci Lett. 2010;468:38–41. doi: 10.1016/j.neulet.2009.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz AN, et al. Quantification of reserve pool dopamine in methionine sulfoxide reductase A null mice. Neuroscience. 2011;177:223–229. doi: 10.1016/j.neuroscience.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce AD, et al. Glucose-activated RUNX2 phosphorylation promotes endothelial cell proliferation and an angiogenic phenotype. J Cell Biochem. 2012;113:218–292. doi: 10.1002/jcb.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pili R, et al. Altered angiogenesis underlying age-dependent changes in tumor growth. J Natl Cancer Inst. 1994;86:1303–1314. doi: 10.1093/jnci/86.17.1303. [DOI] [PubMed] [Google Scholar]
- Potente M, et al. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873–887. doi: 10.1016/j.cell.2011.08.039. [DOI] [PubMed] [Google Scholar]
- Qiao M, et al. Insulin-like growth factor-1 regulates endogenous RUNX2 activity in endothelial cells through a PI3K/ERK-dependent and Akt-independent signaling pathway. J Biol Chem. 2004;279:42709–42718. doi: 10.1074/jbc.M404480200. [DOI] [PubMed] [Google Scholar]
- Qiao M, et al. Cell cycle-dependent phosphorylation of the RUNX2 transcription factor by cdc2 regulates endothelial cell proliferation. J Biol Chem. 2006;281:7118–7128. doi: 10.1074/jbc.M508162200. [DOI] [PubMed] [Google Scholar]
- Qutub AA, Popel AS. Reactive oxygen species regulate hypoxia-inducible factor 1alpha differentially in cancer and ischemia. Mol Cell Biol. 2008;28:5106–5119. doi: 10.1128/MCB.00060-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renard P, et al. Development of a sensitive multi-well colorimetric assay for active NFkappaB. Nucleic Acids Res. 2001;29:E21. doi: 10.1093/nar/29.4.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y. Molecular diagnosis of tumor angiogenesis and anti-angiogenic cancer therapy. Int J Clin Oncol. 2003;8:200–206. doi: 10.1007/s10147-003-0342-8. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Orr WC. Role of oxidative stress in senescence. Aging (Milano) 1998;10:149–151. [PubMed] [Google Scholar]
- Srivastava SK, et al. Role of aldose reductase and oxidative damage in diabetes and the consequent potential for therapeutic options. Endocr Rev. 2005;26:380–392. doi: 10.1210/er.2004-0028. [DOI] [PubMed] [Google Scholar]
- Stadtman ER, et al. Oxidation of methionine residues of proteins: biological consequences. Antioxid Redox Signal. 2003;5:577–582. doi: 10.1089/152308603770310239. [DOI] [PubMed] [Google Scholar]
- Stadtman ER, et al. Methionine oxidation and aging. Biochim Biophys Acta. 2005;1703:135–140. doi: 10.1016/j.bbapap.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Sun L, et al. Runt-related gene 2 in endothelial cells: inducible expression and specific regulation of cell migration and invasion. Cancer Res. 2001;61:4994–5001. [PubMed] [Google Scholar]
- Sun L, et al. Regulation of TGFβ1-mediated growth inhibition and apoptosis by RUNX2 isoforms. Oncogene. 2004;23:4722–4734. doi: 10.1038/sj.onc.1207589. [DOI] [PubMed] [Google Scholar]
- Sundaresan M, et al. Regulation of reactive-oxygen-species generation in fibroblasts by Rac1. Biochem J. 1996;318(Pt 2):379–382. doi: 10.1042/bj3180379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahirov TH, et al. Structural analyses of DNA recognition by the AML1/Runx-1 Runt domain and its allosteric control by CBFbeta. Cell. 2001;104:755–767. doi: 10.1016/s0092-8674(01)00271-9. [DOI] [PubMed] [Google Scholar]
- Tsuji K, et al. Aged mice require full transcription factor, Runx2/Cbfa1, gene dosage for cancellous bone regeneration after bone marrow ablation. J Bone Miner Res. 2004;19:1481–1489. doi: 10.1359/JBMR.040601. [DOI] [PubMed] [Google Scholar]
- Underwood KF, et al. Regulation of RUNX2 transcription factor-DNA interactions and cell proliferation by Vitamin D3 (cholecalciferol) prohormone activity. J Bone Miner Res. 2012;27:913–925. doi: 10.1002/jbmr.1504. [DOI] [PubMed] [Google Scholar]
- Underwood KF, et al. A quantitative assay to study protein:DNA interactions, discover transcriptional regulators of gene expression, and identify novel anti-tumor agents. J Vis Exp. 2013 Aug 31;(78) doi: 10.3791/50512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitolo MI, et al. The RUNX2 transcription factor cooperates with the YES-associated protein, YAP65, to promote cell transformation. Cancer Biol Ther. 2007;6:856–863. doi: 10.4161/cbt.6.6.4241. [DOI] [PubMed] [Google Scholar]
- Weissbach H, et al. Methionine sulfoxide reductases: history and cellular role in protecting against oxidative damage. Biochim Biophys Acta. 2005;1703:203–212. doi: 10.1016/j.bbapap.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Wood LG, et al. Biomarkers of lipid peroxidation, airway inflammation and asthma. Eur Respir J. 2003;21:177–186. doi: 10.1183/09031936.03.00017003a. [DOI] [PubMed] [Google Scholar]
- Zelzer E, et al. Tissue specific regulation of VEGF expression during bone development requires Cbfa1/Runx2. Mech Dev. 2001;106:97–106. doi: 10.1016/s0925-4773(01)00428-2. [DOI] [PubMed] [Google Scholar]