

Highlights
-
•
The antipsychotic drug olanzapine inhibits the glucose transport of a GLUT4 homologue.
-
•
Olanzapine binds in a newly discovered site at the cytosolic side of the glucose transporter.
-
•
Olanzapine inhibits the glucose transporter by locking it in the inward-facing conformation.
Abbreviations: GLUT, glucose transporter (SLC2); GlcPSe, Staphylococcus epidermidis glucose/H+ symporter; MOE, Molecular Operating Environment; RSO vesicles, right-side-out vesicles; E. coli, Escherichia coli; KPi, potassium phosphate buffer; DTT, dithiothreitol; EDTA, ethylenediaminetetraacetate; HRP, horseradish peroxidase; DMSO, dimethyl sulfoxide; OLZ, olanzapine
Keywords: Carbohydrate transporter, Sugar transporter, Membrane proteins, Molecular docking, Drug side effect, SLC2, GLUT4, Diabetes, Drug design
Abstract
The antipsychotic drug olanzapine is widely prescribed to treat schizophrenia and other psychotic disorders. However, it often causes unwanted side effects, including diabetes, due to disruption of insulin-dependant glucose metabolism through a mechanism yet to be elucidated. To determine if olanzapine can affect the first step in glucose metabolism – glucose transport inside cells – we investigated the effect of this drug on the transport activity of a model glucose transporter. The glucose transporter from Staphylococcus epidermidis (GlcPSe) is specific for glucose, inhibited by various human glucose transporter (GLUT) inhibitors, has high sequence and structure homology to GLUTs, and is readily amenable to transport assay, mutagenesis, and computational modeling. We found that olanzapine inhibits glucose transport of GlcPSe with an IC50 0.9 ± 0.1 mM. Computational docking of olanzapine to the GlcPSe structure revealed potential binding sites that were further examined through mutagenesis and transport assay to identify residues important for olanzapine inhibition. These investigations suggest that olanzapine binds in a polar region of the cytosolic part of the transporter, and interacts with residues R129, strictly conserved in all GLUTs, and N136, conserved in only a few GLUTs, including the insulin-responsive GLUT4. We propose that olanzapine inhibits GlcPSe by impeding the alternating opening and closing of the substrate cavity necessary for glucose transport. It accomplishes this by disrupting a key salt bridge formed by conserved residues R129 and E362, that stabilizes the outward-facing conformation of the transporter.
1. Introduction
Olanzapine is a widely prescribed second generation antipsychotic drug for the treatment of schizophrenia, anxiety and other mental disorders [1–3]. The use of olanzapine and related drugs such as clozapine carries the risk of potentially life-altering side effects including weight gain [4], increased blood cholesterol levels [5], disruption of insulin-dependent glucose uptake [6], and even diabetes [7,8]. Side effects due to olanzapine appear dose dependent [9,10]. The negative effects of antipsychotic agents can be mitigated by administration of general diabetes treatments and weight-loss programs [11], but these medicines can only partially reverse glucose intolerance and weight gain [12], and they have additional side effects [13].
Glucose transport in cells is a key step in glucose metabolism. In humans, facilitated diffusion of glucose and related carbohydrates is mediated by members of the GLUT (SLC2) family. The fourteen human GLUTs transport various substrates, exhibit distinct substrate affinity or specificity and tissue expression, although they share significant sequence homology [14,15]. Crystal structures are available for only two glucose transporters: the bacterial glucose/H+ symporter from Staphylococcus epidermidis (GlcPSe) [16] and human GLUT1 [17]. These structures are similar, with RMSD for their superposition in the transmembrane helices of less than 1.5 Å (GlcPSe PDB ID: 4LDS, GLUT1 PDB ID: 4PYP). Nevertheless, the molecular determinants that confer substrate specificity are poorly understood.
Insulin-dependent glucose transport in adipocytes and skeletal muscle is carried out by GLUT4 [18,19]. GLUT4 is located in intracellular vesicles, which are translocated to the plasma membrane upon insulin stimulation, where GLUT4 is active [20]. It has been proposed that interfering with GLUT4 trafficking disrupts glucose transport [21], leading to the development of diabetes. In adipocytes, olanzapine has an inhibitory effect on insulin-stimulated glucose transport activity [22]. Interestingly, the side-effects of some antiretroviral drugs, including indinavir, are similar to those of olanzapine: weight-gain, obesity, diabetes and others [23]. Furthermore, GLUT4 glucose transport is inhibited by indinavir [24]. This suggested the possibility that olanzapine may interfere directly with glucose transport.
Here, the effect of olanzapine on glucose transport was examined in the bacterial glucose transporter from S. epidermidis, GlcPSe. It is a convenient glucose transporter model for several reasons: it has high affinity and specificity for glucose, known crystal structure, high sequence and structural homology to human glucose transporters (for instance, sequence identity and similarity to GLUT4 are 31% and 54%, respectively) and it is inhibited by human GLUT inhibitors [16]. Additionally GlcPSe is amenable to transport activity assays, mutagenesis and computational modeling.
We found that olanzapine inhibited glucose transport activity of GlcPSe. By computational modeling, coupled with site-directed mutagenesis and transport assay of mutants, we determined the binding site of olanzapine in GlcPSe and proposed a mechanism of inhibition. We discuss the implications of these findings for human glucose transporters.
2. Materials and methods
2.1. Protein expression
GlcPSe was cloned into the pBAD vector (Invitrogen), containing C-terminal 6×His tag [16]. Required mutations were done by site-directed mutagenesis using QuickChange kit (Agilent Technologies). Proteins were expressed in the glucose transporter deficient Escherichia coli strain JM-1100 (from Yale E. coli Genetic Stock Center; genotype: Hfr (PO2A)], garB10, fhuA22, galK2(Oc), λ−, ompF627(T2R), ptsG23, manXYZ-18, Δ(his-gnd)79, mgl-50, fruA10, fadL701(T2R), relA1, thyA111, galP64, pitA10, and spoT). Cells were grown at 37 °C, in Luria Broth medium, with 100 μg/mL ampicillin. Protein expression was induced with 0.2 mM l-arabinose when O.D.600nm reached 0.6; cells were grown for 3 more hours. Cells were harvested by centrifugation at 2500g for 5 min, washed with 0.1 M Potassium phosphate (KPi) pH 7.5 and 10 mM MgSO4 (buffer A), centrifuged again and finally resuspended in buffer A so that O.D.600nm was 2.0. This cell solution was warmed to room temperature and used for transport assay. Protein expression was checked by Western Blotting, with penta-His HRP conjugate antibody (5 PRIME). Band density of His-tagged proteins in the Western film was analyzed by ImageJ software [25] and Kodak 1D Image Analysis (Eastman Kodak) software, and then the relative mean values to wild-type were calculated.
2.2. Preparation of right-side-out (RSO) vesicles
The right-side-out (RSO) membrane vesicles of JM1100 E. coli cells were prepared as described previously [16,26,27]. Cells were resuspended in 30 mM Tris·HCl, pH 8.0, containing sucrose (30% wt/vol), at a concentration of 1.0 g wet pellet/80 mL, with lysozyme, and incubated at room temperature for 45 min. The spheroplasts were harvested by centrifugation at 5000g for 30 min at 4 °C. They were resuspended and rapidly diluted into pre-warmed 50 mM KPi (pH 7.5) with 5 mM DTT. Then 10 mM K2EDTA was added, and the spheroplasts were incubated for 10 min at 37 °C, followed by the addition of 20 mM MgSO4 and another 10-min incubation. The RSO vesicles were centrifuged at 12,000g for 30 min and then resuspended in ice-cold 0.1 M KPi (pH 7.5) with 10 mM K2EDTA. Finally, RSO vesicles were recovered in two steps. Unbroken spheroplasts and cell pellet were removed by centrifuging at 2500 rpm with a SS-34 rotor for 12 min. The supernatant was centrifuged at 15,000 rpm with a SS-34 rotor for 15 min, and the resulting pellet was resuspended in buffer A. This vesicle suspension was frozen in liquid nitrogen and stored at −80 °C until use.
2.3. Radioactive glucose uptake assay
Transport assay was initiated by the addition of 14C-radiolabeled glucose (Moravek Biochemicals) to 50 μL cells or RSO vesicles (at O.D.600nm of 2.0) in buffer A; after 1 min, the transport was stopped with ice-chilled quench buffer [0.1 M KPi (pH 5.5) and 0.1 M LiCl]. For the assay with RSO vesicles, prior to the addition of glucose, 20 mM ascorbate and 0.2 mM phenazine methosulfate were added [28]. The solution was filtered with a cellulose nitrate membrane filter (Whatman; 0.4 μm pore size), and the filter was washed three times with the quench buffer. The membrane filter was placed into a vial filled with BioSafe II scintillation liquid (Research Products International Corp.), and radioactivity was quantified with LS 6500 scintillation counter (Beckman). Inhibitors were added for 1 min (or other times as specified) before the addition of glucose. Kinetic parameters were determined by nonlinear algorithm plots supplied by Prism (GraphPad Software). Olanzapine (Cayman Chemical Company) was dissolved in anhydrous dimethyl sulfoxide (DMSO) at a stock concentration of 100 mM. DMSO up to 5% concentration in the transport assay did not affect activity or measurements. To check if olanzapine inhibition is reversible, we measured transport in RSO vesicles. 2 mM olanzapine was incubated with 1 mL RSO vesicles of JM1100 E. coli expressing GlcPSe (at O.D.600 nm of 2.0, in buffer A) for 30 min. Then 50 μL of this solution was used to check the relative activity as described above. The remaining 950 μL of RSO vesicles-olanzapine solution was centrifuged (16,000g for 3 min, at room temperature) to pellet the RSO vesicles. Supernatant was carefully discarded and the pellet was resuspended in 950 μL of buffer A. The centrifugation and resuspension were repeated once more. A control sample (RSO vesicles in the absence of olanzapine) was processed similarly to account for loss in activity due to repeated centrifugation and re-suspension of the RSO vesicles. Finally, 50 μL of the washed RSO vesicles was assayed for transport activity as above.
2.4. Modeling of olanzapine interaction with GlcPSe
To model the binding sites of olanzapine, we used the SiteFinder function of Molecular Operation Environment (MOE, Chemical Computing Group) based on the three-dimensional crystal structure of GlcPSe [16] (PDB ID: 4LDS, www.rcsb.org) in the inward-facing conformation, and the modeled outward-facing conformation of GlcPSe based on the E. coli xylose transporter XylE [29] (PDB ID: 4GC0). Prior to virtual docking, we generated a set of possible olanzapine conformations, with the Conformational Generation function of MOE. Virtual docking of olanzapine was carried out using the Dock function of MOE, with all default parameters in Triangle Matcher, retaining 105 poses and Alpha HB rescoring (with equal weights for Hydrogen bonds and Alpha parameter). Potential docking positions were selected if they had sufficient space for olanzapine binding and polar and/or charged residues. Using as a selection criterion the lowest-energy scoring algorithm, olanzapine docked best in three positions on GlcPSe, all for the inward-facing conformation of the transporter.
3. Results
3.1. Olanzapine inhibits GlcPSe
The effect of olanzapine on glucose transport by GlcPSe was examined in RSO vesicles. Olanzapine at different concentrations was incubated with RSO vesicles for 1 min before 14C-glucose addition. Varying the pre-incubation time of olanzapine with RSO vesicles from 15 s to 30 min did not affect the inhibition (Fig. 1). Olanzapine inhibition was concentration dependent, with 1 mM and 8 mM olanzapine decreasing glucose transport to approximately 50% and 10%, respectively, compared to uninhibited GlcPSe. Removal of olanzapine from the reaction solution restored 80% of the activity of GlcPSe, compared to a control sample, consistent with olanzapine inhibition being reversible (Fig. 1).
Fig. 1.
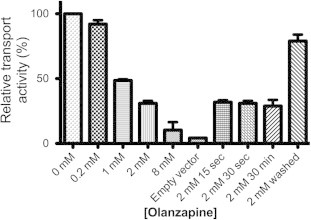
Olanzapine inhibition of GlcPSe. Transport activity was measured in right-side-out (RSO) vesicles of JM1100 E. coli expressing GlcPSe. Final concentration of glucose in the assays was 30 μM. Unless otherwise specified, RSO vesicles were pre-incubated with different concentrations of olanzapine for 1 min before glucose uptake assay. Control is RSO vesicles of JM1100 E. coli cells transformed with empty pBAD vector. Olanzapine at 2 mM was pre-incubated for different time intervals (15 s, 30 s and 30 min). For the 2 mM washed sample, RSO vesicles were incubated with 2 mM olanzapine for 30 min, and then when olanzapine was removed by two washes: RSO vesicles were pelleted by centrifugation (3 min, 16,000g) and resuspensed in the same volume of buffer A. Error bars are standard deviations corresponding to 3 different measurements.
3.2. Computational determination of olanzapine binding sites in GlcPSe
Possible olanzapine binding sites on GlcPSe were determined with Molecular Operating Environment (MOE) software by docking olanzapine to the crystal structure of GlcPSe (PDB ID 4LDS). Docking of olanzapine to GlcPSe in a predicted outward-facing conformation did not show any promising possibilities for interaction. Three binding areas for olanzapine were identified (Fig. 2A), denoted as sites 1, 2 and 3. They are located in the cytosolic part of the substrate cavity, formed by helixes 1, 4, 7 and 10, between the N- and C-terminal 6-helix domains of the transporter (shown as gray in Fig. 2A). Potential olanzapine-interacting residues were searched among charged or polar amino acids that have not been reported to interact with glucose and were within 3.5 Å from olanzapine. The sites included the following residues: R71 and N191 in site 1 (purple); R129 and N136 in site 2 (red); and R367 and R369 in site 3 (cyan) (Fig. 2B). These amino acids display varying levels of sequence conservation compared to human GLUTs (Fig. 2C). Thus R71, R129 and R369 are completely conserved in GlcPSe and human GLUTs, whereas N136, N191 or R367 are less conserved.
Fig. 2.
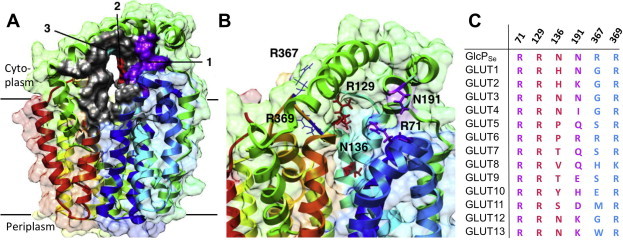
Olanzapine docking in GlcPSe. (A) Predicted olanzapine binding sites are shown in various colors: site 1 (purple), site 2 (red) and site 3 (cyan). (B) Close-up of the predicted binding sites. Residues that may interact with olanzapine are shown (site 1: R71, E191; site 2: R129, N136; site 3: R367, N369). (C) Alignment of olanzapine binding site residues of GlcPSe to human GLUTs. The colors in aminoacid sequence conform to the coloring scheme of the predicted binding sites from (A). Figures were drawn using Chimera [33].
3.3. Expression and transport activity of GlcPSe mutants
To determine which of the computationally identified residues interact with olanzapine, each residue was mutated to alanine, and individual mutants were assessed for glucose transport and subsequent inhibition by olanzapine. All mutants expressed in similar concentrations in the E. coli JM1100 membrane fractions (Fig. 3). Glucose transport activity for each mutant, relative to wild-type GlcPSe, at saturating glucose concentration, is shown in Fig. 3. R71A and N191A from site 1, R129A from site 2, and R369A from site 3 were inactive mutants. Nonetheless, N136A from site 2 and R367A from site 3 retained 57% and 87% of the wild-type activity, respectively, (Fig. 3) and their olanzapine inhibition could be examined. Michaelis–Menten plots of glucose transport by N136A and R367A mutants of GlcPSe, in RSO vesicles, are shown in Fig. 4. For both mutants, Km for glucose transport is similar to that of wild-type (30 μM, [16]): Km,N136A = 53 ± 5 μM, Km,R367A = 51 ± 6 μM.
Fig. 3.
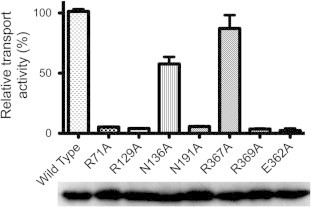
Glucose transport activity of GlcPSe mutants. Glucose uptake of GlcPSe mutants relative to wild-type, at 1 mM glucose, in E. coli JM1100 whole-cells assay. Error bars are standard deviations from 3 different experiments. Western blot of membranes extracted from cells expressing wild-type and mutants of GlcPSe is shown below. Difference in band density in the Western film was within 30%: 100%, 109%, 117%, 116%, 128%, 104%, 123% and 118%, for wild-type, R71A, R129A, N136A, N191A, R367A, R369A and E362A GlcPSe, respectively.
Fig. 4.
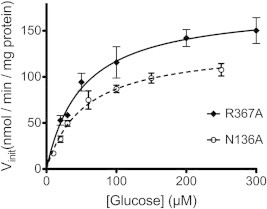
Michaelis–Menten plots for glucose uptake by N136A and R376A GlcPSe. The experiments were conducted in RSO vesicles. Km,N136A = 53 ± 3 μM and Km,R376A = 51 ± 6 μM. Error bar is standard deviation from 3 different measurements.
3.4. N136 is involved in olanzapine-induced inhibition of glucose transport by GlcPSe
Olanzapine inhibition of glucose transport by N136A and R367A GlcPSe is shown in Fig. 5. Compared to wild-type, there was no significant change in olanzapine inhibition of R367A GlcPSe glucose transport (IC50,R367A = 0.8 ± 0.1 mM, IC50,wild-type = 0.9 ± 0.1 mM). In contrast, N136A GlcPSe was less sensitive to olanzapine inhibition (IC50 = 1.5 ± 0.2 mM). Furthermore, at high concentrations of olanzapine, the inhibition was also less efficient for N136A GlcPSe (∼25% of glucose uptake activity remained compared to 10% activity remaining in wild-type GlcPSe). Hence, in N136A GlcPSe both efficacy and potency of olanzapine inhibition were reduced suggesting that N136 may interact with olanzapine.
Fig. 5.
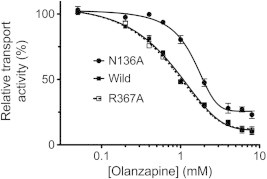
Olanzapine inhibition of glucose transport by N136A, R367A and wild-type GlcPSe. Wild-type and N136A GlcPSe RSO vesicles or R367A GlcPSe cells were pre-incubated with different concentrations of olanzapine for 1 min, before measuring glucose uptake. IC50,wild-type = 0.9 ± 0.1 mM; IC50,N136A = 1.5 ± 0.2 mM; IC50,R367A = 0.8 ± 0.1 mM. Error bar is standard deviation from 3 different measurements.
4. Discussion
The side effects of olanzapine treatment are disruptive, ranging from weight gain to metabolic disorders. The life altering nature of these side effects can negatively influence drug compliance, leading to poor outcomes for mental health patients [30]. Understanding the molecular basis of these side effects could lead to the development of therapeutic agents with fewer side effects and improve patient health and outcomes.
This work shows that olanzapine inhibits glucose transport by GlcPSe, a model for human GLUTs. GlcPSe is a bacterial glucose transporter with high affinity and specificity for glucose, inhibited by human GLUT inhibitors, with high sequence homology to GLUTs, and known protein structure. Besides its homology to GLUTs, GlcPSe is an attractive glucose transport model system because it is tractable to various methods for probing the mechanism of glucose transport inhibition: transport assay in whole cells or RSO vesicles, mutagenesis, and computational ligand docking based on crystal structure.
By whole cells or RSO vesicles transport assay, we found that olanzapine inhibits glucose transport of GlcPSe with IC50 of 0.9 ± 0.1 mM (Fig. 5). Apparently olanzapine easily penetrates the bacterial cell membrane, as the incubation time of the drug with the cells or RSO vesicles had no effect on the transport inhibition (Fig. 1). Computational docking of olanzapine to the GlcPSe structure indicated that the drug may bind to a polar region in the cytosolic part of the transporter, in 3 possible areas (Fig. 2A and B). To identify the olanzapine binding site, we mutated residues from all 3 putative sites and checked for changes in olanzapine inhibition. Among the mutants tested, only N136A and R367A maintained glucose transport activity (Fig. 3). The rest of the possible interacting residues could be involved in olanzapine binding, but are essential to transporter function and could not be further investigated. Olanzapine inhibition of R367A transporter was undistinguishable from that of the wild-type GlcPSe (Fig. 5). As for N136A transporter, olanzapine inhibited glucose transport both less potently and less efficiently than in wild-type GlcPSe (Fig. 5). Hence, N136 is involved in olanzapine inhibition of GlcPSe.
Interaction of olanzapine with GlcPSe was modeled in the computationally identified binding site 2, containing R129 and N136 (Fig. 6), which were within hydrogen bonding distance from olanzapine. The twelve transmembrane helices of GlcPSe are organized into two 6-helix bundles, with the N- and C-domains related by pseudo-twofold symmetry. During transport, GlcPSe undergoes significant conformational rearrangements between the inward- and outward-conformations, with the N- and C-domains moving relative to each other. In the modeled outward-facing conformation of GlcPSe, based on the homologous protein structure PDB ID 4GC0, R129 (from N-domain) forms a salt bridge with E362 (from C-domain) (Fig. 6B). Both R129 and E362 are strictly conserved among all human GLUTs and their mutation to alanine abolishes glucose transport in GlcPSe (Fig. 3). We propose that the salt bridge between R129 and E362 stabilizes the outward-facing conformation and its disruption interferes with glucose transport by trapping the transporter in the inward-facing conformation. Olanzapine binding in the proximity of R129, prevents the interaction between R129 and R362, thus, hampering the normal alternating opening and closing of the substrate cavity, necessary for glucose transport. Essentially olanzapine locks the transporter in the inward-facing conformation (Fig. 6C).
Fig. 6.
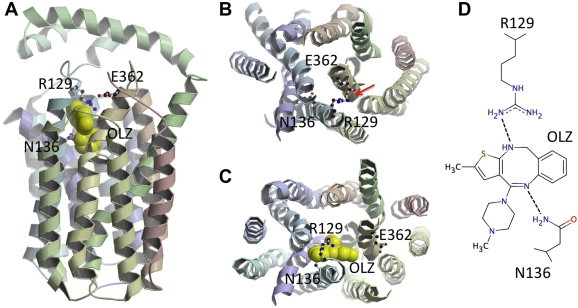
Olanzapine binding site. (A) Predicted olanzapine (OLZ) binding in GlcPSe, corresponding to site 2 from Fig. 2; R129 and N136 are in olanzapine binding site. (B) Outward-facing conformation of GlcPSe; R129 and E362 form a salt bridge shown as a yellow line, pointed by the red arrow. (C) Inward-facing conformation of GlcPSe with olanzapine interacting with R129 and N136. Olanzapine prevents the formation of the salt bridge between R129 and E362 that stabilizes the outward-facing conformation, thus locking the transporter in the inward-facing conformation. (D) Cartoon diagram of olanzapine binding site showing presumed interactions with N136 and R129. Figures were drawn using Molscript [34] and raster3D [35].
This proposed mechanism of olanzapine inhibition allows for the development of a new drug design strategy. While the sequence similarity within the transmembrane region among GLUTs is over 60%, the soluble loops that form the cytoplasmic and periplasmic faces are variable [16]. Binding of small molecules to these poorly conserved regions can block the cycling between the inward-facing and outward-facing conformations, providing the opportunity to target one member of a highly conserved family, like the GLUTs. The structure of a small molecule that binds to the cytoplasmic or periplasmic poorly conserved regions of the transporter can be tweaked to interact specifically with a particular GLUT member. So, the proposed olanzapine inhibition mechanism can be an inspiration for future drug design that target a certain GLUT.
Olanzapine inhibition of glucose transport by GlcPSe is reversible and dose dependent. While metabolic side effects related to olanzapine treatment are dose dependent [9,10], it is unclear if these effects persist after cessation of olanzapine. It was claimed that olanzapine does not affect glucose metabolism in adipocytes at therapeutic concentrations [31], however, it is not known if this is true for other types of cells. Since in some organs, mainly liver and spleen, olanzapine is accumulated at much higher concentrations than in plasma [32] (up to 46-fold difference), it is possible for the drug to reach levels sufficient for binding to glucose transporters and substantial inhibition of glucose transport in certain tissues.
This work presents evidence for glucose transport inhibition by olanzapine, a broadly used antipsychotic. This inhibition may affect glucose metabolism and lead to related diseases, such as diabetes. Accordingly, malfunction of GLUT4 (which has asparagine in N136 position of GlcPSe) due to olanzapine inhibition would result in less glucose uptake in muscle or adipocytes cells, eventually leading to diabetes. This provides a working molecular model of the causes of olanzapine’s life-altering side effects. Our future efforts include solving the crystal structure of GlcPSe complexed with olanzapine, and the study of olanzapine inhibition in various human glucose transporters. Nevertheless, for drugs that have as side-effect disruption in glucose metabolism, it is advisable to examine their potential for interaction with glucose transporters.
Acknowledgements
This work was supported by NIH – United States Grant R01-DK091754 (to JC). JC designed experiments, PB, AMGT and JC acquired the data. All authors analyzed and interpreted the data, and wrote the manuscript.
References
- 1.Bhana N., Foster R.H., Olney R., Plosker G.L. Olanzapine: an updated review of its use in the management of schizophrenia. Drugs. 2001;61:111–161. doi: 10.2165/00003495-200161010-00011. [DOI] [PubMed] [Google Scholar]
- 2.Tollefson G.D., Sanger T.M. Anxious-depressive symptoms in schizophrenia: a new treatment target for pharmacotherapy? Schizophr. Res. 1999;35(Suppl.):S13–21. doi: 10.1016/s0920-9964(98)00164-9. [DOI] [PubMed] [Google Scholar]
- 3.Van Brunt D.L., Gibson P.J., Ramsey J.L., Obenchain R. Outpatient use of major antipsychotic drugs in ambulatory care settings in the United States, 1997–2000. MedGenMed Medscape Gen. Med. 2003;5:16. [PubMed] [Google Scholar]
- 4.Gothelf D., Falk B., Singer P., Kairi M., Phillip M., Zigel L., Poraz I., Frishman S., Constantini N., Zalsman G., Weizman A., Apter A. Weight gain associated with increased food intake and low habitual activity levels in male adolescent schizophrenic inpatients treated with olanzapine. Am. J. Psychiatry. 2002;159:1055–1057. doi: 10.1176/appi.ajp.159.6.1055. [DOI] [PubMed] [Google Scholar]
- 5.Lindenmayer J.-P., Czobor P., Volavka J., Citrome L., Sheitman B., McEvoy J.P., Cooper T.B., Chakos M., Lieberman J.A. Changes in glucose and cholesterol levels in patients with schizophrenia treated with typical or atypical antipsychotics. Am. J. Psychiatry. 2003;160:290–296. doi: 10.1176/appi.ajp.160.2.290. [DOI] [PubMed] [Google Scholar]
- 6.Albaugh V.L., Henry C.R., Bello N.T., Hajnal A., Lynch S.L., Halle B., Lynch C.J. Hormonal and metabolic effects of olanzapine and clozapine related to body weight in rodents. Obesity (Silver Spring) 2006;14:36–51. doi: 10.1038/oby.2006.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koller E.A., Doraiswamy P.M. Olanzapine-associated diabetes mellitus. Pharmacotherapy. 2002;22:841–852. doi: 10.1592/phco.22.11.841.33629. [DOI] [PubMed] [Google Scholar]
- 8.Wirshing D.A., Spellberg B.J., Erhart S.M., Marder S.R., Wirshing W.C. Novel antipsychotics and new onset diabetes. Biol. Psychiatry. 1998;44:778–783. doi: 10.1016/s0006-3223(98)00100-0. [DOI] [PubMed] [Google Scholar]
- 9.Weston-Green K., Huang X.-F., Deng C. Olanzapine treatment and metabolic dysfunction: a dose response study in female Sprague Dawley rats. Behav. Brain Res. 2011;217:337–346. doi: 10.1016/j.bbr.2010.10.039. [DOI] [PubMed] [Google Scholar]
- 10.Kinon B.J., Volavka J., Stauffer V., Edwards S.E., Liu-Seifert H., Chen L., Adams D.H., Lindenmayer J.-P., McEvoy J.P., Buckley P.F., Lieberman J.A., Meltzer H.Y., Wilson D.R., Citrome L. Standard and higher dose of olanzapine in patients with schizophrenia or schizoaffective disorder: a randomized, double-blind, fixed-dose study. J. Clin. Psychopharmacol. 2008;28:392–400. doi: 10.1097/JCP.0b013e31817e63a5. [DOI] [PubMed] [Google Scholar]
- 11.Hasnain M., Vieweg W.V.R., Fredrickson S.K. Metformin for atypical antipsychotic-induced weight gain and glucose metabolism dysregulation: review of the literature and clinical suggestions. CNS Drugs. 2010;24:193–206. doi: 10.2165/11530130-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 12.Milano W., Grillo F., Del Mastro A., De Rosa M., Sanseverino B., Petrella C., Capasso A. Appropriate intervention strategies for weight gain induced by olanzapine: a randomized controlled study. Adv. Ther. 2007;24:123–134. doi: 10.1007/BF02850000. [DOI] [PubMed] [Google Scholar]
- 13.Loke Y.K., Kwok C.S., Singh S. Comparative cardiovascular effects of thiazolidinediones: systematic review and meta-analysis of observational studies. BMJ. 2011;342:d1309. doi: 10.1136/bmj.d1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uldry M., Thorens B. The SLC2 family of facilitated hexose and polyol transporters. Pflüg. Arch. Eur. J. Physiol. 2004;447:480–489. doi: 10.1007/s00424-003-1085-0. [DOI] [PubMed] [Google Scholar]
- 15.Thorens B., Mueckler M. Glucose transporters in the 21st century. Am. J. Physiol. Endocrinol. Metab. 2010;298:E141–145. doi: 10.1152/ajpendo.00712.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iancu C.V., Zamoon J., Woo S.B., Aleshin A., Choe J. Crystal structure of a glucose/H+ symporter and its mechanism of action. Proc. Natl. Acad. Sci. USA. 2013;110:17862–17867. doi: 10.1073/pnas.1311485110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deng D., Xu C., Sun P., Wu J., Yan C., Hu M., Yan N. Crystal structure of the human glucose transporter GLUT1. Nature. 2014;510:121–125. doi: 10.1038/nature13306. [DOI] [PubMed] [Google Scholar]
- 18.Olson A.L., Pessin J.E. Structure, function, and regulation of the mammalian facilitative glucose transporter gene family. Annu. Rev. Nutr. 1996;16:235–256. doi: 10.1146/annurev.nu.16.070196.001315. [DOI] [PubMed] [Google Scholar]
- 19.Ciaraldi T.P., Abrams L., Nikoulina S., Mudaliar S., Henry R.R. Glucose transport in cultured human skeletal muscle cells. Regulation by insulin and glucose in nondiabetic and non-insulin-dependent diabetes mellitus subjects. J. Clin. Invest. 1995;96:2820–2827. doi: 10.1172/JCI118352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carvalho E., Schellhorn S.E., Zabolotny J.M., Martin S., Tozzo E., Peroni O.D., Houseknecht K.L., Mundt A., James D.E., Kahn B.B. GLUT4 overexpression or deficiency in adipocytes of transgenic mice alters the composition of GLUT4 vesicles and the subcellular localization of GLUT4 and insulin-responsive aminopeptidase. J. Biol. Chem. 2004;279:21598–21605. doi: 10.1074/jbc.M312269200. [DOI] [PubMed] [Google Scholar]
- 21.Garvey W.T., Maianu L., Zhu J.H., Brechtel-Hook G., Wallace P., Baron A.D. Evidence for defects in the trafficking and translocation of GLUT4 glucose transporters in skeletal muscle as a cause of human insulin resistance. J. Clin. Invest. 1998;101:2377–2386. doi: 10.1172/JCI1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vestri H.S., Maianu L., Moellering D.R., Garvey W.T. Atypical antipsychotic drugs directly impair insulin action in adipocytes: effects on glucose transport, lipogenesis, and antilipolysis. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2007;32:765–772. doi: 10.1038/sj.npp.1301142. [DOI] [PubMed] [Google Scholar]
- 23.Carr A., Samaras K., Chisholm D.J., Cooper D.A. Pathogenesis of HIV-1-protease inhibitor-associated peripheral lipodystrophy, hyperlipidaemia, and insulin resistance. Lancet. 1998;351:1881–1883. doi: 10.1016/S0140-6736(98)03391-1. [DOI] [PubMed] [Google Scholar]
- 24.Hresko R.C., Kraft T.E., Tzekov A., Wildman S.A., Hruz P.W. Isoform-selective inhibition of facilitative glucose transporters: elucidation of the molecular mechanism of HIV protease inhibitor binding. J. Biol. Chem. 2014;289:16100–16113. doi: 10.1074/jbc.M113.528430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaback H.R. Bacterial membranes. Methods Enzymol. 1971;22:99–120. [Google Scholar]
- 27.Short S.A., Kaback H.R., Kohn L.D. Localization of d-lactate dehydrogenase in native and reconstituted Escherichia coli membrane vesicles. J. Biol. Chem. 1975;250:4291–4296. [PubMed] [Google Scholar]
- 28.Robertson D.E., Kaczorowski G.J., Garcia M.L., Kaback H.R. Active transport in membrane vesicles from Escherichia coli: the electrochemical proton gradient alters the distribution of the lac carrier between two different kinetic states. Biochemistry. 1980;19:5692–5702. doi: 10.1021/bi00566a005. [DOI] [PubMed] [Google Scholar]
- 29.Sun L., Zeng X., Yan C., Sun X., Gong X., Rao Y., Yan N. Crystal structure of a bacterial homologue of glucose transporters GLUT1-4. Nature. 2012;490:361–366. doi: 10.1038/nature11524. [DOI] [PubMed] [Google Scholar]
- 30.Allison D.B., Casey D.E. Antipsychotic-induced weight gain: a review of the literature. J. Clin. Psychiatry. 2001;62(Suppl. 7):22–31. [PubMed] [Google Scholar]
- 31.Robinson K.A., Yacoub Wasef S.Z., Buse M.G. At therapeutic concentrations, olanzapine does not affect basal or insulin-stimulated glucose transport in 3T3-L1 adipocytes. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2006;30:93–98. doi: 10.1016/j.pnpbp.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 32.Aravagiri M., Teper Y., Marder S.R. Pharmacokinetics and tissue distribution of olanzapine in rats. Biopharm. Drug Dispos. 1999;20:369–377. doi: 10.1002/1099-081x(199911)20:8<369::aid-bdd200>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 33.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera – a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 34.Kraulis P.J. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]
- 35.Merritt E.A., Bacon D.J. Raster3D: photorealistic molecular graphics. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]