

Abstract
Biomaterials for bone tissue engineering must be able to instruct cell behavior in the presence of the complex biophysical and biomolecular environments encountered in vivo. While soluble supplementation strategies have been identified to enhance osteogenesis, they are subject to significant diffusive loss in vivo or the need for frequent re-addition in vitro. This investigation therefore explored whether biophysical and biochemical properties of a mineralized collagen-GAG scaffold were sufficient to enhance human mesenchymal stem cell (hMSC) osteogenic differentiation and matrix remodeling in the absence of supplementation. We examined hMSC metabolic health, osteogenic and matrix gene expression profiles, as well as matrix remodeling and mineral formation as a function of scaffold mineral content. We found that scaffold mineral content enhanced long term hMSC metabolic activity relative to non-mineralized scaffolds. While osteogenic supplementation or exogenous BMP-2 could enhance some markers of hMSC osteogenesis in the mineralized scaffold, we found the mineralized scaffold was itself sufficient to induce osteogenic gene expression, matrix remodeling, and mineral formation. Given significant potential for unintended consequences with the use of mixed media formulations and potential for diffusive loss in vivo, these findings will inform the design of instructive biomaterials for regenerative repair of critical-sized bone defects, as well as for applications where non-uniform responses are required, such as in biomaterials to address spatially-graded interfaces between orthopedic tissues.
Introduction
Critical-sized bone defects present unique challenges for tissue engineering. Large in size and often irregular in shape, these defects often occur as a result of acute trauma or surgical resection, and can be marked by significant potential for infection as well as severe functional deficits.1–4 Allogenic or autogenic bone repair remain the current gold-standard.5–7 However, concerns remain about the resultant secondary wound site, supply and disease transmission, as well as the need for shaping of donor bone to fit often complex defects. With world demand topping two million bone replacement procedures per year,8, 9 tissue engineering approaches offer the tantalizing possibility of generating a mechanically robust, patient-customized implant to enhance bone regeneration. However, the development of appropriate biomaterial substrates requires balancing structural, mechanical, and biomolecular design concerns as well as considering the appropriate selection of clinically-relevant cell populations.10–13
Given the well-described composition of bone, many tissue engineering approaches have explored mineral constructs consisting entirely or in part of a calcium phosphate (CaP),14, 15 polymeric (synthetic or natural) scaffolds,16, 17 or, increasingly, polymer-mineral composites.9, 18–20 Such biomaterial substrates offer the advantages of tunability, namely allowing the manipulation of biomaterial morphology, such as fiber alignment;21 topology, like surface roughness;22 and substrate stiffness.23, 24 However, the inclusion of growth factors such as BMP-2 or BMP-7 (known commercially as OP-1)25, 26 or full-biochemical supplementation strategies such as osteogenic media14, 27–29 remain a common, if not ubiquitous, practice of soluble supplementation. Such strategies are particularly common in approaches utilizing adipose or marrow derived mesenchymal stem cells (MSCs).25, 26, 28, 30 However, soluble supplementation strategies – defined here as addition of an exogenous factor to the media – introduce two major concerns. The first is the need for repeated supplementation due to short biomolecule half-lives and diffusive loss of the added factor away from the biomaterial,31 a requirement that introduces expense as well as significantly complexity for clinical applications. Second, soluble supplementation methods limit the potential for inducing spatially-selective responses, such as for applications looking to repair orthopedic interfaces between hard and soft tissues. Soluble supplementation also presents a number of concerns for clinical translation. Notably, while BMP-2 has received FDA approval for use, the current need of supra-physiological doses, frequent need for repeated doses due to diffusive loss, and extensive off-label use has raised a range of safety concerns.9
Our laboratory has recently developed a series of collagen-glycosaminoglycan (CG) scaffolds for musculoskeletal tissue engineering applications. These efforts have included optimization of scaffold pore structure (porosity and pore anisotropy),32, 33 soluble factor cocktails to balance proliferation and phenotypic stability,34, 35 glycosaminoglycan (GAG) content to mediate transient sequestration of growth factors, and structural reinforcement via the incorporation of CG membranes structures.36 Recently we described a calcium phosphate mineralized CG (CGCaP) scaffold with potential application for bone tissue engineering and as a component of an osteotendinous repair biomaterial.33 In this formulation, CaP crystallites are precipitated with collagen and GAG into a precursor solution which is subsequently lyophilized to form the final CGCaP scaffold.37 While we have previously reported the impact of addition of a CaP mineral phase on scaffold biophysical properties (mechanics, permeability),33 we had not explored the osteogenic potential of the scaffold itself, notably the potential to avoid the need for osteogenic or BMP-2 media supplementation. Here, the CGCaP scaffold design offers two potential routes to impact MSC osteogenesis. First, given the known impact of substrate stiffness on MSC osteogenesis,23 the increased stiffness of CGCaP (vs. non-mineralized CG) scaffolds has the potential to impact MSC osteogenesis.33 Second, the precipitation-based method for introducing CaP into the scaffold offers the potential for long term release of Ca and P ions during cell culture, which have previously been shown to impact osteoblast bioactivity.14, 15, 38, 39
This study therefore examines the potential for a mineralized collagen-GAG scaffold platform to bias osteogenic differentiation of human bone marrow derived MSCs in the absence of conventional media supplementation. We compared the metabolic health and osteogenic potential of hMSCs in two scaffold groups, a non-mineralized CG scaffold control and a 40wt% CaP mineralized CGCaP scaffold in the presence of conventional growth media. We also examined hMSC osteogenic capacity in mineralized CGCaP scaffolds in the presence of conventional osteogenic media and BMP-2 supplemented growth media to determine whether addition of such factors enhanced osteogenesis. MSCs were maintained in scaffolds for 8 weeks in culture, with osteogenic differentiation assessed via periodic assessment of metabolic activity, gene expression, and new matrix synthesis (histology, microCT, mechanics) metrics. Using this approach we asked whether scaffold mineral content was sufficient to promote hMSC osteogenesis.
B. Materials and Methods
B.1. Fabrication of non-mineralized and mineralized scaffolds
Non-mineralized (CG) and mineralized (CGCaP) scaffolds were fabricated via lyophilization from non-mineralized and mineralized CG precursor suspensions. Briefly, the non-mineralized precursor suspension was created by homogenizing type I collagen from bovine Achilles tendon (0.5 w/v%; Sigma-Aldrich, St. Louis, MO) and chondroitin sulfate from shark cartilage (GAG; 0.04 w/v%, Sigma-Aldrich, St. Louis, MO) in acetic acid (Sigma-Aldrich) at a collagen:GAG ratio of 11.25:1.33, 40 The mineralized suspension was fabricated from collagen (1.9 w/v%) and GAG (0.84 w/v%) as before along with calcium salts (0.9 w/v% Ca(OH)2, 0.4 w/v% Ca(NO3)2·4H2O, Sigma-Aldrich) in phosphoric acid.33 Precursor suspensions were stored at 4°C and degassed prior to use. Lyophilization was performed in a Genesis freeze-dryer (VirTis, Gardener, NY) using a custom 3″ × 3″ polysulfone trays. Briefly, the suspensions were solidified via cooling at a constant cooling rate of 1°C/min to a final freezing temperature of −10°C.41 After allowing 2 h at the final freezing temperature to complete solidification, the frozen suspension was then sublimated at 0°C and 200 mTorr. CG scaffold variants were then lightly crosslinked using a dehydrothermal treatment at <25 Torr and 105°C for 24 h in a vacuum oven (Welch, Niles, IL).42 Cylindrical CG and CGCaP scaffold specimens were then cut from the resulting 4 mm thick sheets using an 8 mm biopsy punch (Integra-Miltex, York, PA). Unless otherwise noted, all scaffolds were then sterilized in ethanol for 1 h, hydrated in PBS overnight, crosslinked in a solution of 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC) and N-hydroxysulfosuccinimide (NHS) at a molar ratio of 5:2:1 EDC:NHS:COOH where COOH represents the amount of collagen in the scaffold, then washed prior to use.42, 43
B.2. Calcium and phosphate ion release from mineralized collagen scaffolds
Ca and P ion release profiles from the CGCaP scaffolds were determined via Calcium Colorimetric and Phosphate Colorimetric Assay Kits (BioVision, Milpitas, CA). Acellular CG and CGCaP scaffolds were cultured in complete mesenchymal stem cell growth media (Lonza, Walkersville, MD) at 37°C and 5% CO2 for up to 8 weeks. Media was changed regularly, with the removed media (n = 6 specimens per timepoint) stored at 4°C for analysis. All media specimens were then analyzed together. Media aliquots from each time point were analyzed separately via a Tecan M200 fluorometer (Tecan, Männedorf, Switzerland) and compared to series of prepared calcium and phosphate standards to compute total calcium and phosphate concentration. Media cultured with non-mineralized CG scaffolds was used as a negative control.
B.3. Human mesenchymal stem cell culture
Human mesenchymal stem cells (hMSCs, Lonza) were expanded in standard flasks in complete mesenchymal stem cell growth media (Low glucose Dulbecco’s Modified Eagle Medium; 10% MSC Fetal bovine serum; 1% Penicillin-Streptomycin; 1% L-glutamine; Invitrogen, Carlsbad, CA) at 37°C and 5% CO2. After expansion to passage 6, hMSCs were seeded onto 8 mm diameter (4 mm thick) scaffold specimens. A total of 7.5 ×104 cells were seeded onto the scaffold disc via a previously described static seeding method.32, 41 MSC seeded scaffolds were subsequently cultured at 37°C and 5% CO2 for up to 56 days. CGCaP scaffolds were cultured in one of three media formulations throughout: (1) complete MSC growth media (CGCaP Growth); (2) complete MSC growth media supplemented with 100 ng/mL BMP2 (CGCaP BMP2); or (3), complete MSC osteogenic media (CGCaP Osteo). Human recombinant BMP-2 was obtained from ProSpec (Israel). Osteogenic media contained 50μM ascorbic acid, 0.1μM Dexamethasone, and 10mM β-glycerophosphate added to growth media. MSC-seeded CG scaffolds were cultured in standard MSC growth media (CG Growth) as a control. The chosen dose of BMP-2 (100ng/mL) was based on previous studies by our group that documented a pro-osteogenic effect in CG scaffolds.32, 34, 44 All cell seeded scaffold groups were fed their respective media every 3 days.
B.4. Quantification of hMSC metabolic activity
The mitochondrial metabolic activity of MSCs seeded within the CG and CGCaP scaffolds was quantified using the alamarBlue® assay.45 Briefly, cell-seeded scaffolds were washed in PBS to remove non-attached and dead cells. Cell-seeded scaffolds were incubated in alamarBlue® solution (Invitrogen) for 105 min at 37°C under mild shaking. The reduction of resazurin to the fluorescent byproduct resorufin by metabolically active cells was measured on a F200 spectrophotometer (Tecan) at 540(52)/580(20) nm (excitation/emission). Results were compared to a prepared standard to compute equivalent cell number. Results (n = 6 per timepoint) were reported as the relative metabolic activity normalized to the number of originally seeded cells.
B.5. hMSC gene expression profiles
The gene expression of osteogenic and matrix synthesis markers was determined via PCR using previously described methods 46. Briefly, RNA was isolated from scaffolds using 1% β-mercaptoethanol lysis solution and the RNeasy Plant Mini Kit (Qiagen, Valencia, CA). RNA was then reverse transcribed into cDNA using a Bio-Rad S1000 thermal cycler (BioRad, Hercules, CA) via QuantiTect Reverse Transcription Kit (Qiagen). A QuantiTect SYBR Green PCR Kit (Qiagen) and an Applied Biosystems 7900HT Fast Real-Time PCR System (Carlsbad, CA) were used to perform real time PCR. Gene expression profiles were obtained for Collagen 1 (COL1A1; matrix related marker), BSP (bone sialoprotein), OPN (osteopontin), and RUNX2 (runt-related transcription factor 2). GAPDH (glyceraldehyde 3-phosphate dehydrogenase) was used as a housekeeping gene. Previously validated primer sequences were chosen from the literature (Supp. Table 1) then synthesized by Integrated DNA Technologies (Coraville, IA). All markers were quantified after 14, 28, and 56 days in culture and analyzed using Sequence Detection Systems software v2.4 (Applied Biosystems, Carlsbad, CA) via the delta-delta Ct method. Results were expressed as fold changes normalized to MSCs cultured in the non-mineralized CG scaffolds at day 14.
B.6. Mechanical behavior of hMSC seeded scaffolds
The elastic modulus of hMSC-seeded CG and CGCaP scaffolds was determined after 14, 28, and 56 days in culture via hydrated unconfined compression using a TA.XTplus Texture Analyzer (StableMicro Systems Ltd., Surrey, UK). Briefly, samples were compressed to 50% strain at a rate of 0.05 % strain/s to capture the linear elastic response of the scaffold. Cell-seeded scaffolds behaved as low-density open cell foams, with elastic moduli obtained from the linear elastic regime of the resulting stress-strain plots.42, 47 The estimated densification (ρcells/ρacellular) of each scaffold was subsequently calculated from differences in hMSC-seeded vs. unseeded scaffold moduli at each time point (Supp. Table 2). Here, an established cellular solids theory method that relates changes in scaffold elastic modulus to changes in scaffold density was used to determine the relative change in scaffold densification through cellular remodeling (ρcells) as compared to the acellular scaffold control (ρacellular) based on changes in elastic modulus (Ecells and Eacellular respectively): Ecells / Eacellular = (ρcells / ρacellular)2.42, 47, 48
B.7. Histology
Histological assessment of scaffold mineral content was performed via Alizarin red. Scaffold specimens were obtained post fabrication (day 0, control) as well as after 14, 28, and 56 days in culture. Scaffolds were fixed in 10% formalin (Polyscience). Scaffolds were then embedded in paraffin; 5 μm thick transverse sections were then stained with Alizarin Red (Sigma-Aldrich). Representative sections of each scaffold were captured using an optical microscope (Leica; Buffalo Grove, IL). Histograms of average red pixel intensity calculated via Matlab for each scaffold group and time point.
B.8. Quantification of mineral content and distribution in hMSC-seeded scaffolds
The mineral content of CG and CGCaP scaffolds was assessed post-fabrication (day 0) via micro-computed tomography (microCT). Additionally, hMSC-seeded scaffold specimens were removed from culture at days 14, 28, and 56 for microCT analysis. Scaffolds were stored in 10% formalin (Polysciences Inc., Warrington, PA) at 4°C prior to analysis. Subsequently, scaffolds were washed in de-ionized water to remove the formalin then re-lyophilized to obtain a dry scaffold specimen. Mineral content and distribution was assessed from microCT data gathered using an Xradia MicroXCT-400. Scans consisted of 793 projections performed at 25kEv and 5 watts (corresponding voxel size, 20 μm). The image z-stacks were analyzed via a defined image processing sequence: (1) selection of image file from the z-stack; (2) identification of the scaffold edge and surrounding margin; (3) removal of image background (calculated from the maximum of the non-scaffold margin region); (4) selection of the scaffold edge; (5) partitioning the scaffold into 15 concentric rings of equal width; and (6) calculating the average pixel intensity within each concentric ring (Fig. 1). This analysis was performed through the image z-stack, with results reported as either mean mineral content for the entire scaffold or as a function of radial position.
Figure 1.

Step by step schematic of microCT z-stack imaging process. Process proceeds by: (1) original image is obtained from the z-stack; (2) user selects a large ROI containing both scaffold and background; (3) background not selected in step 2 is subtracted from the image; (4) user selects only scaffold ROI; (5) the scaffold ROI is divided into 15 equally spaced concentric rings; (6) voxels within each concentric ring are averaged; (7) process is repeated for each image within the z-stack. Example image is dimmed to 80% intensity to better define outlined ROIs. Scale bar: 2mm.
B.9. Statistics
Statistical analysis was performed via two-way analysis of variance (ANOVA) tests after which a Tukey-HSD post-hoc test was used. Independent factors included time, scaffold type (CG vs. CGCaP), and treatment (growth media, osteogenic media, BMP-2 supplemented growth media). Mechanical characterization and gene expression experiments used at least n = 3 scaffolds per group while metabolic activity and scaffold compositional analyses used n = 6 scaffolds per group. Significance was set at p < 0.05. Error bars are reported as standard deviation unless otherwise noted.
C. Results
C.1 Release of calcium and phosphate ions from the mineralized scaffold
The release of Ca and P ions from acellular CGCaP scaffolds (vs. non-mineralized scaffold control) was tracked through 56 days in culture. A significant (p < 0.05) increase in Ca and P ion release was observed from mineralized (vs. CG) scaffolds as early as day 2 (phosphate) or day 5 (calcium) (Fig. 2). Ion release appeared to reach an asymptote, with little change after day 21 in culture. Total ion release corresponded to ~80% of the mineral content incorporated during fabrication. The heavy washing associated with EDC crosslinking steps leads to a burse release of mineral (Supp. Fig. 1A, days 1 – 3). However, after this initial burst release, mineral release during subsequent periods of culture culture was unchanged as a result of EDC crosslinking (Supp. Fig. 1B, days 4–7).
Figure 2.

Cumulative release of (A) calcium and (B) phosphate ions in CGCaP relative to CG scaffolds. *: significant (p < 0.05) difference at given time point between scaffold groups.
C.2. Metabolic activity of hMSCs within non-mineralized and mineralized scaffolds
Metabolic activity of hMSCs in all scaffold-media combinations increased throughout the 56 day culture period (Fig. 3). When cultured in growth media, the metabolic activity of hMSCs within mineralized scaffolds initially lagged behind hMSCs within non-mineralized scaffolds over the first three weeks of culture, likely due to differences in scaffold permeability.33 However, long term results suggest that the mineralized scaffold supports long term hMSC metabolic health at the level of non-mineralized scaffolds. Addition of BMP-2 to the media did not impact the metabolic health of the MSCs in mineralized collagen scaffolds. However, use of osteogenic media led to a significant (p < 0.001) reduction in the metabolic health of hMSCs compared to growth media in mineralized scaffolds as early as day 1.
Figure 3.
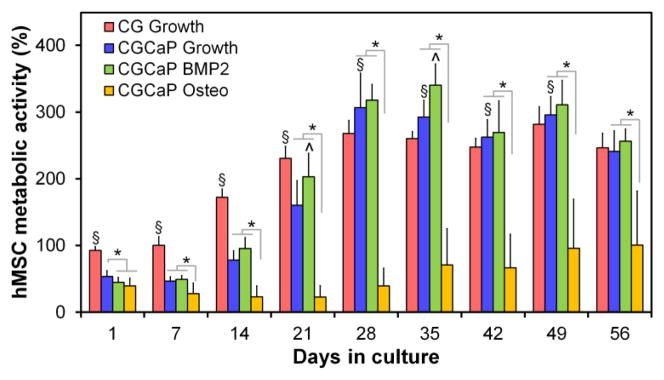
Metabolic activity of hMSCs within non-mineralized CG scaffolds in growth media (CG Growth), mineralized scaffolds in growth media (CGCaP Growth), mineralized scaffolds in BMP2 supplemented growth media (CGCaP BMP2), and mineralized scaffolds in osteogenic media (CGCaP Osteo). *: significant (p < 0.05) difference between mineralized scaffold groups. ^: significant (p < 0.05) up-regulation compared to all other mineralized groups at the same time point. §: significant (p < 0.05) difference between non-mineralized and mineralized (growth media) scaffold groups at a given time point. Metabolic activity results normalized to the number of originally seeded cells.
C.3. hMSC gene expression profiles
Trends of increasing expression levels with time for all genes tested (COL1A1, BSP, OPN, RUNX2) were found across all treatment groups (Fig. 4). Significant increases in BSP expression was observed after 8 weeks in culture for all scaffold conditions, but were significantly (p < 0.05) greater for hMSCs in mineralized versus non-mineralized scaffolds (maintained in growth media). Interestingly, hMSCs cultured in mineralized scaffolds with BMP-2 supplemented media showed signs of earlier osteogenesis, with a significant increase (p < 0.05) in BSP expression versus all other scaffold groups by 4 weeks.
Figure 4.

Gene expression levels of (A) bone sialoprotein (BSP), (B) collagen 1 (COL1A1), (C) osteopontin (OPN), and (D) runt-related transcription factor 2 (RUNX2). Expression levels were normalized to MSCs in non-mineralized scaffolds at day 14 to compare the effects of both scaffold mineralization and time. *: significant (p < 0.05) difference at between scaffold groups at a single time point. ^: significant (p < 0.05) difference versus day 14 for a given scaffold group. §: significant (p < 0.05) difference between non-mineralized and mineralized scaffolds (growth media) at a given time point.
C.4. Changes in scaffold modulus with hMSC culture
The modulus of hMSC-seeded mineralized collagen scaffolds increased significantly (p < 0.0005) with culture time for all media formulations relative to the unseeded scaffold control (CGCaP blank) (Fig. 5). Changes in elastic moduli of scaffolds at the end of culture versus unseeded scaffolds at the end of culture (1.83 ± 0.5 kPa, 23.00 ± 0.6 kPa for CG, CGCaP scaffolds, respectively), suggest a 50 to 80 percent increase in the density of the construct over 8 weeks of culture (Supp. Table 2). No significant increase in modulus was observed in hMSC-seeded non-mineralized scaffolds, suggesting that addition of hMSCs did not result in appreciable net matrix production. hMSC-seeded mineralized scaffolds were also significantly (p < 0.05) stiffer than the hMSC seeded non-mineralized scaffold at all time points (Fig. 5). The modulus of unseeded non-mineralized or mineralized scaffolds remained unchanged over the entire experimental period, consistent with previous observations that EDC-crosslinked CG scaffolds have greater than 6 month half-lives in vivo.49 Addition of pro-osteogenic supplements impacted the modulus of hMSC-seeded mineralized scaffolds. Mineralized scaffolds maintained in osteogenic media consistently demonstrated a significantly (p < 0.05) increased modulus compared to mineralized scaffolds maintained in growth or BMP-2 supplemented media (Fig. 5).
Figure 5.

Elastic moduli of hMSC seeded mineralized and non-mineralized collagen scaffolds in compression as a function of time and media supplementation.*: significant (p < 0.05) difference at given time point compared to scaffold groups. ^: significant (p < 0.05) increase compared to other time points of the same scaffold group. §: significant (p < 0.05) difference between non-mineralized and mineralized scaffolds (growth media) at a given time point.
C.5. Analysis of mineral via Alizarin red staining
Histological sections were taken after days 14, 28, and 56 to evaluate new mineral formation via Alizarin red (Fig. 6; Supp. Table 1). Significant mineral content was observed in mineralized scaffolds regardless of media (growth, osteogenic, BMP-2). However, limited to no mineral content was observed in the hMSC-seeded non-mineralized CG scaffolds.
Figure 6.
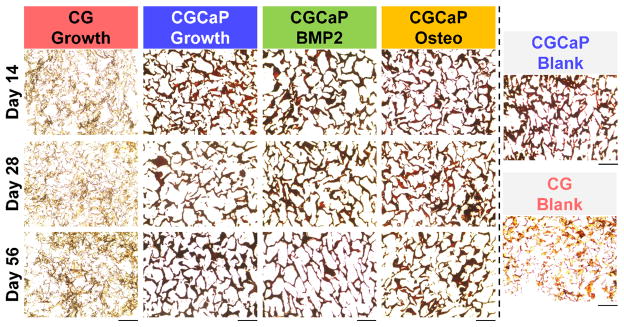
Alizarin red analysis of mineral synthesis. Representative histology sections from days 14, 28, and 56 showing increased mineral content in the mineralized scaffold groups (versus non-mineralized scaffold). Scale bar: 200μm.
C.6. CaP mineral content and distribution
The relative mineral content of hMSC seeded non-mineralized and mineralized scaffold groups was calculated via microCT (Fig. 7). All analyses were performed at identical scanner settings and thresholded to reveal mineral content. Depth-averaged results of mineral distribution were calculated as a function of radial position, as no significant differences were observed in radial mineral content as a function of vertical position within the scaffold (data not shown). The average scaffold mineral content of hMSC-seeded mineralized CG scaffolds was significantly (p < 0.0005) higher than hMSC-seeded non-mineralized scaffolds for all time points. Choice of media impacted mineral content in hMSC-seeded mineralized scaffolds, but for all conditions significant increases in mineral content were observed with culture time (growth, osteogenic media: day 56 vs. 14; BMP-2 supplemented media: day 56 vs. 28 vs. 14) (Fig. 7B). Analysis of the relative radial distribution of mineral content suggested maximal new mineral formation took place towards the edges of the scaffold for all mineralized scaffold groups, with negligible mineral content found within the non-mineralized scaffold (Supp. Fig. 2).
Figure 7.
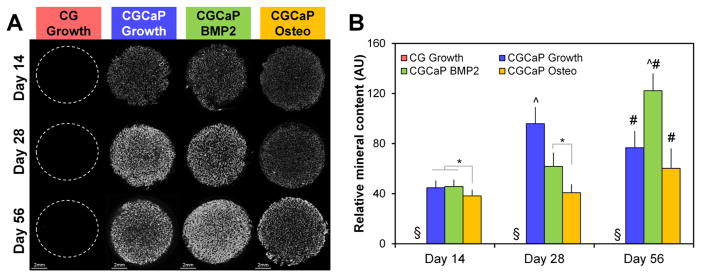
Micro-CT analysis of new mineral formation as a function of time and scaffold culture conditions. (A) Representative micro-CT slices. (B) Average pixel intensity was used as a quantitative metric of relative mineral content within each scaffold at each time point.*: significant difference between groups at a given time point. ^: significant (p < 0.05) increase compared to all other groups at the same time point. §: significantly (p < 0.0005) lower than all other groups. #: significant (p < 0.05) increase compared same group at day 14.
D. Discussion
This study examined the impact of selective incorporation of a CaP mineral component into collagen-GAG scaffolds on the viability, osteogenic differentiation, and matrix biosynthesis capacity of hMSCs. Previously, extensive work in the literature has suggested that calcium phosphate mineral is a critical component of a range of biomaterials for osteogenic repair applications.50–54 Previous work with CG scaffolds suggested that increasing scaffold stiffness and mineral content can resist cell-mediated contraction and promote a more osteogenic phenotype.28, 32, 55, 56 However, these efforts all used supplemented media (osteogenic) formulations. For this study we tested the hypothesis that the mineralized CG scaffold could facilitate osteogenesis independent of osteogenic or BMP-2 supplemented media. And while the main goal of this study was to compare the results of hMSC-seeded mineralized versus non-mineralized scaffolds, we included hMSC-seeded mineralized scaffolds maintained in either BMP-2 supplemented (100ng/mL) or osteogenic media as positive controls. The results of our study support that mineralized collagen-GAG scaffolds can themselves promote hMSC osteogenesis.
While we have previously reported increased mechanical properties due to the precipitation based incorporation of CaP mineral content prior to lyophilization,33 we had not yet examined the long term stability of this mineral content over long term culture as in other material systems.57 We therefore examined the kinetics of release of Ca and P ions into the surrounding culture media and their supporting role in cell behavior.58, 59 We observed a significant release of Ca and P ions into the surrounding media as early as 2 days in culture (Fig. 2). Though release was quantified for a full 8 weeks in vitro, Ca and P release was largely concluded within the first three weeks for mineralized CG scaffolds. Comparing overall phosphate to calcium release, these results further suggested that Ca and P ions were released both due to dissolution of brushite CaP from the scaffold33 as well as release of residual phosphoric acid trapped in the scaffold following lyophilization.37, 60 EDC crosslinking of the mineralized scaffolds had no impact on Ca and P release rate (Supp. Fig. 1) beyond that observed during the extensive washing steps during crosslinking. This suggests the potential to tune scaffold mechanics and degradation rate via crosslinking independent of Ca and P ion release. These results also suggest that this precipitation-based scheme allows fabrication of mineralized collagen scaffolds whose mineral content may contribute to its osteogenic potential in multiple manners: (1) greater initial stiffness; (2) slow release of pro-osteogenic Ca and P ions over time.
While subject to burst diffusive losses like other soluble supplementation methods, scaffold mineral content was able to instruct pro-osteogenic phenotype. Analysis of osteogenic gene expression profiles (Fig. 4) and matrix remodeling (Fig. 5, 6) indicated that the mineralized scaffold significantly contributed to an enhanced pro-osteogenic phenotype at later time points (day 56) in culture and without the need for conventional osteogenic supplements. The kinetics of Ca and P release also suggest the need for future studies to more closely examine mechanisms underlying scaffold-mediated MSC osteogenesis as a function of ion release.
Measures of metabolic activity give a good representation of the overall metabolic health of the cell-seeded biomaterials,32, 34, 61 they are not exact measures of total cell number. Given previous studies favorably comparing explicit measures of cell number versus metabolic activity of cell-seeded CG scaffolds62, 63 as well as the large number of scaffold groups and experimental end-points in this study, we chose not to explicitly determine cell number via destructive assays, but rather tracked construct metabolic activity via non-destructive AlamarBlue. Future efforts will explicitly measure overall MSC number. Regardless, all scaffold variants supported a significant increase in cell metabolic activity over the course of the 8 week in vitro culture (Fig. 3). Examining the impact of scaffold type (mineralized vs. non-mineralized), non-mineralized CG scaffolds supported significantly enhanced MSC metabolic health up to day 28 of culture, while mineralized CG scaffolds supported enhanced metabolic health at later time points (Fig. 3). These results suggest an important trade-off in initial scaffold properties versus cell-mediated remodeling. The increased relative density of the mineralized CG scaffolds reduces construct permeability33 and likely initial cell attachment,64, 65 leading to initially reduced MSC metabolic health in the mineralized scaffold during initial culture (Fig. 3). Cell-mediated contraction can significantly impact CG scaffold microstructural properties as well as resultant cell activity at the functional and genomic level over longer periods of culture.66 However mineralized, highly crosslinked, and high-density variants of CG scaffolds are substantially less subject to significant cell-mediated contraction,32 suggesting that the relative improvement of MSC metabolic activity in mineralized scaffolds at late culture times may be due to reduced contraction-mediated remodeling. Comparing the impact of media supplementation on MSC metabolic health in mineralized CG scaffolds, MSCs supplemented with osteogenic media showed significantly reduced metabolic activity versus growth media or BMP-2 supplemented growth media (Fig. 3), in line with previous findings in the literature regarding tradeoffs in cell proliferation versus phenotype.67–69
Given observed differences in metabolic activity, we therefore examined whether: (1) the mineralized CG scaffold was sufficient to induce osteogenic MSC differentiation in the absence of osteogenic supplementation; and (2) osteogenic media supplementation enhanced the speed of osteogenesis. An analysis of osteogenic gene expression profiles suggested the mineralized scaffold supported higher levels of pro-osteogenic gene expression (BSP) versus the non-mineralized scaffold. Comparing expression profiles within the mineralized scaffold groups, we observed an earlier increase in bone sialoprotein expression for MSCs in BMP-2 supplemented relative to growth or osteogenic media mineralized scaffolds. By day 56, however, hMSCs in mineralized scaffolds without osteogeneic supplementation showed the highest expression levels of BSP (Fig. 4). We also observed the increased expression of COL1A1 within both non-mineralized and mineralized CG scaffolds in growth media versus scaffolds maintained in osteogenic of BMP-2 supplemented media, suggesting that hMSCs may be more inclined to deposit or remodel their matrix without supplementation (Fig. 4). Recent efforts have shown enhanced osteogenesis and osteogenic gene expression notably increased COL1A1 and RUNX2 expression, in presence of calcium phosphate,70, 71 suggesting that the phase of mineral and the mode of incorporating it into a collagen scaffold network may alter MSC response. Ongoing efforts are looking to identify mechanisms associated with MSC bioactivity within these mineralized collagen scaffolds.
To better understand if both the mineral content and/or media supplementation enhanced osteogenic cell remodeling we examined changes in scaffold mechanical properties (Fig. 5) as well as changes in scaffold mineral content via histological and microCT analysis (Figs. 6–7). Mechanical analysis provided insight into changes within the matrix such as protein and CaP mineral content deposition. We observed no significant difference in elastic modulus between acellular CG scaffolds and those cultured in growth media with hMSCs. While some cell-mediated degradation of the CG scaffold is expected given the long culture time, these results suggest little to no excess ECM deposition occurred over the 8 week period of culture. In contrast, all mineralized CG scaffolds seeded with MSCs exhibited significant increases in elastic modulus relative to the acellular control. Consistent with the concept of a tradeoff in metabolic expansion and functional activity, while hMSCs maintained in osteogenic media showed relatively little increases in metabolic activity, these scaffolds showed the greatest gains in elastic moduli at each time point (Fig. 5). However the mechanical properties of mineralized scaffolds maintained increased significantly over the course of the 8 week experiment regardless of media formulation used.
The observed change in scaffold mechanical properties correlated with analysis of scaffold mineral content via Alizarin Red histological staining (Fig. 6) and microCT (Fig. 7). Despite significant release of initial scaffold mineral content, extensive Alizarin Red staining was observed in all in mineralized scaffolds with little to no staining in non-mineralized scaffolds (Fig. 6). Analysis of the micro-CT scans of all scaffold groups suggested significant new mineral formation in the mineralized CG scaffolds regardless of media supplementation, while again little to no new mineral content was observed in the non-mineralized CG scaffold (Fig. 7). Examining trends in time, it appears that the mineralized CG scaffold was able to support the largest increase in mineral content over the first 4 weeks of culture, while BMP-2 supplementation led to a larger increase in new mineral formation at the later time points (8 weeks). This result suggests that the significant increase in modulus of mineralized CG scaffolds in osteogenic media may be due to a combination of protein and mineral deposition. Given the significant release of initial scaffold mineral content over the course of 8 weeks of culture (~80% of the initial mineral, Fig. 2), the presence of significant amounts of mineral and significant modulus increases suggests that the mineralized scaffold provides the correct microenvironment to support significant new mineral formation and matrix synthesis.
A radial analysis of the mineral content (Supp. Fig. 2) also provided information regarding both mineral deposition and, by proxy, cell distribution and ingrowth. The largest increases in mineral were concentrated towards the edges of the scaffold disks, with radial plots of mineral content suggesting substantial additional mineral content in the external 1.5 mm of the 8 mm diameter disks. In agreement with the average mineral content within a scaffold (Fig. 7) no mineral content was detected within the CG growth scaffolds. Assuming uniform release of mineral content within a fully hydrated scaffold, radial differences in mineral content suggest nonuniform matrix remodeling via hMSCs. Such results motivate future optimization of mineralized scaffold microstructural properties to support enhanced cell penetration as well as subsequent metabolic support via diffusive transport. These results also motivate future efforts to explore strategies such as incorporation of biomolecular agonists (e.g., VEGF) to enhance the speed of vascular ingrowth in vivo. Together these results suggest that the mineralized collagen scaffold described here is sufficient to induce pro-osteogenic behavior of hMSCs in the absence of conventional media supplements. Ongoing efforts are examining both the mechanisms associated with these finding, hypothesized to be mechanotransduction and TGF-β superfamily signal transduction pathway, and their use in vivo for bone regeneration. However, these results have significance regarding the developing of biomaterials for orthopedic insertional repair (e.g., osteotendinous, osteochondral interface). Identifying a mineralized scaffold able to support enhanced hMSC osteogenesis in the absence of media supplementation provides the foundation for developing instructive biomaterials containing spatially-gradated mineral content to locally enhance osteogeneic specification.
Conclusions
Biomaterials for bone tissue engineering must be able to instruct cell behavior in the presence of the complex biophysical and biomolecular environments encountered in vivo. While osteogenic media or exogenous BMP-2 supplementation are often used as an essential element of many bone regeneration studies, diffusive loss and rapid degradation are primary concerns of efforts requiring exogenous supplementation. Using a series of collagen-GAG scaffolds, we investigated whether incorporation of a transient CaP mineral content was sufficient to enhance hMSC osteogenic differentiation and matrix remodeling in the absence of lineages-specific media supplementation. We found that the presence of CaP mineral enhances hMSC osteogenesis compared to non-mineralized scaffold in the absence of traditional osteogenic supplements. While osteogenic media or BMP-2 supplementation may enhance the speed or long term intensity of the response, the mineralized CG scaffold is able to support extensive hMSC osteogenesis, matrix remodeling, and new mineral formation in the absence of lineage specific media. These efforts are informing ongoing work in our lab exploring the regenerative potential of mineralized CG scaffolds in critical sized bone defects as well as methods to selectively immobilize osteo-inductive signals such as BMP-2 in the mineralized CG scaffold to enhance this effect.
Supplementary Material
Acknowledgments
The authors acknowledge Donna Epps for assistance with histology, IGB Core Facilities for assistance with RT-PCR, Dr. Leilei Yin for assistance in micro-computed tomography, as well as Yue Wang and Dr. Michael Insana for assistance with mechanical testing of cell-seeded scaffolds. This material is based upon work supported by the National Science Foundation under Grant No. 1105300. Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Numbers R21 AR063331. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. DWW was funded at UIUC from National Science Foundation (NSF) Grant 0965918 IGERT: Training the Next Generation of Researchers in Cellular & Molecular Mechanics and BioNanotechnology. We are also grateful for funding for this study provided by the Chemistry-Biology Interface Training Program NIH NIGMS T32GM070421 (SRC). This research was carried out in part in the Microscopy Suite, Imaging Technology Group, Beckman Institute for Advanced Science and Technology, University of Illinois at Urbana-Champaign.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/b000000x/
References
- 1.Miller MJ, Goldberg DP, Yasko AW, Lemon JC, Satterfield WC, Wake MC, Mikos AG. Tissue Eng. 1996;2:51–59. doi: 10.1089/ten.1996.2.51. [DOI] [PubMed] [Google Scholar]
- 2.Krishnan L, Willett NJ, Guldberg RE. Ann Biomed Eng. 2014;42:432–444. doi: 10.1007/s10439-014-0969-9. [DOI] [PubMed] [Google Scholar]
- 3.Mountziaris PM, Mikos AG. Tissue Eng Part B Rev. 2008;14:179–186. doi: 10.1089/ten.teb.2008.0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christenson EM, Anseth KS, van den Beucken JJ, Chan CK, Ercan B, Jansen JA, Laurencin CT, Li WJ, Murugan R, Nair LS, Ramakrishna S, Tuan RS, Webster TJ, Mikos AG. J Orthop Res. 2007;25:11–22. doi: 10.1002/jor.20305. [DOI] [PubMed] [Google Scholar]
- 5.Smith MH, Flanagan CL, Kemppainen JM, Sack JA, Chung H, Das S, Hollister SJ, Feinberg SE. Int J Med Robot. 2007;3:207–216. doi: 10.1002/rcs.143. [DOI] [PubMed] [Google Scholar]
- 6.Dodson TB, Bays RA, Pfeffle RC, Barrow DL. J Oral Maxillofac Surg. 1997;55:260–267. doi: 10.1016/s0278-2391(97)90539-5. [DOI] [PubMed] [Google Scholar]
- 7.Zimmermann G, Moghaddam A. Injury. 2011;42:S16–21. doi: 10.1016/j.injury.2011.06.199. [DOI] [PubMed] [Google Scholar]
- 8.Laurencin C, Khan Y, El-Amin SF. Expert Rev Med Devices. 2006;3:49–57. doi: 10.1586/17434440.3.1.49. [DOI] [PubMed] [Google Scholar]
- 9.Lyons FG, Gleeson JP, Partap S, Coghlan K, O’Brien FJ. Clin Orthop Relat Res. 2014;472:1318–1328. doi: 10.1007/s11999-013-3438-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hutmacher DW. J Biomater Sci Polym Ed. 2001;12:107–124. doi: 10.1163/156856201744489. [DOI] [PubMed] [Google Scholar]
- 11.Place ES, Evans ND, Stevens MM. Nat Mater. 2009;8:457–470. doi: 10.1038/nmat2441. [DOI] [PubMed] [Google Scholar]
- 12.Grayson WL, Fröhlich M, Yeager K, Bhumiratana S, Chan ME, Cannizzaro C, Wan LQ, Liu XS, Guo XE, Vunjak-Novakovic G. Proc Natl Acad Sci U S A. 2010;107:3299–3304. doi: 10.1073/pnas.0905439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu XL, Lou J, Tang TT, Ng KW, Zhang JH, Yu CF, Dai KR. J Biomed Mater Res B Appl Biomater. 2005;75B:289–303. doi: 10.1002/jbm.b.30299. [DOI] [PubMed] [Google Scholar]
- 14.Muller P, Bulnheim U, Diener A, Luthen F, Teller M, Klinkenberg ED, Neumann HG, Nebe B, Liebold A, Steinhoff G, Rychly J. J Cell Mol Med. 2008;12:281–291. doi: 10.1111/j.1582-4934.2007.00103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakamura S, Matsumoto T, Sasaki J, Egusa H, Lee KY, Nakano T, Sohmura T, Nakahira A. Tissue Eng Part A. 2010;16:2467–2473. doi: 10.1089/ten.TEA.2009.0337. [DOI] [PubMed] [Google Scholar]
- 16.Musah S, Morin SA, Wrighton PJ, Zwick DB, Jin S, Kiessling LL. ACS Nano. 2012;6:10168–10177. doi: 10.1021/nn3039148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu J, Sun X, Ma H, Xie C, Chen YE, Ma PX. Biomaterials. 2010;31:7971–7977. doi: 10.1016/j.biomaterials.2010.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Al-Munajjed AA, Plunkett NA, Gleeson JP, Weber T, Jungreuthmayer C, Levingstone T, Hammer J, O’Brien FJ. J Biomed Mater Res B Appl Biomater. 2009 doi: 10.1002/jbm.b.31320. [DOI] [PubMed] [Google Scholar]
- 19.Kim HW, Song JH, Kim HE. J Biomed Mater Res A. 2006;79:698–705. doi: 10.1002/jbm.a.30848. [DOI] [PubMed] [Google Scholar]
- 20.Liao SS, Cui FZ, Zhang W, Feng QL. J Biomed Mater Res B Appl Biomater. 2004;69B:158–165. doi: 10.1002/jbm.b.20035. [DOI] [PubMed] [Google Scholar]
- 21.Bashur CA, Shaffer RD, Dahlgren LA, Guelcher SA, Goldstein AS. Tissue Eng Part A. 2009;15:2435–2445. doi: 10.1089/ten.tea.2008.0295. [DOI] [PubMed] [Google Scholar]
- 22.Massumi M, Abasi M, Babaloo H, Terraf P, Safi M, Saeed M, Barzin J, Zandi M, Soleimani M. Tissue Eng Part A. 2012;18:609–620. doi: 10.1089/ten.tea.2011.0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Engler AJ, Sen S, Sweeney HL, Discher DE. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 24.Sun Y, Villa-Diaz LG, Lam RH, Chen W, Krebsbach PH, Fu J. PLoS ONE. 2012;7:e37178. doi: 10.1371/journal.pone.0037178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pountos I, Georgouli T, Henshaw K, Bird H, Jones E, Giannoudis PV. J Orthop Trauma. 2010;24:552–556. doi: 10.1097/BOT.0b013e3181efa8fe. [DOI] [PubMed] [Google Scholar]
- 26.Shen W, Chen X, Chen J, Yin Z, Heng BC, Chen W, Ouyang HW. Biomaterials. 2010;31:7239–7249. doi: 10.1016/j.biomaterials.2010.05.040. [DOI] [PubMed] [Google Scholar]
- 27.You MH, Kwak MK, Kim DH, Kim K, Levchenko A, Kim DY, Suh KY. Biomacromolecules. 2010;11:1856–1862. doi: 10.1021/bm100374n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farrell E, O’Brien FJ, Doyle P, Fischer J, Yannas I, Harley BA, O’Connell B, Prendergast PJ, Campbell VA. Tissue Eng. 2006;12:459–468. doi: 10.1089/ten.2006.12.459. [DOI] [PubMed] [Google Scholar]
- 29.Lee JH, Rhie JW, Oh DY, Ahn ST. Biochem Biophys Res Commun. 2008;370:456–460. doi: 10.1016/j.bbrc.2008.03.123. [DOI] [PubMed] [Google Scholar]
- 30.Solorio LD, Fu AS, Hernandez-Irizarry R, Alsberg E. J Biomed Mater Res A. 2010;92:1139–1144. doi: 10.1002/jbm.a.32440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen YH, Shoichet MS, Radisic M. Acta Biomater. 2008;4:477–489. doi: 10.1016/j.actbio.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 32.Caliari SR, Harley BAC. Advanced healthcare materials. 2014;3:1086–1096. doi: 10.1002/adhm.201300646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weisgerber DW, Kelkhoff DO, Caliari SR, Harley BAC. J Mech Behav Biomed Mater. 2013;28:26–36. doi: 10.1016/j.jmbbm.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caliari SR, Harley BAC. Tissue Eng A. 2014;20:2463–2472. doi: 10.1089/ten.tea.2013.0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caliari SR, Harley BA. Tissue Eng Part A. 2013;19:1100–1112. doi: 10.1089/ten.tea.2012.0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caliari SR, Mozdzen LC, Armitage O, Oyen ML, Harley BAC. J Biomed Mater Res A. 2014;102:917–927. doi: 10.1002/jbm.a.35058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harley BA, Lynn AK, Wissner-Gross Z, Bonfield W, Yannas IV, Gibson LJ. J Biomed Mater Res A. 2010;92:1066–1077. doi: 10.1002/jbm.a.32361. [DOI] [PubMed] [Google Scholar]
- 38.An S, Ling J, Gao Y, Xiao Y. J Periodontal Res. 2012;47:374–382. doi: 10.1111/j.1600-0765.2011.01443.x. [DOI] [PubMed] [Google Scholar]
- 39.Zhou Y, Lewis TL, Robinson LJ, Brundage KM, Schafer R, Martin KH, Blair HC, Soboloff J, Barnett JB. J Cell Physiol. 2011;226:1082–1089. doi: 10.1002/jcp.22423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yannas IV, Lee E, Orgill DP, Skrabut EM, Murphy GF. Proc Natl Acad Sci USA. 1989;86:933–937. doi: 10.1073/pnas.86.3.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Brien FJ, Harley BA, Yannas IV, Gibson LJ. Biomaterials. 2005;26:433–441. doi: 10.1016/j.biomaterials.2004.02.052. [DOI] [PubMed] [Google Scholar]
- 42.Harley BA, Leung JH, Silva EC, Gibson LJ. Acta Biomater. 2007;3:463–474. doi: 10.1016/j.actbio.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 43.Olde Damink LH, Dijkstra PJ, van Luyn MJ, van Wachem PB, Nieuwenhuis P, Feijen J. Biomaterials. 1996;17:765–773. doi: 10.1016/0142-9612(96)81413-x. [DOI] [PubMed] [Google Scholar]
- 44.Caliari SR, Gonnerman EA, Grier WK, Weisgerber DW, Banks JM, Alsop AJ, Lee J-S, Bailey RC, Harley BAC. Advanced healthcare materials. 2014 doi: 10.1002/adhm.201400252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tierney CM, Jaasma MJ, O’Brien FJ. J Biomed Mater Res A. 2009;91:92–101. doi: 10.1002/jbm.a.32207. [DOI] [PubMed] [Google Scholar]
- 46.Duffy GP, McFadden TM, Byrne EM, Gill SL, Farrell E, O’Brien FJ. Eur Cell Mater. 2011;21:15–30. doi: 10.22203/ecm.v021a02. [DOI] [PubMed] [Google Scholar]
- 47.Kanungo BP, Gibson LJ. Acta Biomater. 2009;5:1006–1018. doi: 10.1016/j.actbio.2008.11.029. [DOI] [PubMed] [Google Scholar]
- 48.Kanungo BP, Gibson LJ. Acta Biomater. 2009;6:344–353. doi: 10.1016/j.actbio.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 49.Harley BA, Spilker MH, Wu JW, Asano K, Hsu HP, Spector M, Yannas IV. Cells Tissues Organs. 2004;176:153–165. doi: 10.1159/000075035. [DOI] [PubMed] [Google Scholar]
- 50.Barradas AM, Monticone V, Hulsman M, Danoux C, Fernandes H, Tahmasebi Birgani Z, Barrere-de Groot F, Yuan H, Reinders M, Habibovic P, van Blitterswijk C, de Boer J. Integr Biol (Camb) 2013;5:920–931. doi: 10.1039/c3ib40027a. [DOI] [PubMed] [Google Scholar]
- 51.Brovarone CV, Verne E, Appendino P. J Mater Sci Mater Med. 2006;17:1069–1078. doi: 10.1007/s10856-006-0533-8. [DOI] [PubMed] [Google Scholar]
- 52.Lynn AK, Nakamura T, Patel N, Porter AE, Renouf AC, Laity PR, Best SM, Cameron RE, Shimizu Y, Bonfield W. Journal of Biomedical Materials Research Part A. 2005;74:447–453. doi: 10.1002/jbm.a.30373. [DOI] [PubMed] [Google Scholar]
- 53.Polak SJ, Levengood SK, Wheeler MB, Maki AJ, Clark SG, Johnson AJ. Acta Biomater. 2011;7:1760–1771. doi: 10.1016/j.actbio.2010.12.030. [DOI] [PubMed] [Google Scholar]
- 54.Cunniffe GM, O’Brien FJ, Partap S, Levingstone TJ, Stanton KT, Dickson GR. J Biomed Mater Res A. 2010;95:1142–1149. doi: 10.1002/jbm.a.32931. [DOI] [PubMed] [Google Scholar]
- 55.Curtin CM, Cunniffe GM, Lyons FG, Bessho K, Dickson GR, Duffy GP, O’Brien FJ. Adv Mater. 2012;24:749–754. doi: 10.1002/adma.201103828. [DOI] [PubMed] [Google Scholar]
- 56.Murphy CM, Matsiko A, Haugh MG, Gleeson JP, O’Brien FJ. J Mech Behav Biomed Mater. 2012;11:53–62. doi: 10.1016/j.jmbbm.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 57.Seol YJ, Park JY, Jung JW, Jang J, Girdhari R, Kim SW, Cho DW. Tissue engineering Part A. 2014;20:2840–2849. doi: 10.1089/ten.tea.2012.0726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barradas AM, Fernandes HA, Groen N, Chai YC, Schrooten J, van de Peppel J, van Leeuwen JP, van Blitterswijk CA, de Boer J. Biomaterials. 2012;33:3205–3215. doi: 10.1016/j.biomaterials.2012.01.020. [DOI] [PubMed] [Google Scholar]
- 59.Jung GY, Park YJ, Han JS. J Mater Sci Mater Med. 2010;21:1649–1654. doi: 10.1007/s10856-010-4011-y. [DOI] [PubMed] [Google Scholar]
- 60.Lynn AK, Best SM, Cameron RE, Harley BA, Yannas IV, Gibson LJ, Bonfield W. J Biomed Mater Res A. 2010;92:1057–1065. doi: 10.1002/jbm.a.32415. [DOI] [PubMed] [Google Scholar]
- 61.Tierney CM, Haugh MG, Liedl J, Mulcahy F, Hayes B, O’Brien FJ. J Mech Behav Biomed Mater. 2009;2:202–209. doi: 10.1016/j.jmbbm.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 62.Caliari SR, Weisgerber DW, Ramirez MA, Kelkhoff DO, Harley BAC. J Mech Behav Biomed Mater. 2012;11:27–40. doi: 10.1016/j.jmbbm.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Caliari SR, Harley BAC. Biomaterials. 2011;32:5330–5340. doi: 10.1016/j.biomaterials.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Murphy CM, Haugh MG, O’Brien FJ. Biomaterials. 2010;31:461–466. doi: 10.1016/j.biomaterials.2009.09.063. [DOI] [PubMed] [Google Scholar]
- 65.O’Brien FJ, Harley BA, Waller MA, Yannas IV, Gibson LJ, Prendergast PJ. Technol Health Care. 2007;15:3–17. [PubMed] [Google Scholar]
- 66.Vickers SM, Gotterbarm T, Spector M. J Orthop Res. 2010;28:1184–1192. doi: 10.1002/jor.21113. [DOI] [PubMed] [Google Scholar]
- 67.Wang H, Pang B, Li Y, Zhu D, Pang T, Liu Y. Cytotherapy. 2012;14:423–430. doi: 10.3109/14653249.2011.652735. [DOI] [PubMed] [Google Scholar]
- 68.Thomopoulos S, Harwood FL, Silva MJ, Amiel D, Gelberman RH. J Hand Surg Am. 2005;30:441–447. doi: 10.1016/j.jhsa.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 69.Malladi P, Xu Y, Chiou M, Giaccia AJ, Longaker MT. Am J Physiol Cell Physiol. 2006;290:C1139–1146. doi: 10.1152/ajpcell.00415.2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.