

Abstract
Background
Rapid industrialization is discharging toxic heavy metals into the environment, disturbing human health in many ways and causing various neurologic, cardiovascular, and dermatologic abnormalities and certain types of cancer. The presence of arsenic in drinking water from different urban and rural areas of the major cities of Pakistan, for example, Lahore, Faisalabad, and Kasur, was found to be beyond the permissible limit of 10 parts per billion set by the World Health Organization. Therefore the present study was initiated to examine the effects of arsenite (As+3) on DNA biosynthesis and cell death.
Methods
After performing cytotoxic assays on a human epithelial carcinoma cell line, expression analysis was done by quantitative polymerase chain reaction, western blotting, and flow cytometry.
Results
We show that As+3 ions have a dose- and time-dependent cytotoxic effect through the activation of the caspase-dependent apoptotic pathway. In contrast to previous research, the present study was designed to explore the early cytotoxic effects produced in human cells during exposure to heavy dosage of As+3 (7.5 µg/ml). Even treatment for 1 h significantly increased the mRNA levels of p21 and p27 and caspases 3, 7, and 9. It was interesting that there was no change in the expression levels of p53, which plays an important role in G2/M phase cell cycle arrest.
Conclusion
Our results indicate that sudden exposure of cells to arsenite (As+3) resulted in cytotoxicity and mitochondrial-mediated apoptosis resulting from up-regulation of caspases.
Keywords: apoptosis, epithelial carcinoma, cytotoxicity, arsenite, caspases, Pakistan
Arsenic exists ubiquitously in soil, water, and air. It is a metalloid that occurs in four different forms: arsenite (As+3), arsenate (As+5), mono-methyl arsenic acid, and di-methyl arsenic acid, but the +3 and +5 oxidation states occur more frequently (1, 2). Epidemiological studies report As+3 as a potent carcinogen (3–7). Humans are mostly exposed to arsenic through contaminated drinking water, either through natural means or industrial pollution (5). Arsenite has also been employed as a medicinal agent since ancient Greek and Roman times. The therapeutic effects of arsenite in the treatment of acute myelogenous leukemia are well recognized (3).
In living cells, As+3 is involved in a series of drastic chemical reactions. It strongly reacts with macromolecules and leads to aberrations in DNA structure (8–10). It has been associated with an increased rate of micronuclei formation and sister chromatid exchange in humans and lab animals (3). Although arsenic does not affect DNA directly, it intensifies the toxic effects of other physiochemical agents through inhibition of DNA repair mechanisms, shifting cell redox potential and altering proteins that control the cell cycle (3). Arsenic alters signal transduction pathways, resulting in activation or inhibition of different regulatory proteins, such as transcription factors that bind to DNA and regulate gene expression (11). Generally, p53 is activated in response to DNA damage, which plays a fundamental role in regulation of the cell cycle. Furthermore, ataxia telangiectasia mutated gene product is also involved in activation of p53; p53 can arrest the cell cycle at various stages: G1/S, G2, G2/M, and M (12, 13).
Although the use of arsenic has been reduced, still elevated levels of arsenic were found in the drinking water of various regions of the Asian subcontinent and Canada. Increase in the ingestion or inhalation of arsenic results in the development of various types of cancer (14). Rapidly growing industries are causing arsenic toxicity in Pakistan. Arsenic levels of 0.05 mg/L of water (50 ppb) are considered safe for human consumption, as regulated by the Pakistan Standards and Quality Control Authority. According to a survey conducted by the Pakistan Environmental Protection Agency, arsenic contamination was found in more than 253 of 392 tube wells.
The present study was designed to analyze the genotoxicity induced by As+3 in human cervical epithelioid carcinoma cells. Unlike earlier studies, the study was extended to explore at the molecular level (transcriptomic level) the acute response of cells to a sudden increased concentration of As+3.
Materials and methods
Mammalian cell culture
HeLa cells (human cervical epithelial carcinoma cells) from the American Type Culture Collection were generously donated by the Veterinary Research Institute, Lahore. The cells were cultivated in Corning T-75 culture flasks (Corning Life Sciences, MA, USA) in Dulbecco's modified Eagle's medium (DMEM) supplemented with high glucose (Invitrogen, CA, USA), 10% fetal bovine serum (Invitrogen), and 1% antibiotic-antimycotic solution (Anti-Anti 100X, Invitrogen). Following trypsinization with 0.05% trypsin-EDTA solution (Invitrogen, CA, USA) using standard protocol, cells were seeded at 3–4 million cells per flask. The flasks were incubated in a CO2 water-jacketed incubator (Thermo Fisher Scientific, MA, USA) at 37°C in 5% CO2 and 95% air. The flask was confluent with a monolayer of cells within 48–72 h, after which growth medium was replaced with DMEM (supplemented with 2% Fetal Bovine Serum [FBS]) as maintenance media.
Cell viability assay
The trypan blue dye exclusion assay was performed to measure viability and proliferation of HeLa cells in the presence of As+3 (sodium meta arsenite, Sigma Aldrich, MO, USA). Dead cells are stained blue as a result of perforations in the cell membrane, whereas the intact membranes of viable cells prevent the dye from entering and the cells remain unstained. HeLa cells were seeded in six-well plates in the maintenance medium at a density of 1.5×104 cells per well. The cells were grown overnight. The following day, cells from one well were trypsinized, stained with 0.2% trypan Blue Solution (Nalgene Packing, Life Technologies, Thermo Fisher Scientific, USA), and the number of viable cells and total cells were counted in a hemocytometer chamber (Hausser Scientific, PA, USA) as the Day 0 count. The other wells were washed and fresh maintenance medium was added, excluding the control well. All wells, except the control, were treated with different concentrations of sodium meta arsenite for 24 h. The next day, the cells were trypsinized and stained with 0.2% trypan blue, and the viable cells were counted in a hemocytometer.
Cytotoxic effects of arsenite
The neutral red (NR) uptake assay was applied to measure the level of cytotoxicity (15). The NR assay is a cell viability test that is based on the ability of living cells to take up NR dye within their lysosomes. The uptake of NR is directly proportional to cellular viability. HeLa cells were grown in a T-75 flask, trypsinized, and counted in a hemocytometer. Cell viability was confirmed up to be ≥95% by trypan blue exclusion. Then cells were diluted in the cell culture medium, gently agitated, and placed in a sterile reservoir, and 200 µl of the cell suspension was dispensed per well of a 96-well tissue culture microtiter plate (about 1×104 cells per well). The cells were allowed to grow and adhere to the surface of the plate in maintenance medium. After 24 h, the medium was aspirated from each well and 200 µl of fresh medium containing different concentrations of sodium meta arsenite were added to each sample well. The plate was covered and incubated as described. After 24 h, digital images were taken using a phase contrast inverted microscope (Inverted Microscope System IX53, Olympus, Japan), to record the morphological changes of cells (e.g. rounding, vacuolization, detachment, and lysis) prior to the addition of medium containing NR. The NR medium was prepared by dissolving 50 µg/ml NR dye (Neutral Rot, Act. 1369, Germany) in DMEM medium (for each well) and centrifuging at 1,800 rpm for 10 min to remove any crystals of NR. After 24 h, the medium of all wells was decanted; and replaced it with 100 µl of NR medium (in each well) using a multichannel pipette and incubated at 37°C for 3 h to allow the uptake of neutral red by the cells. Cells were washed and destained with 150 µl of destaining solution containing 50% H2O, 49% C2H5OH (99.8%, Riedel-de Haën, Germany) and 1% CH3COOH (Sigma Aldrich, Saint Louis, Missouri, USA). A microtiter plate shaker (Camlab Choice Microplate Shaker, Camlab, UK) was used for shaking the plate at room temperature for 10 min. Optical density (OD) was monitored using a microtiter plate reader spectrophotometer (xMark™, BioRad, USA) at a wavelength of 540 nm. All assays were done in triplicate. The percentage cellular viability was expressed as OD540 nm values derived from the cells treated with arsenite, divided by that from untreated samples (100% represented zero cytotoxicity and lower values indicated the presence of cytotoxicity).
Microscopic evaluation of cytotoxic effects
HeLa cells were grown to 80–90% confluence in T-25 flasks. Maintenance medium (5 ml) treated with 2.5 µg/ml of As+3 was added, and the flask was incubated at 37°C in 5% CO2. A phase contrast microscope was used to monitor the cultures for morphological changes after 6, 12, and 24 h. The digital images of all arsenite-treated flasks were acquired with an attached Charge-Coupled Device (CCD) SP 480 H color camera (Olympus, Japan).
Quantitative polymerase chain reaction analysis
HeLa cells grown in T-75 flasks (after treatment with 7.5 µg/ml As+3 for 30 and 60 min) were trypsinized and harvested by centrifugation at 1,500 rpm. The total RNA was extracted using an RNeasy Mini Kit (Qiagen, California, USA) according to the manufacturer's protocol. To avoid any traces of residual DNA, the isolated total RNA was subjected to DNase treatment (Turbo DNase Kit, Ambion, TX, USA). DNase was later inactivated according to the kit protocol. About 5 µg of RNA was used for a reverse transcriptase polymerase chain reaction (PCR), which was performed using a SuperScript III RT Kit (Invitrogen). First-strand cDNA was synthesized using random hexamers, in accordance with the manufacturer's protocol.
Quantitative PCR was carried out to quantify the mRNA levels of cyclins (A, B, and E), Cyclin Dependent Kinases (CDKs) (1 and 2), CDK inhibitors (p21, p27, p53, and p57), and caspases (3, 7, and 9), while GAPDH (glyceraldehydes-3-phosphate dehydrogenase) was employed as an internal control. The primers were designed using Primer3web version 4.0.0 and were chemically synthesized by Macrogen, Korea (Table 1). Each PCR reaction of 20 µl (performed in duplicate) contained 10 µl of 2× SYBR Green PCR Master Mix (Life Technologies, UK), 0.2 µl of each forward (20 pmol) and reverse (20 pmol) primer and 100 ng of template cDNA. The qPCR was performed on an Applied Biosystem 7500/7500 Fast Real-Time PCR system (CA, USA). Thermal cycling was performed using a machine preset two-step cycling protocol. It consisted of 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min.
Table 1.
Sequences of primers used for quantitative (real-time) PCR
| No. | Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|---|
| 1 | GAPDH | TGATGACATCAAGAAGGTGGTGAAG | TCCTTGGAGGCCATGTGGGCCAT |
| 2 | CyclinA | TAGACACCGGCAACTCAAG | TCTTCAGACTGGGAGAGGAGA |
| 3 | CyclinB1 | GAACAACTGCAGGCCAAAAT | CACTGGCACCAGCATAGG |
| 4 | CyclinE1 | GAAATGGCCAAAATCGACAG | TGTCAGGTGTGGGGATCA |
| 5 | CDK1 | TGGAGAAGGTACCTATGGAGTTG | AGGAACCCCTTCCTCTTCAC |
| 6 | CDK2 | AAAGCCAGAAACAAGTTGACG | GAGATCTCTCGGATGGCAGT |
| 7 | p21 | CTGGGGATGTCCGTCAGAAC | GCCATTAGCGCATCACAGT |
| 8 | p27 | AAACGTGCGAGTGTCTAACG | GTCCCGGGTTAACTCTTCGT |
| 9 | p53 | AGGTTGGCTCTGACTGTACC | GATTCTCTTCCTCTGTGCGC |
| 10 | p57 | CAGGAGCCTCTCGCTGAC | CCGGACAGCTTCTTGATCG |
| 11 | Caspase-3 | TGAGCCATGGTGAAGAAGGA | TCGGCCTCCACTGGTATTTT |
| 12 | Caspase-7 | ACCGGTCCTCGTTTGTACC | TTGTACTGATATGTAGGCACTCG |
| 13 | Caspase-9 | CTCTTGAGAGTTTGAGGGGAAA | ACTCACGGCAGAAGTTCACA |
GAPDH: glyceraldehydes-3-phosphate dehydrogenase.
Western blotting analysis
HeLa cells grown in T-75 flasks (after 1-day treatment with 2.5 µg/ml As+3) were trypsinized and harvested by centrifugation at 1,500 rpm and rinsed with 1× PBS buffer. The cells were lysed by sonication in HEGMT buffer (25 mM HEPES with a pH of 7.9, 12.5 mM MgCl2, 150 mM NaCl, 10% glycerol, 0.1 mM EDTA, 0.5% Tween-20, 1× Protease Inhibitor Cocktail) at 10% amplitude for 1 min, followed by a quick freeze-thawing step and centrifugation at 11,000 rpm for 15 min at 4°C. The supernatants were collected and used for western blotting. The protein concentration of cell extracts was measured by using a BCA kit (Pierce, Illinois, USA) according to the kit manual.
After adjusting the protein concentrations, the lysates were boiled with loading buffer at 95°C for 5 min, run in 12% SDS-polyacrylamide gels, and transferred for western blotting to a polyvinylidene difluoride (PVDF) membrane (Invitrogen) at room temperature by employing a semidry transfer technique. The PVDF membrane was blocked with 2% non-fat milk (prepared in 1× PBS containing 0.01% Tween-20) for 45 min at room temperature. Tubulin and cleaved caspase-3 primary antibodies were prepared in 1.5% non-fat milk. The PVDF membrane was incubated in the primary antibody solution for 2 h, followed by three subsequent washings with wash buffer (1× PBS buffer containing 0.01% Tween-20). After washing for an hour, the membrane was incubated in a solution of secondary antibodies, also prepared in 1.5% non-fat milk. After three washes with wash buffer, the blot was developed on x-ray films (GE Healthcare, UK) by using Enhanced chemiluminescence (ECL) Reagent (GE Healthcare) chemiluminescence.
Flow cytometric analysis of cell cycle
Cell cycle distribution was studied by FACS (fluorescence activated cell sorting). Briefly, HeLa cells were grown in maintenance medium and then treated with 2.5 µg/ml of As+3 for 12 and 24 h. After posttreatment, cells were harvested by trypsinization, washed twice with 1× PBS, and fixed with 70% ethanol overnight at 4°C. The next day, the cells were harvested by centrifugation at 1,500 rpm for 5 min, resuspended in 0.5 ml 1× PBS, and treated with RNaseA (20 µg/ml), followed by staining with propidium iodide solution (0.5 mg/ml) for 30 min. The cell count was performed in multiple batches with a cell count of 104 in each. The percentage of apoptotic cells was measured and documented by plotting propidium iodide fluorescence versus cell number using the preinstalled internal software system of the flow cytometer (FACSCalibur, BD Bioscience California, USA).
Results
Arsenite affected the proliferation and morphology of cells
Initial experiments were conducted to check the effect of As+3 on the proliferation of HeLa cells. The HeLa cells were incubated with different concentrations of sodium meta arsenite for 24 h and cellular viability was quantified by trypan blue dye exclusion assay. As+3 exhibited a dose-dependent inhibitory effect on HeLa cell proliferation (Fig. 1). Cytotoxic effects of As+3 on HeLa cells were screened by NR assay. About 5 µg/ml of As+3 decreased cellular viability up to 70%, whereas 10 µg/ml of As+3 resulted in only 50% live cells. Phase-contrast microscopy showed that As+3 is cytotoxic to HeLa cells. Our results indicate loss of cellular viability in a dose-dependent fashion (Fig. 2).
Fig. 1.

The effect of As+3 on HeLa cells (trypan blue assay). The values represent the means of triplicate experiments. Error bars represent standard deviation from the mean of triplicate experiments.
Fig. 2.

Kinetics of viability of HeLa cells (neutral red assay) treated with different concentrations of As+3. One hundred percent represents zero cytotoxicity (untreated control cells) and lower values indicate presence of cytotoxicity. Error bars represent standard deviation from the mean of triplicate experiments.
The goal of cytotoxicity studies is to define the toxic effects of arsenite on cells, which might include oxidative stress and inflammatory responses leading to apoptosis. Arsenite resulted in a decrease of cell proliferation. Long-term exposure to arsenite resulted in the shrinking and rounding of cells and in cell death (Fig. 3).
Fig. 3.
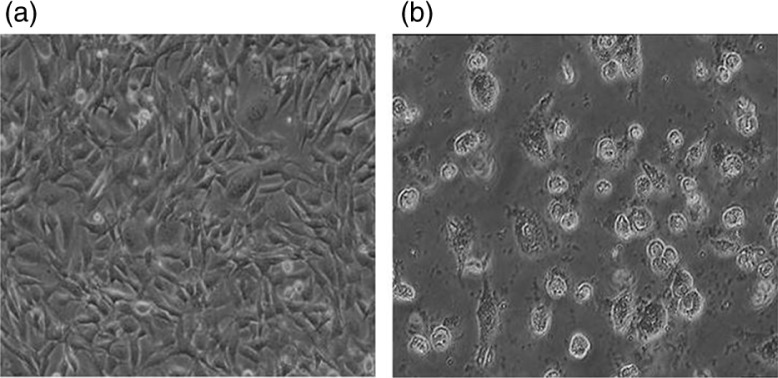
Arsenite-induced morphological deformities in HeLa cell lines. a, untreated cells; b, arsenite-treated cells.
At mRNA level p53 is not involved in As+3-induced G2/M cell cycle arrest
The molecular mechanism of As+3-induced cytotoxicity was analyzed by quantitative PCR. Cells treated with 7.5 µg/ml As+3 for 30 or 60 min did not reveal any fluctuation in the levels of cyclins and CDKs. It has been reported that p53 plays a critical role in G2/M phase cell cycle arrest; however, in this study, arsenic did not significantly affect p53 at the mRNA level (Fig. 4).
Fig. 4.

The relative expression of cyclins (A, B, and E), CDKs (1 and 2), CDK inhibitors (p21, p27, p53, and p57), and caspases (3, 7, and 9) was determined by quantitative PCR. The test was performed in duplicate.
Arsenite induces the expression of DNA damage response genes
Even exposing HeLa cells to As+3 for 30 min led to an increase in mRNA levels of the CDK inhibitory proteins p21 and p27, in a time-dependent manner (Fig. 4). Generally, p21 stops cell proliferation in response to DNA damage or replication stress by blocking CDK2 (G1/S-CDK and S-CDK) and binding to PCNA (proliferating cell nuclear antigen), an auxiliary component of DNA polymerases δ and ɛ (16, 17). Interestingly, in mammals p21 is transcriptionally activated by p53, although its mRNA levels did not alter very much. Likewise, p27 also suppresses the same CDKs targeted by p21 and helps cells withdraw from the cell cycle when they terminally differentiate (18). Arsenic does not directly damage DNA, but it targets the enzymes involved innucleotide excision repair and base excision repair mechanisms (10, 19), which results in DNA damage and activation of p53 (Fig. 7).
Fig. 7.

Molecular mechanism of As+3-induced cytotoxicity in HeLa cells (based on 21, 22). According to real-time PCR data, CDK inhibitory proteins p21 and p27 are overexpressed to stop the cell cycle, while caspases are triggered to initiate mitochondria-dependent (intrinsic) apoptosis.
As+3 modulates mRNA levels of apoptosis-related genes
The mechanism of As+3-induced apoptosis was studied in HeLa cells by real-time PCR analysis. Caspases 3, 7, and 9 were overexpressed and supported the induction of apoptosis by triggering intracellular stimuli (cytochrome c) from mitochondria (as shown in Fig. 5). Usually DNA damage leads to alteration in the levels of BCL-2 family proteins, the main regulators of mitochondria-dependent apoptosis (Fig. 7).
Fig. 5.

Western blot analysis. Arsenite toxicity induced overexpression of cleaved caspase-3. Lane 1: treated cells; lane 2: control (untreated cells).
As+3 induced slight G2 accumulation of cell cycle
Cells were treated with As+3 for 24 and 48 h and their cell cycles were examined by FACS. As shown in Fig. 6, there was slight increase of G1 and S phases at 24 h and accumulation of the G2/M phase at 48 h after As+3treatment, suggesting that As+3 alters the cell cycle by impairing DNA repair and replication. After 48 h of As+3 treatment, the appearance of cells with sub-G1 level DNA are indicative of recent apoptotic events.
Fig. 6.

Untreated HeLa cells were grown for 24 h (a) and 48 h (c) in standard medium and compared to posttreatment with arsenite for 24 h (b) and 48 h (d). Histogram of propidium iodide uptake versus cell count to depict DNA fragmentation in HeLa cells.
Discussion
Arsenite is a ubiquitous and toxic environmental pollutant associated with an increased risk of human cancers (4). In the current study, the underlying mechanisms associated with As+3-mediated toxicity, DNA damage, and proliferation were explored in depth. According to the present study, As+3 can affect the proliferation rate of cervical cancer cells in a dosage- and time-dependent manner. Overexpression of DNA damage-response elements is clear evidence of DNA damage. The mode of this genotoxicity may be due to blockage of DNA-repair enzymes as well as to direct damage in DNA caused by reactive oxygen species (ROS) generated by As+3. ROS typically includes hydrogen peroxide (H2O2), superoxide (O2 −), and hydroxyl (OH) free radicals, which cause severe indirect and direct damage to cellular components, including DNA, and ultimately lead to apoptosis (3, 20). According to previous reports, arsenic trioxide (As2O3) induces production of ROS in vitro as well as in vivo. Chinese hamster ovary (CHO) cells resistant to H2O2 are also resistant to As+3, whereas CHO cells deficient in antioxidant enzymes are hypersensitive to arsenite (3).
When DNA is damaged, protein kinases involved in phosphorylation of p53 are activated. Normally, MDM2 binds to p53 and promotes its proteasome-mediated degradation. Accumulation of phosphorylated p53 facilitates production of p21 (21); p21 binds and inactivates CDK2 (G1/S-CDK and S-CDK), which results in arrest of the cell cycle in the G1 phase (Fig. 7).
As+3 induced mitochondria-dependent apoptosis in cervical cancer cells. Generally, the intrinsic or mitochondria-dependent apoptotic pathway is regulated by Bax and Bak proteins. Their translocation, oligomerization, and insertion into the outer membrane of the mitochondrion is followed by the release of cytochrome c in the mitochondrial intermembrane space, which generates a cytosolic apoptosome complex with APAF-1 (apoptotic protease activating factor 1) and procaspase-9. This leads to the activation of procaspase-9, which triggers the caspase cascade by activation of procaspase-3 (22).
In conclusion, the current study has demonstrated that arsenite functions in mitochondrial-mediated apoptosis by activation of caspases 3 and 9. Although p53 expression levels vary frequently during DNA damage, it remained unaltered in our study. More investigations are needed to elucidate the mechanisms involved.
Conflict of interest and funding
The authors have not received any funding or benefits from industry or elsewhere to conduct this study.
References
- 1.Hughes MF, Beck BD, Chen Y, Lewis AS, Thomas DJ. Arsenic exposure and toxicology: a historical perspective. Toxicol Sci. 2011;123:305–32. doi: 10.1093/toxsci/kfr184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petrick JS, Ayala-Fierro F, Cullen WR, Carter DE, Vasken Aposhian H. Monomethylarsonous acid (MMA(III)) is more toxic than arsenite in Chang human hepatocytes. Toxicol Appl Pharmacol. 2000;163:203–7. doi: 10.1006/taap.1999.8872. [DOI] [PubMed] [Google Scholar]
- 3.Hughes MF. Arsenic toxicity and potential mechanisms of action. Toxicol Lett. 2002;133:1–16. doi: 10.1016/s0378-4274(02)00084-x. [DOI] [PubMed] [Google Scholar]
- 4.Rossman TG. Mechanism of arsenic carcinogenesis: an integrated approach. Mutat Res. 2003;533:37–65. doi: 10.1016/j.mrfmmm.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 5.Hindmarsh JT, McCurdy RF. Clinical and environmental aspects of arsenic toxicity. Crit Rev Clin Lab Sci. 1986;23:315–47. doi: 10.3109/10408368609167122. [DOI] [PubMed] [Google Scholar]
- 6.Jha AN, Noditi M, Nilsson R, Natarajan AT. Genotoxic effects of sodium arsenite on human cells. Mutat Res. 1992;284:215–21. doi: 10.1016/0027-5107(92)90005-m. [DOI] [PubMed] [Google Scholar]
- 7.Stevens JJ, Graham B, Walker AM, Tchounwou PB, Rogers C. The effects of arsenic trioxide on DNA synthesis and genotoxicity in human colon cancer cells. Int J Environ Res Public Health. 2010;7:2018–32. doi: 10.3390/ijerph7052018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmad A, Toor RH, Aftab S, Shakoori AR. Anti-proliferative and genotoxic effect of arsenic and lead on human brain cancer cell line. Pakistan J Zool. 2014;46:1069–76. [Google Scholar]
- 9.Chou WC, Hawkins AL, Barrett JF, Griffin CA, Dang CV. Arsenic inhibition of telomerase transcription leads to genetic instability. J Clin Invest. 2001;108:1541–7. doi: 10.1172/JCI14064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen S, Wang C, Weinfeld M, Le XC. Inhibition of nucleotide excision repair by arsenic. Chin Sci Bull. 2013;58:214–21. [Google Scholar]
- 11.Druwe IL, Vaillancourt RR. Influence of arsenate and arsenite on signal transduction pathways: an update. Arch Toxicol. 2010;84:585–96. doi: 10.1007/s00204-010-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–31. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 13.Magnelli L, Ruggiero M, Chiarugi V. The old and the new in p53 functional regulation. Biochem Mol Med. 1997;62:3–10. doi: 10.1006/bmme.1997.2616. [DOI] [PubMed] [Google Scholar]
- 14.Bates MN, Smith AH, Hopenhayn-Rich C. Arsenic ingestion and internal cancers: a review. Am J Epidemiol. 1992;135:462–76. doi: 10.1093/oxfordjournals.aje.a116313. [DOI] [PubMed] [Google Scholar]
- 15.Repetto G, del Peso A, Zurita JL. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat Protoc. 2008;3:1125–31. doi: 10.1038/nprot.2008.75. [DOI] [PubMed] [Google Scholar]
- 16.Coqueret O. New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment? Trends Cell Biol. 2003;13:65–70. doi: 10.1016/s0962-8924(02)00043-0. [DOI] [PubMed] [Google Scholar]
- 17.de Renty C, DePamphilis ML, Ullah Z. Cytoplasmic localization of p21 protects trophoblast giant cells from DNA damage induced apoptosis. PLoS One. 2014;9:e97434. doi: 10.1371/journal.pone.0097434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding J, He G, Gong W, Wen W, Sun W, Ning B, et al. Effects of nickel on cyclin expression, cell cycle progression and cell proliferation in human pulmonary cells. Cancer Epidemiol Biomarkers Prev. 2009;18:1720–9. doi: 10.1158/1055-9965.EPI-09-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ebert F, Weiss A, Bultemeyer M, Hamann I, Hartwig A, Schwerdtle T. Arsenicals affect base excision repair by several mechanisms. Mutat Res. 2011;715:32–41. doi: 10.1016/j.mrfmmm.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 20.Ahmad A, Muneer B, Shakoori AR. Effect of chromium, cadmium and arsenic on growth and morphology of HeLa cells. J Basic Appl Sci. 2012;8:53–8. [Google Scholar]
- 21.Moll UM, Petrenko O. The MDM2-p53 interaction. Mol Cancer Res. 2003;1:1001–8. [PubMed] [Google Scholar]
- 22.Estaquier J, Vallette F, Vayssiere JL, Mignotte B. The mitochondrial pathways of apoptosis. Adv Exp Med Biol. 2012;942:157–83. doi: 10.1007/978-94-007-2869-1_7. [DOI] [PubMed] [Google Scholar]