

Abstract
Defects in Complex I assembly is one of the emerging underlying causes of severe mitochondrial disorders. The assembly of Complex I has been difficult to understand due to its large size, dual genetic control and the number of proteins involved. Mutations in Complex I subunits as well as assembly factors have been reported to hinder its assembly and give rise to a range of mitochondria disorders. In this review, we summarize the recent progress made in understanding the Complex I assembly pathway. In particularly, we focus on the known as well as novel assembly factors and their role in assembly of Complex I and human disease.
Keywords: Complex I, assembly, assembly factors, mitochondrial disorders
Introduction
Compared to our knowledge of the assembly of other respiratory complexes, investigation of Complex I assembly has lagged behind, primarily due to a lack of powerful genetic systems, since the conventional model S. cerevisiae does not have Complex I but another type of single subunit NADH-Q oxidoreductase that is distinct from complex I and collectively called NDH-2 (Boumans et al., 1998). Despite of this technical difficulty, significant progress in understanding the regulation of complex I assembly pathway has been made in recent years. Here we try to give an update focusing on the players involved in this important process.
1. Respiratory Complex I Structure
In mammalian cells, Complex I is the largest respiratory enzyme, weighing almost 1000KDa and consists of 44 sub-units; 7 of which are encoded by the mitochondrial genome and the rest are encoded by the nuclear genome (Carroll et al., 2003, 2006; Chomyn et al., 1996). Most of the studies regarding the structure and composition of Complex I have been carried out on fungi Neurospora crassa or bovines mitochondria (Efremov et al., 2010; Guénebaut et al., 1998). While the crystal structure of intact Complex I from bacterium Thermus thermophilus was published recently (Baradaran et al., 2013), the same has not been achieved for the mammalian Complex I largely due to its huge size. The observations from both, the fungal Complex I as well as the bovine Complex I, suggest an L shaped structure with a hydrophobic membrane arm embedded in the inner mitochondrial membrane and a hydrophilic peripheral matrix arm which juts out in the mitochondrial matrix (Friedrich and Böttcher, 2004; Schultz and Chan, 2001, Hoffhaus et al., 1991). The iron sulfur centers responsible for electron transport are present in the matrix arm while the proton pumping takes place through the membrane arm (Belevich et al., 2006; Sazanov and Hinchliffe, 2006). Recent studies show that conformational changes in the structure of Complex I may allow electron transfer from NADH to the iron sulfur centers and couple the transfer of electrons to the proton translocations (Hunte et al., 2010; Schultz and Chan, 2001). The mitochondrial DNA encoded subunits are all part of the membrane arm while the matrix arm is made up of the nuclear encoded subunits. At least 7 of the nuclear encoded subunits, namely, NDUFV1, NDUFV2, NDUFS1, NDUFS2, NDUFS3, NDUFS7 and NDUFS8 represent the ‘core’ subunits which are conserved across genus along with the 7 mtDNA encoded subunits (Carroll et al., 2002, 2003; Potluri et al., 2004). These sub-units are involved in electron transfer and oxidation of NADH. The rest of the subunits of the peripheral arm are thought to be important for structural stability. The other 30 ‘supernumerary’ subunits, which have been hypothesized to have evolved with the mammalian mitochondria, play a role in the assembly or stability of Complex I or preventing oxidative damage by ROS. Post translational modifications of some of these subunits have been hypothesized to play a role in the regulation of Complex I (Carroll et al., 2013; Papa et al., 2012; Rhein et al., 2013). ND1 was found to have a quinone binding site and may be binding to ubiquinone while ND2, ND4 and ND5 resemble sodium and potassium antiporters and may be responsible for proton pumping activity (Carroll et al., 2013; Fearnley et al.).
2. Complex I Assembly Pathway
Deciphering the Complex I assembly pathway has been very difficult, complicated by its large size and dual genomic control that must coordinate the incorporation of subunits encoded by the nuclear genome with the subunits encoded by the mtDNA. Most of the detailed knowledge of Complex I assembly pathway is derived either from study of model systems such as N.crassa or by study of patient cells carrying disassembling Complex I mutations affecting the stability or assembly of Complex I thus leading to a Complex I deficiency, using blue native electrophoresis and pulse chase after labeling the mtDNA encoded subunits (Antonicka et al., 2003; Chomyn, 1996; Wittig et al., 2006). In the N.crassa model, it was observed that mutations in the subunits belonging to matrix arm led to a complete loss of the matrix arm and an accumulation of the hydrophobic membrane arm (Tuschen et al., 1990),(Friedrich and Weiss, 1997) indicating that the two arms of Complex I could assemble independently. Even in mammalian mitochondria, it was observed that Complex I membrane arm could assemble separately from the matrix arm. In the presence of mutations in the membrane arm subunits, the levels of assembled matrix arm did not change, indicating that the membrane arm subunits do not interfere with the assembly of the matrix arm (Bourges et al., 2004). This gave rise to the modular assembly pathway for respiratory Complex I where in the membrane arm and matrix arm assembled independently of each other and were joined to form the complete enzyme later in the assembly pathway (Ugalde et al., 2004).
The current model for Complex I assembly pathway gained by combining the knowledge of Complex I assembly from both fungi as well as patient cells is as follows (Gershoni et al., 2010; Lazarou et al., 2007; Nehls et al., 1992; Perales-Clemente et al., 2010; Tuschen et al., 1990; Vogel et al., 2007; Yadava et al., 2004). The entry points for the mtDNA encoded subunits of the membrane arm of Complex I have been a little difficult to understand. Recent reports suggest that membrane arm formation begins with ND1 subunit forming a 400KDa subcomplex, making it the first mtDNA encoded subunit to be incorporated in Complex I. This intermediate is also thought to contain the matrix arm subunits NDUFA9, NDUSF2, NDUFS3, NDUFS7 and NDUFS8. Another membrane arm intermediate, ∼460KDa, containing ND2, ND3 and ND6 then joins the ∼400KDa ND1 containing subcomplex to form a ∼650KDa subcomplex along with nuclear encoded subunits NDUFB6, NDUFB8 and NDUFA13. The next entry point for mtDNA encoded subunits is ND4 and ND5. They are hypothesized to be part of a single subcomplex and are thought to enter the Complex I assembly process to form a ∼830KDa intermediate which then forms a complete Complex I with the addition of the remaining matrix arm subunits. The presence of ND6 in the ∼400KDa sub complex and subsequently its entry early on has been debated (Perales-Clemente et al., 2010). This model thus differs slightly from the N.crassa model wherein the assembly of membrane arm and matrix arm remains independent throughout the assembly while in the mammalian model the intermediates of membrane arm and matrix arm assemble separately but the intermediates come together early on in the assembly process. Recent studies have shown that assembly of Complex III and Complex IV assemble with the ∼830KDa sub complex to form supercomplexes, with the final Complex I subunits assembling after that indicating that the assembly of Complex I is linked intricately with the assembly of respiratory supercomplexes (Moreno-Lastres et al., 2012)
There are some knowledge gaps in the current model of Complex I assembly. Because most of the studies have been conducted in fungi or patients with mutations in the subunits of Complex I, no study has been conducted looking at Complex I assembly in real time. As such it is difficult to differentiate between intermediates that accumulate in patients as actual assembly subcomplexes vs. just artifacts of defective Complex I assembly. As a result, there are contradicting reports on the entry points of different mtDNA encoded subunits. The roles of these individual mtDNA subunits in the assembly are also unclear. While it is believed that loss of ND4, ND5 and ND6 stalls the assembly process, it is still not clearly known at what stage each subunit is important to the assembly pathway. The other major loophole in Complex I assembly is the limited number of assembly factors known to assist Complex I assembly process. In a lot of cases where patients present with Complex I deficiency no known mutations are detected indicating that proteins involved in the assembly process haven't yet been identified (Fassone and Rahman, 2012; Ugalde et al., 2004). With a large size, it is hypothesized that more than the known 10 assembly factors may be involved in the assembly process.
3. Complex I Assembly Factors
Most of the assembly factors identified for Complex I have been identified in patients carrying mutations for them that lead to Complex I deficiency. Most of the assembly factors were identified as part of Complex I subcomplexes that accumulated in patients with mutations. The following summarizes each of the known Complex I assembly factors and their roles in Complex I assembly as well as mitochondrial disorders. Figure 1 shows the steps in which each of the assembly factor is involved in the Complex I assembly pathway.
Figure 1.
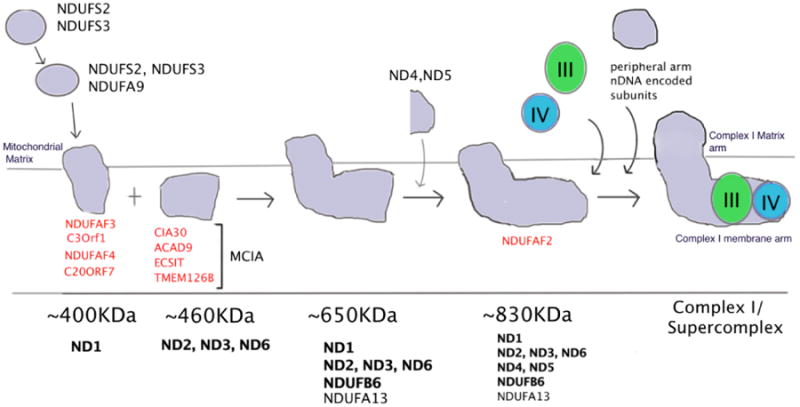
Schematic representation of the Complex I assembly pathway: The different sub complexes formed during the assembly and the various assembly factors involved in the assembly of Complex I. The assembly of Complex I starts with the initial sub complexes ∼400Kda and ∼460Kda to which additional sub complexes are attached to form the entire Complex I with the membrane and matrix arm. Roles of different assembly factors have been indicated at each stage.
C20ORF7: This assembly factor was identified in 2 families with severe neonatal Complex I deficiency disorder resulting from a homozygous missense mutation in the C20ORF7 protein (Gerards et al., 2010; Sugiana et al., 2008). Analysis of the Complex I subunits from mitochondria of patients revealed a loss of the mtDNA encoded ND1 subunit. There was also a decrease in the intensity of the 400KDa subcomplex band containing ND1 subunit indicating that C20ORF7 was involved in the stability of the 400 KDa subunit and the incorporation of ND1 was hindered in the absence of the assembly factor (Sugiana et al., 2008). Further studies revealed that C20orf7 has a S-adenosylmethionine (SAM)-dependent methyltransferase fold which methylates complex I subunit NDUFB3 as a requiste step in the process of complex I assembly. (Gerards et al., 2010).
NDUFAF1 (CIA30): CIA30 was first identified in N.crassa as a Complex I assembly factor (Küffner et al., 1998). Its homologue in mammalian cells, NDUFAF1 was subsequently identified in patients with cardioencephalomyopathies with Complex I deficiency (Dunning et al., 2007). NDUFAF1 was shown to be imported into mitochondrial matrix and is required for correct complex I stabilization along with another assembly factor, Ecsit (Vogel et al., 2007). Knockdown of either NDUFAF1 or Ecsit in human fibroblasts led to a decrease in Complex I levels and accumulation of the ∼460KDa sub complex and disappearance of the ∼830KDa sub complex indicating that NDUFAF1 might be essential for the ∼460KDa subcomplex to assemble into the ∼830KDa subcomplex. In another study, NDUFAF1 was found to associate with newly synthesized mtDNA encoded ND1, ND2 and ND3 subunits of the membrane arm indicating that it may also play a role in stabilizing the subunits to be incorporated into sub complexes (Dunning et al., 2007).
NDUFAF2 (B17.2L): Null mutations in the gene encoding B17.2 L or NDUFAF2 were found in patients suffering from progressive encephalopathy (Ogilvie et al., 2005). B17.2L is a molecular chaperone that is essential for the assembly of complex I and for normal functioning of the nervous system. B17.2L is not found with the mature holoenzyme but is associated with a specific subassembly of complex I ∼830kDa. It is required for stabilizing large complex I subassembly containing elements of both membrane and peripheral arms of the enzyme in which junction has not fully matured. B17.2L was found to be required until the late stages of the assembly of ∼980 kDa holoenzyme. In the absence of B17.2L, there is accumulation of ∼380kDa and ∼480kDa subassemblies but it does not lead to formation of ∼830kDa complex I halting the formation of the complex I.
Ecsit: Ecsit exists in two isoforms, 50 kDa (exists in cytoplasm) and 45 kDa (localized in mitochondria). Studies showed that the 45kDa form of Ecsit gets localized into the mitochondria through its N-terminal targeting sequence (Vogel et al., 2007). The same group also showed that Ecsit interact with NDUFAF1 in three Complex I assembly intermediates: ∼460KDa, ∼600KDa and ∼830KDa. Knockdown of Ecsit alone decreased the stability of NDUFAF1 in these assembly intermediates indicating that Ecsit might act as a co-chaperone for NDUFAF1.
NDUFAF3 and NDUFAF4: Pathogenic mutation in both NDUFAF3 and NDUFAF4 were identified in cases of fatal neonatal mitochondrial disease with sever complex I deficiency (Saada et al., 2008, 2009). Both these assembly factors were shown to interact with early subcomplexes containing ND1 subunit and were though to play a role in anchoring the early ND1 containing Complex I intermediate to the mitochondrial inner membrane.
ACAD9: Acyl CoA aldehyde dehydrogenase 9 s a member of the acyl dehydrogenase family thought to be involved in the mitochondrial fatty acid beta oxidation pathway. Pathogenic mutations in ACAD9 were identified in patients suffering from exercise intolerance and cardiomyopathies, symptoms characteristic of mitochondrial disorders (Haack T, et al., 2010). The study also showed that the patients suffered from a severe Complex I deficiency. ACAD9 was found to interact with NDUFAF1 and Ecsit in the same assembly intermediates previously mentioned. Knockdown of ACAD9 resulted in decreased levels of both NDUFAF1 and ecsit indicating that it might be needed for stability of the two chaperones (Nouws J et al., 2010).
TMEM126B and C3ORF1: Using Large pore gel electrophoresis and proteomics to identify the proteins present in rat mitochondrial complexome, i.e. complexes from rat mitochondria isolated on a native gel, TMEM126B was identified as a part of the MCIA (mitochondrial complex I assembly complex) along with CIA30, ACAD9 and Ecsit (Heide H et al., 2012). The MCIA complex was proposed to compose of the mentioned assembly factors which may lead to increased stability of the assembly complex. Knockdown of TMEM126B, a protein of currently unknown function, abolished Complex I assembly leading to accumulation of low molecular weight complexes. The study suggested that recruitment of CIA30, ACAD9 and Ecsit to the MCIA may be one of the critical functions of TMEM126B. Suppression of NDUFA11, a structural subunit of Complex I necessary for assembly, also led to accumulation of sub complexes ranging from 550KDa to 850KDa molecular weights. Associated with these sub complexes were components of the MCIA complex as well as other assembly factors and a novel protein C3ORF1. Further studies employing knockdown and co-immunoprecipitation methods showed that C3ORF1 was associated with the ∼400KDa ND1 containing subcomplex (Andrews B et al., 2013). Considering the fact that both TMEM126B and C3ORF1 are hydrophobic proteins, the rationale that they help in the assembly of membrane arm is valid.
Other assembly factors: Four additional assembly factors have also been hypothesized for Complex I assembly. These include: Ind1, AIF and FoxRed1 (Fassone et al., 2010; Sheftel et al., 2009; Vahsen et al., 2004). These were either discovered because of pathogenic mutation in patients or with the help of bioinformatics. While the exact role of these assembly factors in Complex I is not understood, knockdown of these proteins leads to decrease in Complex I levels.
4. Complex I assembly defects and disease
Among the mtDNA encoded subunits, ND1 is thought to have a quinone binding site and may be binding to ubiquinone while ND2, ND4 and ND5 resemble sodium and potassium antiporters and may be responsible for proton pumping activity (Carroll et al., 2013; Fearnley et al.). Structurally, mutations in either ND4, ND5 or ND6 have been observed to have effects on the assembly of Complex I. Pathogenic mutations in ND4 subunit causing Leber's hereditary optic neuropathy, abolish the Complex I assembly pathway causing a deficiency of Complex I (Bai et al., 2001; Hofhaus and Attardi, 1993; Huoponen et al., 1990). Mutations in ND6 are thought to abolish assembly of the membrane arm leading to a Complex I deficiency in patients with Leber's optic neuropathy (Bai and Attardi, 1998; Bai et al., 2004; Chinnery et al., 2001). Mutations in ND5 have also been known to cause Complex I deficiency and are frequently seen in patients with Leber's neuropathy and other disorders of oxidative phosphorylation (Bai et al., 2004; Blok et al., 2007; Brown et al., 1992; Hofhaus and Attardi, 1995). Mutations in ND1, however, do not cause a deficiency of Complex I but are known to affect the function of Complex I and are seen in Leber's neuropathy as well as MELAS syndrome (Pätsi et al., 2012). While mutations in ND4, ND5 and ND6 all cause Complex I deficiency, the difference in the severity of the disease phenotypes has not been understood very well. For instance, loss of ND5 subunit gives rise to a milder phenotype than ND4 and sponataneous recovery of disease phenotype is often observed in patients with ND6 mutations. Thus understanding the assembly of Complex I and the contributions of the different subunits has become of paramount importance.
Among the nuclear encoded subunits mutations in almost all core subunits and some of the supernumerary subunits as well as other protein participating in OXPHOS assembly have been encountered in mitochondrial disorders with assembly defects occurring due to mutations in NDUFV1, NDUFV2, NDUFS1, NDUFS2, NDUFS3, NDUFS7 and NDUFS8 as well as Complex I assembly factors. (Dunning et al., 2007; Fernandez-Vizarra et al., 2007; Benit, et al., 2001, 2003, 2004). Understanding Complex I assembly is therefore of significant importance and relevance to human health.
In summary, considering the significance of Complex assembly and assembly factors in human pathology, greater understanding is needed of this complicated process. It is particularly exciting that researchers are moving beyond the traditional approaches and are now relying on sophisticated proteomic approaches to identify proteins involved in the Complex I assembly pathway. Understanding the expanding role of respiratory super complexes in assembly of Complex I and thus human diseases will be the focus of next phase of Complex I research.
Acknowledgments
The related work carried out in the authors' lab has been supported by grants from National Institute of Health (R21 NS072777, and R01 GM109434) to YB.
References
- Andrews B, Carroll J, Ding S, Fearnley I, Walker JE. (2013). Assembly factors for human Complex I membrane arm. Proc Natl Acad Sci U S A. 2013 Nov 19;110(47):18934–9. doi: 10.1073/pnas.1319247110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonicka H, Ogilvie I, Taivassalo T, Anitori RP, Haller RG, Vissing J, Kennaway NG, Shoubridge EA. Identification and characterization of a common set of complex I assembly intermediates in mitochondria from patients with complex I deficiency. J Biol Chem. 2003;278:43081–43088. doi: 10.1074/jbc.M304998200. [DOI] [PubMed] [Google Scholar]
- Attardi G, Schatz G. Biogenesis of mitochondria. Annu Rev Cell Biol. 1988;4:289–333. doi: 10.1146/annurev.cb.04.110188.001445. [DOI] [PubMed] [Google Scholar]
- Bai Y, Attardi G. The mtDNA-encoded ND6 subunit of mitochondrial NADH dehydrogenase is essential for the assembly of the membrane arm and the respiratory function of the enzyme. EMBO J. 1998;17:4848–4858. doi: 10.1093/emboj/17.16.4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Shakeley RM, Attardi G. Tight control of respiration by NADH dehydrogenase ND5 subunit gene expression in mouse mitochondria. Mol Cell Biol. 2000;20:805–815. doi: 10.1128/mcb.20.3.805-815.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Hájek P, Chomyn A, Chan E, Seo BB, Matsuno-Yagi A, Yagi T, Attardi G. Lack of complex I activity in human cells carrying a mutation in MtDNA-encoded ND4 subunit is corrected by the Saccharomyces cerevisiae NADH-quinone oxidoreductase (NDI1) gene. J Biol Chem. 2001;276:38808–38813. doi: 10.1074/jbc.M106363200. [DOI] [PubMed] [Google Scholar]
- Bai Y, Hu P, Park JS, Deng JH, Song X, Chomyn A, Yagi T, Attardi G. Genetic and functional analysis of mitochondrial DNA-encoded complex I genes. Ann N Y Acad Sci. 2004;1011:272–283. doi: 10.1007/978-3-662-41088-2_26. [DOI] [PubMed] [Google Scholar]
- Baradaran R, Berrisford JM, Minhas GS, Sazanov LA. Crystal structure of the entire respiratory complex I. Nature. 2013;494:443–448. doi: 10.1038/nature11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belevich I, Verkhovsky MI, Wikström M. Proton-coupled electron transfer drives the proton pump of cytochrome c oxidase. Nature. 2006;440:829–832. doi: 10.1038/nature04619. [DOI] [PubMed] [Google Scholar]
- Benit P, Chretien D, Kadhom N, de Lonlay-Debeney P, Cormier-Daire V, Cabral A, Peudenier S, Rustin P, Munnich A, Rotig A. Large-scale deletion and point mutations of the nuclear NDUFV1 and NDUFS1 genes in mitochondrial complex I deficiency. Am J Hum Genet. 2001;68:1344–1352. doi: 10.1086/320603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benit P, Beugnot R, Chretien D, Giurgea I, de Lonlay-Debeney P, Issartel JP, Corral-Debrinski M, Kerscher S, Rustin P, Rotig A, Munnich A. Mutant NDUFV2 subunit of mitochondrial complex I causes early onset hypertrophic cardiomyopathy and encephalopathy. Hum Mutat. 2003;21:582–586. doi: 10.1002/humu.10225. [DOI] [PubMed] [Google Scholar]
- Benit P, Slama A, Cartault F, Giurgea I, Chretien D, Lebon S, Marsac C, Munnich A, Rotig A, Rustin P. Mutant NDUFS3 subunit of mitochondrial complex I causes Leigh syndrome. J Med Genet. 2004;41:14–17. doi: 10.1136/jmg.2003.014316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blok MJ, Spruijt L, de Coo IFM, Schoonderwoerd K, Hendrickx A, Smeets HJ. Mutations in the ND5 subunit of complex I of the mitochondrial DNA are a frequent cause of oxidative phosphorylation disease. J Med Genet. 2007;44:e74. doi: 10.1136/jmg.2006.045716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourges I, Ramus C, Mousson de Camaret B, Beugnot R, Remacle C, Cardol P, Hofhaus G, Issartel JP. Structural organization of mitochondrial human complex I: role of the ND4 and ND5 mitochondria-encoded subunits and interaction with prohibitin. Biochem J. 2004;383:491–499. doi: 10.1042/BJ20040256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MD, Voljavec AS, Lott MT, Torroni A, Yang CC, Wallace DC. Mitochondrial DNA complex I and III mutations associated with Leber's hereditary optic neuropathy. Genetics. 1992;130:163–173. doi: 10.1093/genetics/130.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll J, Shannon RJ, Fearnley IM, Walker JE, Hirst J. Definition of the nuclear encoded protein composition of bovine heart mitochondrial complex I. Identification of two new subunits. J Biol Chem. 2002;277:50311–50317. doi: 10.1074/jbc.M209166200. [DOI] [PubMed] [Google Scholar]
- Carroll J, Fearnley IM, Shannon RJ, Hirst J, Walker JE. Analysis of the subunit composition of complex I from bovine heart mitochondria. Mol Cell Proteomics MCP. 2003;2:117–126. doi: 10.1074/mcp.M300014-MCP200. [DOI] [PubMed] [Google Scholar]
- Carroll J, Fearnley IM, Skehel JM, Shannon RJ, Hirst J, Walker JE. Bovine Complex I Is a Complex of 45 Different Subunits. J Biol Chem. 2006;281:32724–32727. doi: 10.1074/jbc.M607135200. [DOI] [PubMed] [Google Scholar]
- Carroll J, Ding S, Fearnley IM, Walker JE. Post-translational modifications near the quinone binding site of mammalian complex I. J Biol Chem. 2013;288:24799–24808. doi: 10.1074/jbc.M113.488106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery PF. Mitochondrial Disorders Overview. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Smith RJ, Stephens K, editors. GeneReviews™. Seattle (WA): University of Washington, Seattle; 1993. [PubMed] [Google Scholar]
- Chomyn A. In vivo labeling and analysis of human mitochondrial translation products. Methods Enzymol. 1996;264:197–211. doi: 10.1016/s0076-6879(96)64020-8. [DOI] [PubMed] [Google Scholar]
- Chomyn A, Mariottini P, Cleeter MWJ, Ragan CI, Matsuno-Yagi A, Hatefi Y, Doolittle RF, Attardi G. (1985). Six unidentified reading frames of human mitochondrial DNA encode components of the respiratory-chain NADH dehydrogenase. 1985 Apr 18;314:592–597. doi: 10.1038/314592a0. Publ. Online. doi:101038314592a0. [DOI] [PubMed] [Google Scholar]
- Dunning CJR, McKenzie M, Sugiana C, Lazarou M, Silke J, Connelly A, Fletcher JM, Kirby DM, Thorburn DR, Ryan MT. Human CIA30 is involved in the early assembly of mitochondrial complex I and mutations in its gene cause disease. EMBO J. 2007;26:3227–3237. doi: 10.1038/sj.emboj.7601748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremov RG, Baradaran R, Sazanov LA. The architecture of respiratory complex I. Nature. 2010;465:441–445. doi: 10.1038/nature09066. [DOI] [PubMed] [Google Scholar]
- Fassone E, Rahman S. Complex I deficiency: clinical features, biochemistry and molecular genetics. J Med Genet. 2012;49:578–590. doi: 10.1136/jmedgenet-2012-101159. [DOI] [PubMed] [Google Scholar]
- Fassone E, Duncan AJ, Taanman JW, Pagnamenta AT, Sadowski MI, Holand T, Qasim W, Rutland P, Calvo SE, Mootha VK, et al. FOXRED1, encoding an FAD-dependent oxidoreductase complex-I-specific molecular chaperone, is mutated in infantile-onset mitochondrial encephalopathy. Hum Mol Genet. 2010;19:4837–4847. doi: 10.1093/hmg/ddq414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearnley IM, Carroll J, Walker JE. Cardiovascular Proteomics. New Jersey: Humana Press; Proteomic Analysis of the Subunit Composition of Complex I (NADH:Ubiquinone Oxidoreductase) From Bovine Heart Mitochondria; pp. 103–126. [DOI] [PubMed] [Google Scholar]
- Fernandez-Vizarra E, Bugiani M, Goffrini P, Carrara F, Farina L, Procopio E, Donati A, Uziel G, Ferrero I, Zeviani M. Impaired complex III assembly associated with BCS1L gene mutations in isolated mitochondrial encephalopathy. Hum Mol Genet. 2007;16:1241–1252. doi: 10.1093/hmg/ddm072. [DOI] [PubMed] [Google Scholar]
- Friedrich T, Böttcher B. The gross structure of the respiratory complex I: a Lego System. Biochim Biophys Acta. 2004;1608:1–9. doi: 10.1016/j.bbabio.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Friedrich T, Weiss H. Modular evolution of the respiratory NADH:ubiquinone oxidoreductase and the origin of its modules. J Theor Biol. 1997;187:529–540. doi: 10.1006/jtbi.1996.0387. [DOI] [PubMed] [Google Scholar]
- Gerards M, Sluiter W, van den Bosch BJC, de Wit LEA, Calis CMH, Frentzen M, Akbari H, Schoonderwoerd K, Scholte HR, Jongbloed RJ, et al. Defective complex I assembly due to C20orf7 mutations as a new cause of Leigh syndrome. J Med Genet. 2010;47:507–512. doi: 10.1136/jmg.2009.067553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershoni M, Fuchs A, Shani N, Fridman Y, Corral-Debrinski M, Aharoni A, Frishman D, Mishmar D. Coevolution predicts direct interactions between mtDNA-encoded and nDNA-encoded subunits of oxidative phosphorylation complex i. J Mol Biol. 2010;404:158–171. doi: 10.1016/j.jmb.2010.09.029. [DOI] [PubMed] [Google Scholar]
- Guénebaut V, Schlitt A, Weiss H, Leonard K, Friedrich T. Consistent structure between bacterial and mitochondrial NADH:ubiquinone oxidoreductase (complex I) J Mol Biol. 1998;276:105–112. doi: 10.1006/jmbi.1997.1518. [DOI] [PubMed] [Google Scholar]
- Haack TB, Danhauser K, Haberberger B, Hoser J, Strecker V, Boehm D, Uziel G, Lamantea E, Invernizzi F, Poulton J, Rolinski B, Iuso A, Biskup S, Schmidt T, Mewes HW, Wittig I, Meitinger T, Zeviani M, Prokisch H. Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nature Genetics. 2010;42:1131–1134. doi: 10.1038/ng.706. [DOI] [PubMed] [Google Scholar]
- Heide H1, Bleier L, Steger M, Ackermann J, Dröse S, Schwamb B, Zörnig M, Reichert AS, Koch I, Wittig I, Brandt U. Complexome profiling identifies TMEM126B as a component of the mitochondrial complex I assembly complex. Cell Metab. 2012 Oct 3;16(4):538–49. doi: 10.1016/j.cmet.2012.08.009. [DOI] [PubMed] [Google Scholar]
- Hofhaus G, Attardi G. Lack of assembly of mitochondrial DNA-encoded subunits of respiratory NADH dehydrogenase and loss of enzyme activity in a human cell mutant lacking the mitochondrial ND4 gene product. EMBO J. 1993;12:3043–3048. doi: 10.1002/j.1460-2075.1993.tb05973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofhaus G, Attardi G. Efficient selection and characterization of mutants of a human cell line which are defective in mitochondrial DNA-encoded subunits of respiratory NADH dehydrogenase. Mol Cell Biol. 1995;15:964–974. doi: 10.1128/mcb.15.2.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofhaus G, Weiss H, Leonard K. Electron microscopic analysis of the peripheral and membrane parts of mitochondrial NADH dehydrogenase (Complex I) J Mol Biol. 1991;221:1027–1043. doi: 10.1016/0022-2836(91)80190-6. [DOI] [PubMed] [Google Scholar]
- Huoponen K, Vilkki J, Savontaus ML, Aula P, Nikoskelainen EK. Analysis of mitochondrial ND4 gene DNA sequence in Finnish families with Leber hereditary optic neuroretinopathy. Genomics. 1990;8:583–585. doi: 10.1016/0888-7543(90)90049-z. [DOI] [PubMed] [Google Scholar]
- Keeney PM, Xie J, Capaldi RA, Bennett JP., Jr Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci Off J Soc Neurosci. 2006;26:5256–5264. doi: 10.1523/JNEUROSCI.0984-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Küffner R, Rohr A, Schmiede A, Krüll C, Schulte U. Involvement of two novel chaperones in the assembly of mitochondrial NADH:Ubiquinone oxidoreductase (complex I) J Mol Biol. 1998;283:409–417. doi: 10.1006/jmbi.1998.2114. [DOI] [PubMed] [Google Scholar]
- Lazarou M, McKenzie M, Ohtake A, Thorburn DR, Ryan MT. Analysis of the Assembly Profiles for Mitochondrial- and Nuclear-DNA-Encoded Subunits into Complex I. Mol Cell Biol. 2007;27:4228–4237. doi: 10.1128/MCB.00074-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffen JL, Smeitink JA, Trijbels JM, Janssen AJ, Triepels RH, Sengers RC, van den Heuvel LP. Isolated complex I deficiency in children: clinical, biochemical and genetic aspects. Hum Mutat. 2000;15:123–134. doi: 10.1002/(SICI)1098-1004(200002)15:2<123::AID-HUMU1>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Mimaki M, Wang X, McKenzie M, Thorburn DR, Ryan MT. Understanding mitochondrial complex I assembly in health and disease. Biochim Biophys Acta BBA - Bioenerg. 2012;1817:851–862. doi: 10.1016/j.bbabio.2011.08.010. [DOI] [PubMed] [Google Scholar]
- Moreno-Lastres D, Fontanesi F, García-Consuegra I, Martín MA, Arenas J, Barrientos A, Ugalde C. Mitochondrial complex I plays an essential role in human respirasome assembly. Cell Metab. 2012;15:324–335. doi: 10.1016/j.cmet.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehls U, Friedrich T, Schmiede A, Ohnishi T, Weiss H. Characterization of assembly intermediates of NADH:ubiquinone oxidoreductase (complex I) accumulated in Neurospora mitochondria by gene disruption. J Mol Biol. 1992;227:1032–1042. doi: 10.1016/0022-2836(92)90519-p. [DOI] [PubMed] [Google Scholar]
- Nouws J, Nijtmans L, Houten SM, van den Brand M, Huynen M, Venselaar H, Hoefs S, Gloerich J, Kronick J, Hutchin T, et al. Acyl-CoA dehydrogenase 9 is required for the biogenesis of oxidative phosphorylation complex I. Cell Metab. 2010;12:283–294. doi: 10.1016/j.cmet.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Ogilvie I, Kennaway NG, Shoubridge EA. A molecular chaperone for mitochondrial complex I assembly is mutated in a progressive encephalopathy. J Clin Invest. 2005;115:2784–2792. doi: 10.1172/JCI26020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa S, Rasmo DD, Technikova-Dobrova Z, Panelli D, Signorile A, Scacco S, Petruzzella V, Papa F, Palmisano G, Gnoni A, et al. Respiratory chain complex I, a main regulatory target of the cAMP/PKA pathway is defective in different human diseases. FEBS Lett. 2012;586:568–577. doi: 10.1016/j.febslet.2011.09.019. [DOI] [PubMed] [Google Scholar]
- Pätsi J, Maliniemi P, Pakanen S, Hinttala R, Uusimaa J, Majamaa K, Nyström T, Kervinen M, Hassinen IE. LHON/MELAS overlap mutation in ND1 subunit of mitochondrial complex I affects ubiquinone binding as revealed by modeling in Escherichia coli NDH-1. Biochim Biophys Acta. 2012;1817:312–318. doi: 10.1016/j.bbabio.2011.10.014. [DOI] [PubMed] [Google Scholar]
- Perales-Clemente E, Fernández-Vizarra E, Acín-Pérez R, Movilla N, Bayona-Bafaluy MP, Moreno-Loshuertos R, Pérez-Martos A, Fernández-Silva P, Enríquez JA. Five entry points of the mitochondrially encoded subunits in mammalian complex I assembly. Mol Cell Biol. 2010;30:3038–3047. doi: 10.1128/MCB.00025-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potluri P, Yadava N, Scheffler IE. The role of the ESSS protein in the assembly of a functional and stable mammalian mitochondrial complex I (NADH-ubiquinone oxidoreductase) Eur J Biochem FEBS. 2004;271:3265–3273. doi: 10.1111/j.1432-1033.2004.04260.x. [DOI] [PubMed] [Google Scholar]
- Rhein VF, Carroll J, Ding S, Fearnley IM, Walker JE. NDUFAF7 Methylates Arginine 85 in the NDUFS2 Subunit of Human Complex I. J Biol Chem. 2013;288:33016–33026. doi: 10.1074/jbc.M113.518803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saada A, Edvardson S, Rapoport M, Shaag A, Amry K, Miller C, Lorberboum-Galski H, Elpeleg O. C6ORF66 is an assembly factor of mitochondrial complex I. Am J Hum Genet. 2008;82:32–38. doi: 10.1016/j.ajhg.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saada A, Vogel RO, Hoefs SJ, van den Brand MA, Wessels HJ, Willems PH, Venselaar H, Shaag A, Barghuti F, Reish O, et al. Mutations in NDUFAF3 (C3ORF60), encoding an NDUFAF4 (C6ORF66)-interacting complex I assembly protein, cause fatal neonatal mitochondrial disease. Am J Hum Genet. 2009;84:718–727. doi: 10.1016/j.ajhg.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sazanov LA, Hinchliffe P. Structure of the Hydrophilic Domain of Respiratory Complex I from Thermus thermophilus. Science. 2006;311:1430–1436. doi: 10.1126/science.1123809. [DOI] [PubMed] [Google Scholar]
- Schägger H. Respiratory chain supercomplexes of mitochondria and bacteria. Biochim Biophys Acta BBA - Bioenerg. 2002;1555:154–159. doi: 10.1016/s0005-2728(02)00271-2. [DOI] [PubMed] [Google Scholar]
- Schultz BE, Chan SI. Structures and proton-pumping strategies of mitochondrial respiratory enzymes. Annu Rev Biophys Biomol Struct. 2001;30:23–65. doi: 10.1146/annurev.biophys.30.1.23. [DOI] [PubMed] [Google Scholar]
- Sheftel AD, Stehling O, Pierik AJ, Netz DJA, Kerscher S, Elsässer HP, Wittig I, Balk J, Brandt U, Lill R. Human Ind1, an Iron-Sulfur Cluster Assembly Factor for Respiratory Complex I. Mol Cell Biol. 2009;29:6059–6073. doi: 10.1128/MCB.00817-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain J Neurol. 2003;126:1905–1912. doi: 10.1093/brain/awg170. [DOI] [PubMed] [Google Scholar]
- Sugiana C, Pagliarini DJ, McKenzie M, Kirby DM, Salemi R, Abu-Amero KK, Dahl HHM, Hutchison WM, Vascotto KA, Smith SM, et al. Mutation of C20orf7 disrupts complex I assembly and causes lethal neonatal mitochondrial disease. Am J Hum Genet. 2008;83:468–478. doi: 10.1016/j.ajhg.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuschen G, Sackmann U, Nehls U, Haiker H, Buse G, Weiss H. Assembly of NADH: ubiquinone reductase (complex I) in Neurospora mitochondria. Independent pathways of nuclear-encoded and mitochondrially encoded subunits. J Mol Biol. 1990;213:845–857. doi: 10.1016/S0022-2836(05)80268-2. [DOI] [PubMed] [Google Scholar]
- Ugalde C, Vogel R, Huijbens R, Heuvel Bvanden, Smeitink J, Nijtmans L. Human mitochondrial complex I assembles through the combination of evolutionary conserved modules: a framework to interpret complex I deficiencies. Hum Mol Genet. 2004a;13:2461–2472. doi: 10.1093/hmg/ddh262. [DOI] [PubMed] [Google Scholar]
- Vogel RO, Dieteren CEJ, van den Heuvel LPWJ, Willems PHGM, Smeitink JAM, Koopman WJH, Nijtmans LGJ. Identification of mitochondrial complex I assembly intermediates by tracing tagged NDUFS3 demonstrates the entry point of mitochondrial subunits. J Biol Chem. 2007;282:7582–7590. doi: 10.1074/jbc.M609410200. [DOI] [PubMed] [Google Scholar]
- Wittig I, Braun HP, Schägger H. Blue native PAGE. Nat Protoc. 2006;1:418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- Yadava N, Houchens T, Potluri P, Scheffler IE. Development and characterization of a conditional mitochondrial complex I assembly system. J Biol Chem. 2004;279:12406–12413. doi: 10.1074/jbc.M313588200. [DOI] [PubMed] [Google Scholar]