

Abstract
Migraine is a common, multifactorial, disabling, recurrent, hereditary neurovascular headache disorder. It usually strikes sufferers a few times per year in childhood and then progresses to a few times per week in adulthood, particularly in females. Attacks often begin with warning signs (prodromes) and aura (transient focal neurological symptoms) whose origin is thought to involve the hypothalamus, brainstem, and cortex. Once the headache develops, it typically throbs, intensifies with an increase in intracranial pressure, and presents itself in association with nausea, vomiting, and abnormal sensitivity to light, noise, and smell. It can also be accompanied by abnormal skin sensitivity (allodynia) and muscle tenderness. Collectively, the symptoms that accompany migraine from the prodromal stage through the headache phase suggest that multiple neuronal systems function abnormally. As a consequence of the disease itself or its genetic underpinnings, the migraine brain is altered structurally and functionally. These molecular, anatomical, and functional abnormalities provide a neuronal substrate for an extreme sensitivity to fluctuations in homeostasis, a decreased ability to adapt, and the recurrence of headache. Advances in understanding the genetic predisposition to migraine, and the discovery of multiple susceptible gene variants (many of which encode proteins that participate in the regulation of glutamate neurotransmission and proper formation of synaptic plasticity) define the most compelling hypothesis for the generalized neuronal hyperexcitability and the anatomical alterations seen in the migraine brain. Regarding the headache pain itself, attempts to understand its unique qualities point to activation of the trigeminovascular pathway as a prerequisite for explaining why the pain is restricted to the head, often affecting the periorbital area and the eye, and intensifies when intracranial pressure increases.
Introduction
Migraine is a recurrent headache disorder affecting ∼15% of the population during the formative and most productive periods of their lives, between the ages of 22 and 55 years (Stewart et al., 1994). It frequently starts in childhood, particularly around puberty, and affects women more than men (3:1 female-to-male ratio; Leonardi et al., 2005; Bigal and Lipton, 2009). It tends to run in families and, as such, is considered a genetic disorder (Ferrari et al., 2015).
In some cases, the headache begins with no warning signs and ends with sleep. In other cases, the headache may be preceded by a prodromal phase that includes fatigue; euphoria; depression; irritability; food cravings; constipation; neck stiffness; increased yawning; and/or abnormal sensitivity to light, sound, and smell (Kelman, 2004; Schoonman et al., 2006); and an aura phase that includes a variety of focal cortically mediated neurological symptoms that appear just before and/or during the headache phase (Lashley, 1941; Cutrer et al., 1998; Hansen et al., 2012, 2013). Symptoms of migraine aura develop gradually, feature excitatory and inhibitory phases, and resolve completely (Russell and Olesen, 1996). Positive (gain-of-function) and negative (loss-of-function) symptoms may present as scintillating lights and scotomas when affecting the visual cortex; paresthesia, and numbness of the face and hands when affecting the somatosensory cortex; tremor and unilateral muscle weakness when affecting the motor cortex or basal ganglia; and difficulty saying words (aphasia) when affecting the speech area (Cutrer and Olesen, 2006).
The pursuant headache is commonly unilateral, pulsating, aggravated by routine physical activity, and can last a few hours to a few days (Headache Classification Committee of the International Headache Society, 2013). As the headache progresses, it may be accompanied by a variety of autonomic symptoms (nausea, vomiting, nasal/sinus congestion, rhinorrhea, lacrimation, ptosis, yawning, frequent urination, and diarrhea), affective symptoms (depression and irritability), cognitive symptoms (attention deficit, difficulty finding words, transient amnesia, and reduced ability to navigate in familiar environments), and sensory symptoms (photophobia, phonophobia, osmophobia, muscle tenderness, and cutaneous allodynia; Silberstein, 1995; Lipton et al., 2001; Kelman and Tanis, 2006).
The extent of these diverse symptoms suggests that migraine is more than a headache. It is now viewed as a complex neurological disorder that affects multiple cortical, subcortical, and brainstem areas that regulate autonomic, affective, cognitive, and sensory functions. As such, it is evident that the migraine brain differs from the nonmigraine brain (Borsook et al., 2012a) and that an effort to unravel the pathophysiology of migraine must expand beyond the simplistic view that there are “migraine generator” areas (Borsook and Burstein, 2012). In studying migraine pathophysiology, we must consider how different neural networks interact with each other to allow migraine to commence with stressors such as insufficient sleep, skipping meals, stressful or poststressful periods, hormonal fluctuations, alcohol, certain foods, flickering lights, noise, or certain scents, and why migraine attacks are sometimes initiated by these triggers and sometimes not. We must tackle the enigma of how attacks are resolved on their own or just weaken and become bearable by sleep, relaxation, food, and/or darkness. We must explore the mechanisms by which the frequency of episodic migraine increases over time (from monthly to weekly to daily), and why progression from episodic to chronic migraine is uncommon.
Disease mechanisms
In many cases, migraine attacks are likely to begin centrally, in brain areas capable of generating the classical neurological symptoms of prodromes and aura, whereas the headache phase begins with consequential activation of meningeal nociceptors at the origin of the trigeminovascular system (Noseda and Burstein, 2013). While some clues about how the occurrence of aura can activate nociceptors in the meninges exist, nothing is known about the mechanisms by which common prodromes initiate the headache phase or what sequence of events they trigger that results in activation of the meningeal nociceptors. A mechanistic search for a common denominator in migraine symptomatology and characteristics points heavily toward a genetic predisposition to generalized neuronal hyperexcitability (Ferrari et al., 2015). Mounting evidence for alterations in brain structure and function that are secondary to the repetitive state of headache can explain the progression of disease (Sprenger and Borsook, 2012).
Prodromes
In the context of migraine, prodromes are symptoms that precede the headache by several hours. Examination of symptoms that are most commonly described by patients point to the potential involvement of the hypothalamus (fatigue, depression, irritability, food cravings, and yawning), brainstem (muscle tenderness and neck stiffness), cortex (abnormal sensitivity to light, sound, and smell), and limbic system (depression and anhedonia) in the prodromal phase of a migraine attack (Maniyar et al., 2014). Given that symptoms such as fatigue, yawning, food craving, and transient mood changes occur naturally in all humans, it is critical that we understand how their occurrence triggers a headache; whether the routine occurrence of these symptoms in migraineurs (i.e., when no headache develops) differs mechanistically from their occurrence before the onset of migraine; and why yawning, food craving, and fatigue do not trigger a migraine in healthy subjects. Recently, much attention has been given to the hypothalamus because it plays a key role in many aspects of human circadian rhythms (wake-sleep cycle, body temperature, food intake, and hormonal fluctuations) and in the continuous effort to maintain homeostasis. Because the migraine brain is extremely sensitive to deviations from homeostasis, it seems reasonable that hypothalamic neurons that regulate homeostasis and circadian cycles are at the origin of some of the migraine prodromes.
Unraveling the mechanisms by which hypothalamic and brainstem neurons can trigger a headache is central to our ability to develop therapies that can intercept the headache during the prodromal phase (i.e., before the headache begins; Géraud and Donnet, 2013). The ongoing effort to answer this question focuses on two very different possibilities (Fig. 1). The first suggests that hypothalamic neurons that respond to changes in physiological and emotional homeostasis can activate meningeal nociceptors by altering the balance between parasympathetic and sympathetic tone in the meninges (Burstein and Jakubowski, 2005) toward the predominance of parasympathetic tone (Shechter et al., 2002). Support for such a proposal is based on the following: (1) hypothalamic neurons are in a position to regulate the firing of preganglionic parasympathetic neurons in the superior salivatory nucleus (SSN) and sympathetic preganglionic neurons in the spinal intermediolateral nucleus (Hosoya et al., 1983, 1984; Tucker and Saper, 1985; Loewy, 1990; Dampney, 2011; Fig. 1A); (2) the SSN can stimulate the release of acetylcholine, vasoactive intestinal peptide, and nitric oxide from meningeal terminals of postganglionic parasympathetic neurons in the sphenopalatine ganglion (SPG), leading to dilation of intracranial blood vessels, plasma protein extravasation, and local release of inflammatory molecules capable of activating pial and dural branches of meningeal nociceptors; (3) meningeal blood vessels are densely innervated by parasympathetic fibers (Larsson et al., 1976; Nozaki et al., 1993; Suzuki and Hardebo, 1993); (4) activation of SSN neurons can modulate the activity of central trigeminovascular neurons in the spinal trigeminal nucleus (SpV; Akerman et al., 2012); (5) activation of meningeal nociceptors appears to depend partially on enhanced activity in the SPG (Bolay et al., 2002); (6) enhanced cranial parasympathetic tone during migraine is evident by lacrimation and nasal congestion (Liveing, 1873; Havanka-Kanniainen et al., 1988; Shechter et al., 2002); and, finally, (7) blockade of the sphenopalatine ganglion provides partial or complete relief of migraine pain (Sluder, 1908; Kudrow, 1980; Diamond and Dalessio, 1982; Waldman, 1993; Kudrow et al., 1995; Maizels et al., 1996; Yarnitsky et al., 2003).
Figure 1.
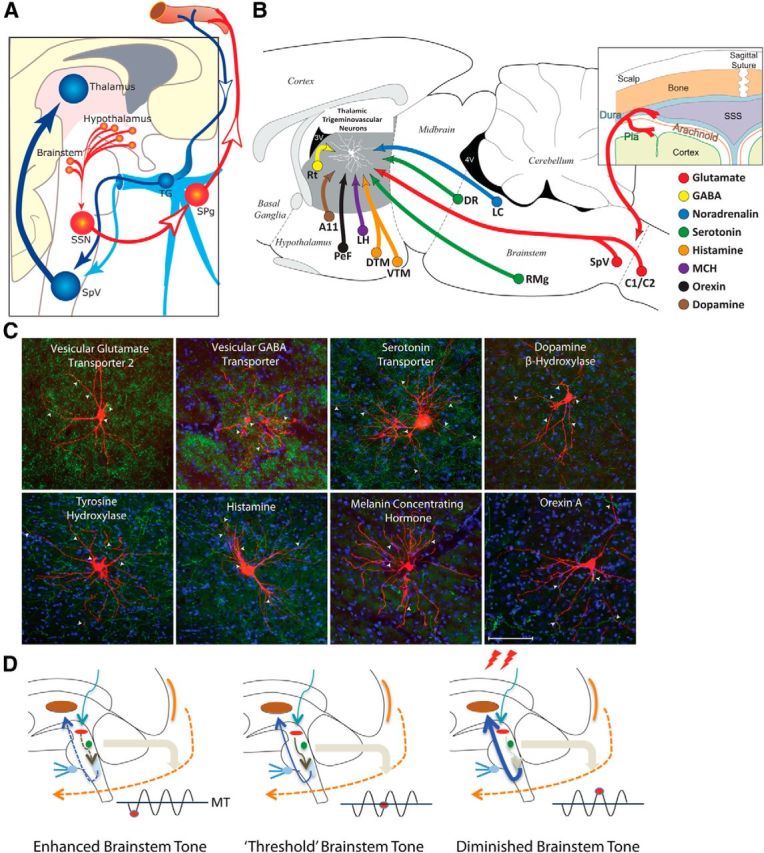
From prodromes to headache: proposed hypothesis for the initiation of headache by the hypothalamus and brainstem. A, Hypothalamic–parasympathetic pathway for the activation of meningeal nociceptors by neurons that regulate homeostasis, circadian rhythms, and autonomic functions (adapted from Burstein and Jakubowski, 2005). Hypothalamically mediated activation of preganglionic parasympathetic neurons in the SSN can trigger the release of acetylcholine, vasoactive intestinal peptide, and nitric oxide from meningeal terminals of postganglionic parasympathetic neurons in the SPG. B, C, Neurochemical pathways capable of modulating the excitability of relay thalamocortical neurons in response to deviation from physiological (food intake, sleep) and emotional (stress, anxiety) homeostasis. The illustration (top right) shows the hypothalamic and brainstem origin of each of the pathways found to converge on thalamic trigeminovascular neurons (adapted from Kagan et al., 2013; Noseda et al., 2014). The photomicrographs show the extent of innervation by vesicular glutamate transporter, vesicular GABA transporter, serotonin transporter, dopamine beta hydroxylase, tyrosine hydroxylase, histamine, melanin-concentrating hormone, and orexin A. D, Conceptual illustration of how brainstem tone (allostatic load) may allow the headache to develop incosistently in response to identical changes in external and internal conditions. Brainstem “state of tone” can limit afferent nociceptive drive in migraine-susceptible individuals (adapted from Borsook and Burstein, 2012). Fluctuation of activity in brainstem neurons is thought to drive adaptive behavior. In the context of migraine, this can apply to the modulation of nociceptive signals from the meninges. The gating of these signals depends on the threshold of the neural networks that modify these afferent signals. Thus, the robustness of the “gate” that allows nociceptive signals to drive central trigeminovascular neurons (and thus headache) is dictated by brainstem tone. When the brainstem tone is high [red dot below line of migraine threshold (MT)], nociceptive signals are inhibited; and when the brainstem tone is low (red dot above MT), afferent signals are not effectively blocked. The model illustrates the following three functional brainstem states: (1) normal state, when cyclical brainstem activity is high, the potency of pain facilitation (enhanced synaptic strength in the dorsal horn) is too high to allow nociceptive signals from the periphery to drive the central neurons into the active state (left); (2) threshold state, at threshold, the system has reached a primed state that could tip into a functional state that would allow nociceptive drive from the dura to activate the central trigeminovascular neurons (middle); and (3) migraine state, when cyclical brainstem activity is low (more sensitive to stimuli), nociceptive signals from the periphery can drive the central neurons into the active state (right). A11, Hypothalamic dopaminergic nucleus; C1/C2, cervical spinal cord segments; DR, dorsal raphe nucleus; DTM, dorsal tuberomammary hypothalamic nucleus; LC, locus ceruleus; LH, lateral hypothalamus, PeF, perifornical area; RMg, nucleus raphe magnus; TG, trigeminal ganglion.
The second proposal suggests that hypothalamic and brainstem neurons that regulate responses to deviation from physiological and emotional homeostasis can lower the threshold for the transmission of nociceptive trigeminovascular signals from the thalamus to the cortex—a critical step in establishing the headache experience (Noseda et al., 2014). This proposal is based on understanding how the thalamus selects, amplifies, and prioritizes information it eventually transfers to the cortex (McCormick, 1992; Sherman and Guillery, 1998; Sherman, 2005), and how hypothalamic and brainstem nuclei regulate relay thalamocortical neurons (Saper et al., 2005, 2010). It is constructed from recent evidence that relay trigeminothalamic neurons in sensory thalamic nuclei receive direct input from hypothalamic neurons that contain dopamine, histamine, orexin, and melanin concentrating hormone (MCH), and brainstem neurons that contain noradrenaline and serotonin (Noseda et al., 2014; Fig. 1B,C). In principle, each of these neuropeptides/neurotransmitters can shift the activity of thalamic neurons from burst to tonic mode if it is excitatory (dopamine, and high concentration of serotonin, noradrenaline, histamine, orexin), and from tonic to burst mode if it is inhibitory (MCH and low concentration of serotonin). The opposing factors that regulate the firing of relay trigeminovascular thalamic neurons provide an anatomical foundation for explaining why prodromes give rise to some migraine attacks but not to others, and why external (e.g., exposure to strong perfume) and internal conditions (e.g., skipping a meal and feeling hungry, sleeping too little and being tired, or simple stress) trigger migraine attacks so inconsistently. In the context of migraine, the convergence of these hypothalamic and brainstem neurons on thalamic trigeminovascular neurons can establish high and low set points for the allostatic load of the migraine brain (Borsook et al., 2012b). The allostatic load, defined as the amount of brain activity required to appropriately manage the level of emotional or physiological stress at any given time (McEwen, 1998, 2004; McEwen and Wingfield, 2003; Peters and McEwen, 2012), can explain why external and internal conditions only trigger headache some of the times, when they coincide with the right circadian phase of cyclic rhythmicity of brainstem, and hypothalamic and thalamic neurons that preserve homeostasis (Fig. 1D).
Cortical spreading depression
Clinical and preclinical studies suggest that migraine aura is caused by cortical spreading depression (CSD), a slowly propagating wave of depolarization/excitation followed by hyperpolarization/inhibition in cortical neurons and glia (Leao, 1944; Sugaya et al., 1975; Cutrer et al., 1998; Hadjikhani et al., 2001). While specific processes that initiate CSD in humans are not known, mechanisms that invoke inflammatory molecules as a result of emotional or physiological stress, such as lack of sleep, may play a role. In the cortex, the initial membrane depolarization is associated with a large efflux of potassium; influx of sodium and calcium; release of glutamate, ATP, and hydrogen ions; neuronal swelling (Hansen and Zeuthen, 1981; Mutch and Hansen, 1984; Schock et al., 2007; Charles and Brennan, 2009; Chang et al., 2010); upregulation of genes involved in inflammatory processing; and a host of changes in cortical perfusion and enzymatic activity that include opening of the megachannel Panx1, activation of caspase-1, and a breakdown of the blood–brain barrier (Karatas et al., 2013). Outside the brain, caspase-1 activation can initiate inflammation by releasing high-mobility group protein B1 and interleukin-1β into the CSF, which then activates nuclear factor-κB in astrocytes, with the consequential release of cyclooxygenase-2 and inducible nitric oxide synthase (iNOS) into the subarachnoid space (Karatas et al., 2013). The introduction into the meninges of these proinflammatory molecules, as well as calcitonin gene-related peptide (CGRP) and nitric oxide (Wahl et al., 1994; Obrenovitch et al., 2002; Russo, 2015), may be the link between aura and headache because the meninges are densely innervated by pain fibers whose activation distinguishes headaches of intracranial origin (e.g., migraine, meningitis, and subarachnoid bleeds) from headaches of extracranial origin (e.g., tension-type headache, cervicogenic headache, or headaches caused by mild trauma to the cranium).
Anatomy and physiology of the trigeminovascular pathway: from activation to sensitization
Anatomical description.
The trigeminovascular pathway conveys nociceptive information from the meninges to the brain. The pathway originates in trigeminal ganglion neurons whose peripheral axons reach the pia, dura, and large cerebral arteries (Uddman et al., 1985), and whose central axons reach the nociceptive dorsal horn laminae of the SpV (Liu et al., 2004). In the SpV, the nociceptors converge on neurons that receive additional input from the periorbital skin and pericranial muscles (Davis and Dostrovsky, 1988). The ascending axonal projections of trigeminovascular SpV neurons transmit monosynaptic nociceptive signals to (1) brainstem nuclei, such as the ventrolateral periaqueductal gray, reticular formation, superior salivatory, parabrachial, cuneiform, and the nucleus of the solitary tract; (2) hypothalamic nuclei, such as the anterior, lateral, perifornical, dorsomedial, suprachiasmatic, and supraoptic; and (3) basal ganglia nuclei, such as the caudate-putamen, globus pallidus, and substantia innominata (Malick et al., 2000). These projections may be critical for the initiation of nausea, vomiting, yawning, lacrimation, urination, loss of appetite, fatigue, anxiety, irritability, and depression by the headache itself (Burstein and Jakubowski, 2005). Additional projections of trigeminovascular SpV neurons are found in the thalamic ventral posteromedial (VPM), posterior (PO), and parafascicular nuclei (Malick et al., 2000). Relay trigeminovascular thalamic neurons that project to the somatosensory, insular, motor, parietal association, retrosplenial, auditory, visual, and olfactory cortices are in a position to construct the specific nature of migraine pain (i.e., location, intensity, and quality) and many of the cortically mediated symptoms that distinguish between migraine headache and other pains. These include transient symptoms of motor clumsiness, difficulty focusing, amnesia, allodynia, phonophobia, photophobia, and osmophobia (Noseda et al., 2011). Figure 2A illustrates the complexity of the trigeminovascular pathway.
Figure 2.
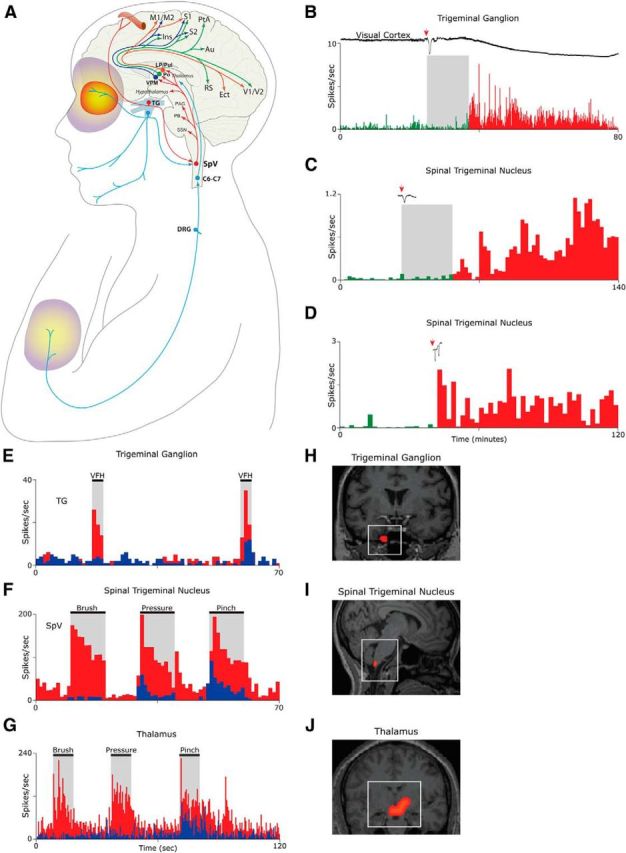
Activation and sensitization of the trigeminovascular pathway provide anatomical and physiological substrates for migraine headache and its associated symptoms. A, Intricate anatomy of the trigeminovascular pathway. B–D, Single-unit recording showing delayed (B, C) and immediate (D) activation of a meningeal nociceptor (B) and two SpV neurons (C, D) following the induction of CSD in the visual cortex of the rat. E, Activation and sensitization of a meningeal nociceptor. Baseline responses to mechanical stimulation of the dura (blue) increase (red) after exposure to inflammatory soup (IS). F, Activation and sensitization of a trigeminovascular neuron in the SpV. Baseline responses to mechanical stimulation of the periorbital skin (blue) increased (red) after a brief exposure of the dura to IS. G, Activation and sensitization of thalamic trigeminovascular neurons. Baseline responses to mechanical stimulation of the lower limb (blue) increased in magnitude and duration (red) following brief application of IS to the dura. H, Contrast analysis of BOLD signals registered in fMRI scans of the human trigeminal ganglion during migraine attacks. I, Contrast analysis of BOLD signals registered in fMRI scans of the human SpV following innocuous mechanical stimulation of the periorbital skin during migraine. J, Contrast analysis of BOLD signals registered in fMRI scans of the human thalamus following innocuous mechanical stimulation of the skin on the dorsum of the hand during migraine. Red/yellow area depicts the periorbital area of referred pain. Purple/yellow areas depict region of cephalic and extracephalic allodynia. Au, Auditory cortex; C6–C7, sixth and seventh spinal cord segments; DRG, dorsal root ganglion; Ins, insular cortex; Ect, ectorhinal cortex; LP, lateral posterior thalamic nucleus; M1/M2, primary and secondary motor cortices; PAG, periaqueductal gray; PB, parabrachial nucleus; PtA, parietal association cortex; Pul, pulvinar; RS, retrosplenial cortex; S1/S2, primary and secondary somatosensory cortices; TG, trigeminal ganglion; V1/V2, primary and secondary visual cortex. Parts of this figure were adapted from Strassman et al., 1996; Burstein et al., 1998, 2010; and Zhang et al., 2010 and 2011.
Activation.
Studies in animals show that CSD initiates delayed activation (Fig. 2B,C) and immediate activation (Fig. 2D) of peripheral and central trigeminovascular neurons in a fashion that resembles the classic delay and occasional immediate onset of headache after aura (Zhang et al., 2010, 2011), and that systemic administration of the M-type potassium channel opener KCNQ2/3 can prevent the CSD-induced activation of the nociceptors (Zhang et al., 2013). These findings support the notion that the onset of the headache phase of migraine with aura coincides with the activation of meningeal nociceptors at the peripheral origin of the trigeminovascular pathway. Whereas the vascular, cellular, and molecular events involved in the activation of meningeal nociceptors by CSD are not well understood, a large body of data suggests that transient constriction and dilatation of pial arteries and the development of dural plasma protein extravasation, neurogenic inflammation, platelet aggregation, and mast cell degranulation (Moskowitz and Macfarlane, 1993; Moskowitz, 1993), many of which may be driven by CSD-dependent peripheral CGRP release (Russo, 2015), can introduce to the meninges proinflammatory molecules, such as histamine, bradykinin, serotonin, and prostaglandins (prostaglandin E2), and a high level of hydrogen ions-thus altering the molecular environment in which meningeal nociceptors exist.
Sensitization.
When activated in the altered molecular environment described above, peripheral trigeminovascular neurons become sensitized (their response threshold decreases and their response magnitude increases) and begin to respond to dura stimuli to which they showed minimal or no response at baseline (Strassman et al., 1996; Fig. 2E). When central trigeminovascular neurons in laminae I and V of SpV (Fig. 2F) and in the thalamic PO/VPM nuclei (Fig. 2G) become sensitized, their spontaneous activity increases, their receptive fields expand, and they begin to respond to innocuous mechanical and thermal stimulation of cephalic and extracephalic skin areas as if it were noxious (Burstein et al., 1998, 2010). The human correlates of the electrophysiological measures of neuronal sensitization in animal studies are evident in contrast analysis of BOLD signals registered in fMRI scans of the human trigeminal ganglion (Fig. 2H), spinal trigeminal nucleus (Fig. 2I), and the thalamus (Fig. 2J), all measured during migraine attacks. The clinical manifestation of peripheral sensitization during migraine, which takes ∼10 min to develop, includes the perception of throbbing headache and the transient intensification of headache while bending over or coughing, activities that momentarily increase intracranial pressure (Blau and Dexter, 1981). The clinical manifestation of sensitization of central trigeminovascular neurons in the SpV, which takes 30–60 min to develop and 120 min to reach full extent, include the development of cephalic allodynia signs such as scalp and muscle tenderness and hypersensitivity to touch (Burstein et al., 2000; Bigal et al., 2008; Lipton et al., 2008). These signs are often recognized in patients reporting that they avoid wearing glasses, earrings, hats, or any other object that come in contact with the facial skin during migraine (Burstein et al., 2000; Bigal et al., 2008; Lipton et al., 2008). The clinical manifestation of thalamic sensitization during migraine, which takes 2–4 h to develop, also includes extracephalic allodynia signs that cause patients to remove tight cloth and jewelry, and avoid being touched, massaged, or hugged (Burstein et al., 2000; Bigal et al., 2008; Lipton et al., 2008). Evidence that triptans, 5HT1B/1D agonists that disrupt communications between peripheral and central trigeminovascular neurons in the dorsal horn (Levy et al., 2004), are more effective in aborting migraine when administered early (i.e., before the development of central sensitization and allodynia) rather than late (i.e., after the development of allodynia; Burstein and Jakubowski, 2004; Burstein et al., 2004) provides further support for the notion that meningeal nociceptors drive the initial phase of the headache. Further support for this concept was provided recently by studies showing that humanized monoclonal antibodies against CGRP, molecules that are too big to penetrate the blood–brain barrier and act centrally (according to the companies that developed them), are effective in preventing migraine (Diener, 2014; Dodick et al., 2014a,b; Russo, 2015). Along this line, it was also reported that drugs that act on central trigeminovascular neurons [e.g., dihydroergotamine (DHE)] are equally effective in reversing an already developed central sensitization (Pozo-Rosich and Oshinsky, 2005)—a possible explanation for DHE effectiveness in aborting migraine after the failure of therapy with triptans.
Genetics and the hyperexcitable brain
Family history points to a genetic predisposition to migraine. A genetic association with migraine was first observed and defined in patients with familial hemiplegic migraine (FHM). The three genes identified with FHM encode proteins that regulate glutamate availability in the synapse. FHM1 (CACNA1A) encodes the pore-forming α1 subunit of the P/Q type calcium channel (Ophoff et al., 1996; Ducros et al., 2001); FHM2 (ATP1A2) encodes the α2 subunit of the Na+/K+-ATPase pump (De Fusco et al., 2003); and the FHM3 (SCN1A) encodes the α1 subunit of the neuronal voltage-gated Nav1.1 channel (Dichgans et al., 2005). Collectively, these genes regulate transmitter release, glial ability to clear (reuptake) glutamate from the synapse, and the generation of action potentials (Ferrari et al., 2015). Since these early findings, large genome-wide association studies have identified 13 susceptibility gene variants for migraine with and without aura (Anttila et al., 2010, 2013; Chasman et al., 2011; Freilinger et al., 2012), three of which regulate glutaminergic neurotransmission (MTDH/AEG-1 downregulates glutamate transporter, LPR1 modulates synaptic transmission through the NMDA receptor, and MEF-2D regulates the glutamatergic excitatory synapse), and two of which regulate synaptic development and plasticity (ASTN2 is involved in the structural development of cortical layers, and FHL5 regulates cAMP-sensitive CREB proteins involved in synaptic plasticity; Anttila et al., 2010, 2013; Chasman et al., 2011; Freilinger et al., 2012). These findings provide the most plausible explanation for the “generalized” neuronal hyperexcitability of the migraine brain.
In the context of migraine, increased activity in glutamatergic systems can lead to excessive occupation of the NMDA receptor, which in turn may amplify and reinforce pain transmission, and the development of allodynia and central sensitization (Burstein et al., 2000). Network-wise, widespread neuronal hyperexcitability may also be driven by thalamocortical dysrhythmia (Llinás, 1988; Steriade and Llinás, 1988; Steriade et al., 1993; Fuggetta and Noh, 2013), defective modulatory brainstem circuits that regulate excitability at multiple levels along the neuraxis (Bahra et al., 2001); and inherently improper regulation/habituation of cortical (Coppola and Schoenen, 2012; Coppola et al., 2013), thalamic (Burstein et al., 2010), and brainstem (Weiller et al., 1995; Moulton et al., 2008) functions by limbic structures, such as the hypothalamus, amygdala, nucleus accumbens, caudate, putamen, and globus pallidus. Given that 2 of the 13 susceptibility genes regulate synaptic development and plasticity, it is reasonable to speculate that some of the networks mentioned above may not be properly wired to set a normal level of habituation throughout the brain, thus explaining the multifactorial nature of migraine. Along this line, it is also tempting to propose that at least some of the structural alterations seen in the migraine brain may be inherited and, as such, may be the “cause” of migraine, rather than being secondary to (i.e., being caused by) the repeated headache attacks. But this concept awaits evidence.
Structural and functional brain alterations
Brain alterations can be categorized into the following two processes: (1) alteration in brain function and (2) alterations in brain structure (Fig. 3). Functionally, a variety of imaging techniques used to measure relative activation in different brain areas in migraineurs (vs control subjects) revealed enhanced activation in the periaqueductal gray (Weiller et al., 1995); red nucleus and substantia nigra (Cao et al., 2002); hypothalamus (Denuelle et al., 2007); posterior thalamus (Burstein et al., 2010); cerebellum, insula, cingulate and prefrontal cortices, anterior temporal pole, and the hippocampus (Afridi et al., 2005; Moulton et al., 2011); and decreased activation in the somatosensory cortex (Tessitore et al., 2011), nucleus cuneiformis (Moulton et al., 2008), caudate, putamen, and pallidum (Maleki et al., 2011b). All of these activity changes occurred in response to nonrepetitive stimuli, and in the cingulate and prefrontal cortex they occurred in response to repetitive stimuli (Aderjan et al., 2010). Collectively, these studies support the concept that the migraine brain lacks the ability to habituate itself and consequently becomes hyperexcitable (Coppola et al., 2007, 2009). It is a matter of debate, however, if such changes are unique to migraine headache. Evidence for nearly identical activation patterns in other pain conditions, such as low back pain, neuropathic pain, fibromyalgia, irritable bowel syndrome, and cardiac pain (Apkarian, 2008; Baliki et al., 2008), raises the possibility that differences between somatic pain and migraine pain are not due to differences in central pain processing.
Figure 3.

Functional (MRIFunct) and morphometric (MRIMorph) changes in the migraine brain. The examples illustrate changes in functional and morphological measures in cortical (somatosensory) and subcortical (basal ganglia) areas, as well as sex differences in male and female migraineurs. Bottom left conceptualizes dendritic tree density (blue = volume loss; red = volume gain). Somatosensory cortex: increased somatosensory activation to a noxious stimulus (pain threshold +1C) applied to the face (forehead) and increased cortical thickness in episodic migraineur. MRIFunct shows bilateral activation in the primary somatosensory cortex (top; yellow-orange). MRIMorph shows significant changes in cortical volume (green) in high vs low episodic migraineurs vs healthy control subjects. Basal ganglia: decreased activation (top) in the caudate (MRIFunct) in high-frequency vs low-frequency episodic migraineurs in response to a noxious heat stimulus (pain threshold +1C) is associated with increased volume (MRIMorph) in the structure (bottom). Sex differences: overlap of disease-related and sex-related functional differences (MRIFunct) in men vs women (top; orange-red) showing decreased activation in episodic migraineurs to a noxious stimulus (pain threshold +1C) applied to the face (forehead). Also shown are decreased activations in female episodic migraineurs (FM) vs healthy control subjects (C). Bottom shows increased cortical thickness in female migraineurs (FM) vs male migraineurs (MM) in the insula. Other areas showing a similar sex difference included the precuneus. The issue of sex-related changes in the migraine brain have been reviewed previously (Borsook et al., 2014). Parts of this figure are adapted from Maleki et al., 2011b, 2012a,b; and Borsook et al., 2013.
Anatomically, voxel-based morphometry and diffusion tensor imaging studies in migraine patients (vs control subjects) have revealed thickening of the somatosensory cortex (DaSilva et al., 2007; Hadjikhani, 2008; Maleki et al., 2012a); increased gray matter density in the caudate (Maleki et al., 2011b); and gray matter volume loss in the superior temporal gyrus, inferior frontal gyrus, precentral gyrus, anterior cingulate cortex, amygdala, parietal operculum, middle and inferior frontal gyrus, inferior frontal gyrus, and bilateral insula (DaSilva et al., 2007; Valfrè et al., 2008). Changes in cortical and subcortical structures may also depend on the frequency of migraine attacks for a number of cortical (Maleki et al., 2011a, 2013) and subcortical regions (Maleki et al., 2012b). As discussed above, it is unclear whether such changes are genetically predetermined or simply a result of the repetitive exposure to pain/stress. Favoring the latter are studies showing that similar gray matter changes occurring in patients experiencing other chronic pain conditions (Apkarian et al., 2004; Baliki et al., 2011) are reversible and that the magnitude of these changes can be correlated with the duration of disease (Rodriguez-Raecke et al., 2009). Further complicating our ability to determine how the migraine brain differs from the brain of a patient experiencing other chronic pain conditions are anatomical findings showing decreased gray matter density in the prefrontal cortex, thalamus, posterior insula, secondary somatosensory cortex, precentral and postcentral gyrus, hippocampus, and temporal pole of chronic back pain patients; anterior insula and orbitofrontal cortex of complex regional pain syndrome patients; and the insula, mid-anterior cingulate cortex, hippocampus, and inferior temporal cortex in osteoarthritis patients with chronic back pain (Apkarian et al., 2004; Baliki et al., 2011). Whereas some of the brain alterations seen in migraineurs depend on the sex of the patient (Maleki et al., 2012b; Borsook et al., 2014), little can be said about the role played by the sex of patients who experience other pain conditions.
Treatments in development
Migraine therapy has two goals: to terminate acute attacks; and to prevent the next attack from happening. The latter can potentially prevent the progression from episodic to chronic state. Regarding the effort to terminate acute attacks, migraine represents one of the few pain conditions for which a specific drug (i.e., triptan) has been developed based on understanding the mechanisms of the disease. In contrast, the effort to prevent migraine from happening is likely to face a much larger challenge given that migraine can originate in an unknown number of brain areas (see above), and is associated with generalized functional and structural brain abnormalities. A number of treatments that attract attention are briefly reviewed below.
Medications
The most exciting drug currently under development is humanized monoclonal antibodies against CGRP. The development of these monoclonal antibodies are directed at both CGRP and its receptors (Diener, 2014; Bigal et al., 2015; Edvinsson, 2015; Russo, 2015). The concept is based on CGRP localization in the trigeminal ganglion and its relevance to migraine pathophysiology (Russo, 2015). In recent phase II randomized placebo-controlled trials (clinical trials NCT0177252, NCT01625988, NCT02025556, and NCT01723514; clinicaltrials.gov), the neutralizing humanized monoclonal antibodies against CGRP (ALD403, LY2951742, LBR-101) administered by injection for the prevention of episodic migraine, showed promising results (Bigal et al., 2013; Diener, 2014; Dodick et al., 2014a,b; Russo, 2015). Remarkably, a single injection may prevent or significantly reduce migraine attacks for 3 months.
Given our growing understanding of the importance of prodromes (likely representing abnormal sensitivity to the fluctuation in hypothalamically regulated homeostasis) and aura (likely representing the inherited cortical hyperexcitability) in the pathophysiology of migraine, drugs that target ghrelin, leptin, and orexin receptors (Hirfanoglu et al., 2009; Peterlin et al., 2010; Sachdev and Marmura, 2012; Chabi et al., 2015; Hoffmann et al., 2015) may be considered for therapeutic development which is based on, their ability to restore proper hypothalamic control of stress, hyperphagia, adiposity, and sleep. All may be critical in reducing allostatic load and, consequently, in initiating the next migraine attack (Borsook and Burstein, 2012; Borsook et al., 2012b).
Brain modification
Neuroimaging studies showing that brain networks, brain morphology, and brain chemistry are altered in episodic and chronic migraineurs (Sprenger and Borsook, 2012; Charles, 2013; Denuelle and Fabre, 2013) justify attempts to develop therapies that widely modify brain networks and their functions. Transcranial magnetic stimulation, which is thought to modify cortical hyperexcitability, is one such approach (Schoenen et al., 2003; Stankewitz and May, 2008; Lipton and Pearlman, 2010; Sánchez-del-Río González, 2013). Another approach for generalized brain modification is cognitive behavioral therapy (Powers et al., 2013; Martin et al., 2014a,b).
Conclusions
Migraine is a common and undertreated disease. For those who suffer, it is a major cause of disability, including missing work or school, and it frequently has associated comorbidities such as anxiety and depression. To put this in context, it is a leading cause of suicide, an indisputable proof of the severity of the distress that the disease may inflict on the individual (Wang et al., 2009; Fuller-Thomson et al., 2013). There is currently no objective diagnosis or treatment that is universally effective in aborting or preventing attacks. As an intermittent disorder, migraine represents a neurological condition wherein systems that continuously evaluate errors (error detection) frequently fail, thus adding to the allostatic load of the disease (Borsook et al., 2013). Given the enormous burden to society (Ferrari, 1998), there is an urgent imperative to focus on better understanding the neurobiology of the disease to enable the discovery of novel treatment approaches.
Footnotes
Editor's Note: Disease Focus articles provide brief overviews of a neural disease or syndrome, emphasizing potential links to basic neural mechanisms. They are presented in the hope of helping researchers identify clinical implications of their research. For more information, see http://www.jneurosci.org/misc/ifa_minireviews.dtl.
This research was supported by National Institutes of Health Grants NS-069847 (R.B.), NS-079687 (R.B.), NS-064050 (D.B.), NS-056195 (D.B.), NS-073997 (D.B.), and AT-007530 (D.B.).
The authors declare no competing financial interests.
References
- Aderjan D, Stankewitz A, May A. Neuronal mechanisms during repetitive trigemino-nociceptive stimulation in migraine patients. Pain. 2010;151:97–103. doi: 10.1016/j.pain.2010.06.024. [DOI] [PubMed] [Google Scholar]
- Afridi SK, Giffin NJ, Kaube H, Friston KJ, Ward NS, Frackowiak RS, Goadsby PJ. A positron emission tomographic study in spontaneous migraine. Archives of Neurology. 2005;62:1270–1275. doi: 10.1001/archneur.62.8.1270. [DOI] [PubMed] [Google Scholar]
- Akerman S, Holland PR, Summ O, Lasalandra MP, Goadsby PJ. A translational in vivo model of trigeminal autonomic cephalalgias: therapeutic characterization. Brain. 2012;135:3664–3675. doi: 10.1093/brain/aws249. [DOI] [PubMed] [Google Scholar]
- Anttila V, Stefansson H, Kallela M, Todt U, Terwindt GM, Calafato MS, Nyholt DR, Dimas AS, Freilinger T, Müller-Myhsok B, Artto V, Inouye M, Alakurtti K, Kaunisto MA, Hämäläinen E, de Vries B, Stam AH, Weller CM, Heinze A, Heinze-Kuhn K, et al. Genome-wide association study of migraine implicates a common susceptibility variant on 8q22.1. Nat Genet. 2010;42:869–873. doi: 10.1038/ng.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anttila V, Winsvold BS, Gormley P, Kurth T, Bettella F, McMahon G, Kallela M, Malik R, de Vries B, Terwindt G, Medland SE, Todt U, McArdle WL, Quaye L, Koiranen M, Ikram MA, Lehtimäki T, Stam AH, Ligthart L, Wedenoja J, et al. Genome-wide meta-analysis identifies new susceptibility loci for migraine. Nat Genet. 2013;45:912–917. doi: 10.1038/ng.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apkarian AV. Pain perception in relation to emotional learning. Curr Opin Neurobiol. 2008;18:464–468. doi: 10.1016/j.conb.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apkarian AV, Sosa Y, Sonty S, Levy RM, Harden RN, Parrish TB, Gitelman DR. Chronic back pain is associated with decreased prefrontal and thalamic gray matter density. J Neurosci. 2004;24:10410–10415. doi: 10.1523/JNEUROSCI.2541-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahra A, Matharu MS, Buchel C, Frackowiak RS, Goadsby PJ. Brainstem activation specific to migraine headache. Lancet. 2001;357:1016–1017. doi: 10.1016/S0140-6736(00)04250-1. [DOI] [PubMed] [Google Scholar]
- Baliki MN, Geha PY, Apkarian AV, Chialvo DR. Beyond feeling: chronic pain hurts the brain, disrupting the default-mode network dynamics. J Neurosci. 2008;28:1398–1403. doi: 10.1523/JNEUROSCI.4123-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baliki MN, Schnitzer TJ, Bauer WR, Apkarian AV. Brain morphological signatures for chronic pain. PLoS One. 2011;6:e26010. doi: 10.1371/journal.pone.0026010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigal ME, Lipton RB. The epidemiology, burden, and comorbidities of migraine. Neurol Clin. 2009;27:321–334. doi: 10.1016/j.ncl.2008.11.011. [DOI] [PubMed] [Google Scholar]
- Bigal ME, Ashina S, Burstein R, Reed ML, Buse D, Serrano D, Lipton RB. Prevalence and characteristics of allodynia in headache sufferers: a population study. Neurology. 2008;70:1525–1533. doi: 10.1212/01.wnl.0000310645.31020.b1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigal ME, Escandon R, Bronson M, Walter S, Sudworth M, Huggins JP, Garzone P. Safety and tolerability of LBR-101, a humanized monoclonal antibody that blocks the binding of CGRP to its receptor: results of the phase 1 program. Cephalalgia. 2013;34:483–492. doi: 10.1177/0333102413517775. [DOI] [PubMed] [Google Scholar]
- Bigal ME, Walter S, Rapoport AM. Therapeutic antibodies against CGRP or its receptor. Br J Clin Pharmacol. 2015 doi: 10.1111/bcp.12591. doi: 10.1111/bcp.12591. Advance online publication. Retrieved April 2, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blau JN, Dexter SL. The site of pain origin during migraine attacks. Cephalalgia. 1981;1:143–147. doi: 10.1046/j.1468-2982.1981.0103143.x. [DOI] [PubMed] [Google Scholar]
- Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, Moskowitz MA. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med. 2002;8:136–142. doi: 10.1038/nm0202-136. [DOI] [PubMed] [Google Scholar]
- Borsook D, Burstein R. The enigma of the dorsolateral pons as a migraine generator. Cephalalgia. 2012;32:803–812. doi: 10.1177/0333102412453952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsook D, May A, Goadsby P, Hargreaves R. The migraine brain. New York: Oxford UP; 2012a. [Google Scholar]
- Borsook D, Maleki N, Becerra L, McEwen B. Understanding migraine through the lens of maladaptive stress responses: a model disease of allostatic load. Neuron. 2012b;73:219–234. doi: 10.1016/j.neuron.2012.01.001. [DOI] [PubMed] [Google Scholar]
- Borsook D, Aasted CM, Burstein R, Becerra L. Migraine mistakes: error awareness. Neuroscientist. 2013;20:291–304. doi: 10.1177/1073858413503711. [DOI] [PubMed] [Google Scholar]
- Borsook D, Erpelding N, Lebel A, Linnman C, Veggeberg R, Grant PE, Buettner C, Becerra L, Burstein R. Sex and the migraine brain. Neurobiol Dis. 2014;68:200–214. doi: 10.1016/j.nbd.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein R, Jakubowski M. Analgesic triptan action in an animal model of intracranial pain: a race against the development of central sensitization. Ann Neurol. 2004;55:27–36. doi: 10.1002/ana.10785. [DOI] [PubMed] [Google Scholar]
- Burstein R, Jakubowski M. Unitary hypothesis for multiple triggers of the pain and strain of migraine. J Comp Neurol. 2005;493:9–14. doi: 10.1002/cne.20688. [DOI] [PubMed] [Google Scholar]
- Burstein R, Yamamura H, Malick A, Strassman AM. Chemical stimulation of the intracranial dura induces enhanced responses to facial stimulation in brain stem trigeminal neurons. J Neurophysiol. 1998;79:964–982. doi: 10.1152/jn.1998.79.2.964. [DOI] [PubMed] [Google Scholar]
- Burstein R, Yarnitsky D, Goor-Aryeh I, Ransil BJ, Bajwa ZH. An association between migraine and cutaneous allodynia. Ann Neurol. 2000;47:614–624. doi: 10.1002/1531-8249(200005)47:5<614::AID-ANA9>3.0.CO%3B2-N. [DOI] [PubMed] [Google Scholar]
- Burstein R, Collins B, Jakubowski M. Defeating migraine pain with triptans: a race against the development of cutaneous allodynia. Ann Neurol. 2004;55:19–26. doi: 10.1002/ana.10786. [DOI] [PubMed] [Google Scholar]
- Burstein R, Jakubowski M, Garcia-Nicas E, Kainz V, Bajwa Z, Hargreaves R, Becerra L, Borsook D. Thalamic sensitization transforms localized pain into widespread allodynia. Ann Neurol. 2010;68:81–91. doi: 10.1002/ana.21994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Aurora SK, Nagesh V, Patel SC, Welch KM. Functional MRI-BOLD of brainstem structures during visually triggered migraine. Neurology. 2002;59:72–78. doi: 10.1212/WNL.59.1.72. [DOI] [PubMed] [Google Scholar]
- Chabi A, Zhang Y, Jackson S, Cady R, Lines C, Herring WJ, Connor KM, Michelson D. Randomized controlled trial of the orexin receptor antagonist filorexant for migraine prophylaxis. Cephalalgia. 2015;35:379–388. doi: 10.1177/0333102414544979. [DOI] [PubMed] [Google Scholar]
- Chang JC, Shook LL, Biag J, Nguyen EN, Toga AW, Charles AC, Brennan KC. Biphasic direct current shift, haemoglobin desaturation and neurovascular uncoupling in cortical spreading depression. Brain. 2010;133:996–1012. doi: 10.1093/brain/awp338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles A. Migraine: a brain state. Curr Opin Neurol. 2013;26:235–239. doi: 10.1097/WCO.0b013e32836085f4. [DOI] [PubMed] [Google Scholar]
- Charles A, Brennan K. Cortical spreading depression-new insights and persistent questions. Cephalalgia. 2009;29:1115–1124. doi: 10.1111/j.1468-2982.2009.01983.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chasman DI, Schürks M, Anttila V, de Vries B, Schminke U, Launer LJ, Terwindt GM, van den Maagdenberg AM, Fendrich K, Völzke H, Ernst F, Griffiths LR, Buring JE, Kallela M, Freilinger T, Kubisch C, Ridker PM, Palotie A, Ferrari MD, Hoffmann W, et al. Genome-wide association study reveals three susceptibility loci for common migraine in the general population. Nat Genet. 2011;43:695–698. doi: 10.1038/ng.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppola G, Schoenen J. Cortical excitability in chronic migraine. Curr Pain Headache Rep. 2012;16:93–100. doi: 10.1007/s11916-011-0231-1. [DOI] [PubMed] [Google Scholar]
- Coppola G, Pierelli F, Schoenen J. Is the cerebral cortex hyperexcitable or hyperresponsive in migraine? Cephalalgia. 2007;27:1427–1439. doi: 10.1111/j.1468-2982.2007.01500.x. [DOI] [PubMed] [Google Scholar]
- Coppola G, Pierelli F, Schoenen J. Habituation and migraine. Neurobiology of learning and memory. 2009;92:249–259. doi: 10.1016/j.nlm.2008.07.006. [DOI] [PubMed] [Google Scholar]
- Coppola G, Di Lorenzo C, Schoenen J, Pierelli F. Habituation and sensitization in primary headaches. J Headache Pain. 2013;14:65. doi: 10.1186/1129-2377-14-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutrer FM, Olesen J. Migraine with aura and their suforms. In: Olesen J, Goadsby PJ, Ramadan N, Tfelt-Hansen J, Welch KM, editors. The headaches. Baltimore, MD: Lippincott, Williams and Wilkins; 2006. pp. 407–421. [Google Scholar]
- Cutrer FM, Sorensen AG, Weisskoff RM, Ostergaard L, Sanchez del Rio M, Lee EJ, Rosen BR, Moskowitz MA. Perfusion-weighted imaging defects during spontaneous migrainous aura. Ann Neurol. 1998;43:25–31. doi: 10.1002/ana.410430108. [DOI] [PubMed] [Google Scholar]
- Dampney RA. The hypothalamus and autonomic regulation: an overview. In: Llewellyn-Smith IJ, Verberne AJM, editors. Central regulation of autonomic functions. New York: Oxford UP; 2011. pp. 47–61. [Google Scholar]
- DaSilva AF, Granziera C, Snyder J, Hadjikhani N. Thickening in the somatosensory cortex of patients with migraine. Neurology. 2007;69:1990–1995. doi: 10.1212/01.wnl.0000291618.32247.2d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis KD, Dostrovsky JO. Responses of feline trigeminal spinal tract nucleus neurons to stimulation of the middle meningeal artery and sagittal sinus. J Neurophysiol. 1988;59:648–666. doi: 10.1152/jn.1988.59.2.648. [DOI] [PubMed] [Google Scholar]
- De Fusco M, Marconi R, Silvestri L, Atorino L, Rampoldi L, Morgante L, Ballabio A, Aridon P, Casari G. Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump alpha2 subunit associated with familial hemiplegic migraine type 2. Nat Genet. 2003;33:192–196. doi: 10.1038/ng1081. [DOI] [PubMed] [Google Scholar]
- Denuelle M, Fabre N. Functional neuroimaging of migraine. Rev Neurol (Paris) 2013;169:380–389. doi: 10.1016/j.neurol.2013.02.002. [DOI] [PubMed] [Google Scholar]
- Denuelle M, Fabre N, Payoux P, Chollet F, Geraud G. Hypothalamic activation in spontaneous migraine attacks. Headache. 2007;47:1418–1426. doi: 10.1111/j.1526-4610.2007.00776.x. [DOI] [PubMed] [Google Scholar]
- Diamond S, Dalessio DJ. The practicing physicians approach to headaches. Ed 3. Baltimore, MD: Williams and Wilkins; 1982. [Google Scholar]
- Dichgans M, Freilinger T, Eckstein G, Babini E, Lorenz-Depiereux B, Biskup S, Ferrari MD, Herzog J, van den Maagdenberg AM, Pusch M, Strom TM. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet. 2005;366:371–377. doi: 10.1016/S0140-6736(05)66786-4. [DOI] [PubMed] [Google Scholar]
- Diener HC. CGRP as a new target in prevention and treatment of migraine. Lancet Neurol. 2014;13:1065–1067. doi: 10.1016/S1474-4422(14)70228-5. [DOI] [PubMed] [Google Scholar]
- Dodick DW, Goadsby PJ, Spierings EL, Scherer JC, Sweeney SP, Grayzel DS. Safety and efficacy of LY2951742, a monoclonal antibody to calcitonin gene-related peptide, for the prevention of migraine: a phase 2, randomised, double-blind, placebo-controlled study. Lancet Neurol. 2014a;13:885–892. doi: 10.1016/S1474-4422(14)70128-0. [DOI] [PubMed] [Google Scholar]
- Dodick DW, Goadsby PJ, Silberstein SD, Lipton RB, Olesen J, Ashina M, Wilks K, Kudrow D, Kroll R, Kohrman B, Bargar R, Hirman J, Smith J. Safety and efficacy of ALD403, an antibody to calcitonin gene-related peptide, for the prevention of frequent episodic migraine: a randomised, double-blind, placebo-controlled, exploratory phase 2 trial. Lancet Neurol. 2014b;13:1100–1107. doi: 10.1016/S1474-4422(14)70209-1. [DOI] [PubMed] [Google Scholar]
- Ducros A, Denier C, Joutel A, Cecillon M, Lescoat C, Vahedi K, Darcel F, Vicaut E, Bousser MG, Tournier-Lasserve E. The clinical spectrum of familial hemiplegic migraine associated with mutations in a neuronal calcium channel. N Engl J Med. 2001;345:17–24. doi: 10.1056/NEJM200107053450103. [DOI] [PubMed] [Google Scholar]
- Edvinsson L. CGRP receptor antagonists and antibodies against CGRP and its receptor in migraine treatment. Br J Clin Pharmacol. 2015 doi: 10.1111/bcp.12618. doi: 10.1111/bcp.12618. Advance online publication. Retrieved April 2, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari MD. The economic burden of migraine to society. Pharmacoeconomics. 1998;13:667–676. doi: 10.2165/00019053-199813060-00003. [DOI] [PubMed] [Google Scholar]
- Ferrari MD, Klever RR, Terwindt GM, Ayata C, van den Maagdenberg AM. Migraine pathophysiology: lessons from mouse models and human genetics. Lancet Neurol. 2015;14:65–80. doi: 10.1016/S1474-4422(14)70220-0. [DOI] [PubMed] [Google Scholar]
- Freilinger T, Anttila V, de Vries B, Malik R, Kallela M, Terwindt GM, Pozo-Rosich P, Winsvold B, Nyholt DR, van Oosterhout WP, Artto V, Todt U, Hämäläinen E, Fernández-Morales J, Louter MA, Kaunisto MA, Schoenen J, Raitakari O, Lehtimäki T, Vila-Pueyo M, et al. Genome-wide association analysis identifies susceptibility loci for migraine without aura. Nat Genet. 2012;44:777–782. doi: 10.1038/ng.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuggetta G, Noh NA. A neurophysiological insight into the potential link between transcranial magnetic stimulation, thalamocortical dysrhythmia and neuropsychiatric disorders. Exp Neurol. 2013;245:87–95. doi: 10.1016/j.expneurol.2012.10.010. [DOI] [PubMed] [Google Scholar]
- Fuller-Thomson E, Schrumm M, Brennenstuhl S. Migraine and despair: factors associated with depression and suicidal ideation among Canadian migraineurs in a population-based study. Depress Res Treat. 2013;2013:401487. doi: 10.1155/2013/401487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Géraud G, Donnet A. Migraine and hypothalamus. Rev Neurol (Paris) 2013;169:372–379. doi: 10.1016/j.neurol.2013.03.005. [DOI] [PubMed] [Google Scholar]
- Hadjikhani N. Relevance of cortical thickness in migraine sufferers. Expert Rev Neurother. 2008;8:327–329. doi: 10.1586/14737175.8.3.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjikhani N, Sanchez Del Rio M, Wu O, Schwartz D, Bakker D, Fischl B, Kwong KK, Cutrer FM, Rosen BR, Tootell RB, Sorensen AG, Moskowitz MA. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A. 2001;98:4687–4692. doi: 10.1073/pnas.071582498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AJ, Zeuthen T. Extracellular ion concentrations during spreading depression and ischemia in the rat brain cortex. Acta Physiol Scand. 1981;113:437–445. doi: 10.1111/j.1748-1716.1981.tb06920.x. [DOI] [PubMed] [Google Scholar]
- Hansen JM, Lipton RB, Dodick DW, Silberstein SD, Saper JR, Aurora SK, Goadsby PJ, Charles A. Migraine headache is present in the aura phase: a prospective study. Neurology. 2012;79:2044–2049. doi: 10.1212/WNL.0b013e3182749eed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JM, Baca SM, Vanvalkenburgh P, Charles A. Distinctive anatomical and physiological features of migraine aura revealed by 18 years of recording. Brain. 2013;136:3589–3595. doi: 10.1093/brain/awt309. [DOI] [PubMed] [Google Scholar]
- Havanka-Kanniainen H, Tolonen U, Myllylä VV. Autonomic dysfunction in migraine: a survey of 188 patients. Headache. 1988;28:465–470. doi: 10.1111/j.1526-4610.1988.hed2807465.x. [DOI] [PubMed] [Google Scholar]
- Headache Classification Committee of the International Headache Society. The international classification of headache disorders, 3rd edition (beta version) Cephalalgia. 2013;33:629–808. doi: 10.1177/0333102413485658. [DOI] [PubMed] [Google Scholar]
- Hirfanoglu T, Serdaroglu A, Gulbahar O, Cansu A. Prophylactic drugs and cytokine and leptin levels in children with migraine. Pediatr Neurol. 2009;41:281–287. doi: 10.1016/j.pediatrneurol.2009.04.019. [DOI] [PubMed] [Google Scholar]
- Hoffmann J, Supronsinchai W, Akerman S, Andreou AP, Winrow CJ, Renger J, Hargreaves R, Goadsby PJ. Evidence for orexinergic mechanisms in migraine. Neurobiol Dis. 2015;74:137–143. doi: 10.1016/j.nbd.2014.10.022. [DOI] [PubMed] [Google Scholar]
- Hosoya Y, Matsushita M, Sugiura Y. A direct hypothalamic projection to the superior salivatory nucleus neurons in the rat. A study using anterograde autoradiographic and retrograde HRP methods. Brain Res. 1983;266:329–333. doi: 10.1016/0006-8993(83)90664-9. [DOI] [PubMed] [Google Scholar]
- Hosoya Y, Matsushita M, Sugiura Y. Hypothalamic descending afferents to cells of origin of the greater petrosal nerve in the rat, as revealed by a combination of retrograde HRP and anterograde autoradiographic techniques. Brain Res. 1984;290:141–145. doi: 10.1016/0006-8993(84)90744-3. [DOI] [PubMed] [Google Scholar]
- Kagan R, Kainz V, Burstein R, Noseda R. Hypothalamic and basal ganglia projections to the posterior thalamus: possible role in modulation of migraine headache and photophobia. Neuroscience. 2013;248:359–368. doi: 10.1016/j.neuroscience.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karatas H, Erdener SE, Gursoy-Ozdemir Y, Lule S, Eren-Koçak E, Sen ZD, Dalkara T. Spreading depression triggers headache by activating neuronal Panx1 channels. Science. 2013;339:1092–1095. doi: 10.1126/science.1231897. [DOI] [PubMed] [Google Scholar]
- Kelman L. The premonitory symptoms (prodrome): a tertiary care study of 893 migraineurs. Headache. 2004;44:865–872. doi: 10.1111/j.1526-4610.2004.04168.x. [DOI] [PubMed] [Google Scholar]
- Kelman L, Tanis D. The relationship between migraine pain and other associated symptoms. Cephalalgia. 2006;26:548–553. doi: 10.1111/j.1468-2982.2006.01075.x. [DOI] [PubMed] [Google Scholar]
- Kudrow L. Cluster headache: mechanism and management. London: Oxford UP; 1980. [Google Scholar]
- Kudrow L, Kudrow DB, Sandweiss JH. Rapid and sustained relief of migraine attacks with intranasal lidocaine: preliminary findings. Headache. 1995;35:79–82. doi: 10.1111/j.1526-4610.1995.hed3502079.x. [DOI] [PubMed] [Google Scholar]
- Larsson LI, Edvinsson L, Fahrenkrug J, Håkanson R, Owman C, Schaffalitzky de Muckadell O, Sundler F. Immunohistochemical localization of a vasodilatory polypeptide (VIP) in cerebrovascular nerves. Brain Res. 1976;113:400–404. doi: 10.1016/0006-8993(76)90951-3. [DOI] [PubMed] [Google Scholar]
- Lashley KS. Patterns of cerebral integration indicated by the scotomas of migraine. Arch Neurol Psychiatry. 1941;46:259–264. [Google Scholar]
- Leao A. Spreading depression of activity in cerebral cortex. J Neurophysiol. 1944;7:359–390. doi: 10.1152/jn.1947.10.6.409. [DOI] [PubMed] [Google Scholar]
- Leonardi M, Steiner TJ, Scher AT, Lipton RB. The global burden of migraine: measuring disability in headache disorders with WHO's classification of functioning, disability and health (ICF) J Headache Pain. 2005;6:429–440. doi: 10.1007/s10194-005-0252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Jakubowski M, Burstein R. Disruption of communication between peripheral and central trigeminovascular neurons mediates the antimigraine action of 5HT 1B/1D receptor agonists. Proc Natl Acad Sci U S A. 2004;101:4274–4279. doi: 10.1073/pnas.0306147101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton RB, Pearlman SH. Transcranial magnetic simulation in the treatment of migraine. Neurotherapeutics. 2010;7:204–212. doi: 10.1016/j.nurt.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton RB, Stewart WF, Diamond S, Diamond ML, Reed M. Prevalence and burden of migraine in the United States: data from the American Migraine Study II. Headache. 2001;41:646–657. doi: 10.1046/j.1526-4610.2001.041007646.x. [DOI] [PubMed] [Google Scholar]
- Lipton RB, Bigal ME, Ashina S, Burstein R, Silberstein S, Reed ML, Serrano D, Stewart WF. Cutaneous allodynia in the migraine population. Ann Neurol. 2008;63:148–158. doi: 10.1002/ana.21211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Broman J, Edvinsson L. Central projections of sensory innervation of the rat superior sagittal sinus. Neuroscience. 2004;129:431–437. doi: 10.1016/j.neuroscience.2004.07.045. [DOI] [PubMed] [Google Scholar]
- Liveing E. On megrim, sick headache. Nijmegen, The Netherlands: Arts and Boeve Publishers; 1873. [Google Scholar]
- Llinás RR. The intrinsic electrophysiological properties of mammalian neurons: insights into central nervous system function. Science. 1988;242:1654–1664. doi: 10.1126/science.3059497. [DOI] [PubMed] [Google Scholar]
- Loewy AD. Central autonomic pathways. In: Loewy AD, Spyer KM, editors. Central regulation of autonomic functions. Oxford: Oxford UP; 1990. [Google Scholar]
- Maizels M, Scott B, Cohen W, Chen W. Intranasal lidocaine for treatment of migraine: a randomized, double-blind, controlled trial. JAMA. 1996;276:319–321. doi: 10.1001/jama.1996.03540040063034. [DOI] [PubMed] [Google Scholar]
- Maleki N, Becerra L, Nutile L, Pendse G, Brawn J, Bigal M, Burstein R, Borsook D. Migraine attacks the basal ganglia. Mol Pain. 2011a;7:71. doi: 10.1186/1744-8069-7-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maleki N, Becerra L, Nutile L, Pendse G, Brawn J, Bigal M, Burstein R, Borsook D. Migraine attacks the basal ganglia. Mol Pain. 2011b;7:71. doi: 10.1186/1744-8069-7-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maleki N, Becerra L, Brawn J, Bigal M, Burstein R, Borsook D. Concurrent functional and structural cortical alterations in migraine. Cephalalgia. 2012a;32:607–620. doi: 10.1177/0333102412445622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maleki N, Linnman C, Brawn J, Burstein R, Becerra L, Borsook D. Her versus his migraine: multiple sex differences in brain function and structure. Brain. 2012b;135:2546–2559. doi: 10.1093/brain/aws175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maleki N, Becerra L, Brawn J, McEwen B, Burstein R, Borsook D. Common hippocampal structural and functional changes in migraine. Brain Struct Funct. 2013;218:903–912. doi: 10.1007/s00429-012-0437-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malick A, Strassman RM, Burstein R. Trigeminohypothalamic and reticulohypothalamic tract neurons in the upper cervical spinal cord and caudal medulla of the rat. J Neurophysiol. 2000;84:2078–2112. doi: 10.1152/jn.2000.84.4.2078. [DOI] [PubMed] [Google Scholar]
- Maniyar FH, Sprenger T, Monteith T, Schankin C, Goadsby PJ. Brain activations in the premonitory phase of nitroglycerin-triggered migraine attacks. Brain. 2014;137:232–241. doi: 10.1093/brain/awt320. [DOI] [PubMed] [Google Scholar]
- Martin PR, Reece J, Callan M, MacLeod C, Kaur A, Gregg K, Goadsby PJ. Behavioral management of the triggers of recurrent headache: a randomized controlled trial. Behav Res Ther. 2014a;61:1–11. doi: 10.1016/j.brat.2014.07.002. [DOI] [PubMed] [Google Scholar]
- Martin PR, Mackenzie S, Bandarian-Balooch S, Brunelli A, Broadley S, Reece J, Goadsby PJ. Enhancing cognitive-behavioural therapy for recurrent headache: design of a randomised controlled trial. BMC Neurol. 2014b;14:233. doi: 10.1186/s12883-014-0233-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA. Neurotransmitter actions in the thalamus and cerebral cortex and their role in neuromodulation of thalamocortical activity. Prog Neurobiol. 1992;39:337–388. doi: 10.1016/0301-0082(92)90012-4. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Stress, adaptation, and disease. Allostasis and allostatic load. Ann N Y Acad Sci. 1998;840:33–44. doi: 10.1111/j.1749-6632.1998.tb09546.x. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Protection and damage from acute and chronic stress: allostasis and allostatic overload and relevance to the pathophysiology of psychiatric disorders. Ann N Y Acad Sci. 2004;1032:1–7. doi: 10.1196/annals.1314.001. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Wingfield JC. The concept of allostasis in biology and biomedicine. Horm Behav. 2003;43:2–15. doi: 10.1016/S0018-506X(02)00024-7. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA. Neurogenic inflammation in the pathophysiology and treatment of migraine. Neurology. 1993;43:S16–S20. [PubMed] [Google Scholar]
- Moskowitz MA, Macfarlane R. Neurovascular and molecular mechanisms in migraine headaches. Cerebrovasc Brain Metab Rev. 1993;5:159–177. [PubMed] [Google Scholar]
- Moulton EA, Burstein R, Tully S, Hargreaves R, Becerra L, Borsook D. Interictal dysfunction of a brainstem descending modulatory center in migraine patients. PLoS One. 2008;3:e3799. doi: 10.1371/journal.pone.0003799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulton EA, Becerra L, Maleki N, Pendse G, Tully S, Hargreaves R, Burstein R, Borsook D. Painful heat reveals hyperexcitability of the temporal pole in interictal and ictal migraine states. Cereb Cortex. 2011;21:435–448. doi: 10.1093/cercor/bhq109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutch WA, Hansen AJ. Extracellular pH changes during spreading depression and cerebral ischemia: mechanisms of brain pH regulation. J Cereb Blood Flow Metab. 1984;4:17–27. doi: 10.1038/jcbfm.1984.3. [DOI] [PubMed] [Google Scholar]
- Noseda R, Burstein R. Migraine pathophysiology: anatomy of the trigeminovascular pathway and associated neurological symptoms, cortical spreading depression, sensitization, and modulation of pain. Pain. 2013;154(Suppl 1):S44–S53. doi: 10.1016/j.pain.2013.07.021. [DOI] [PubMed] [Google Scholar]
- Noseda R, Jakubowski M, Kainz V, Borsook D, Burstein R. Cortical projections of functionally identified thalamic trigeminovascular neurons: implications for migraine headache and its associated symptoms. J Neurosci. 2011;31:14204–14217. doi: 10.1523/JNEUROSCI.3285-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noseda R, Kainz V, Borsook D, Burstein R. Neurochemical pathways that converge on thalamic trigeminovascular neurons: potential substrate for modulation of migraine by sleep, food intake, stress and anxiety. PLoS One. 2014;9:e103929. doi: 10.1371/journal.pone.0103929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki K, Moskowitz MA, Maynard KI, Koketsu N, Dawson TM, Bredt DS, Snyder SH. Possible origins and distribution of immunoreactive nitric oxide synthase-containing nerve fibers in cerebral arteries. J Cereb Blood Flow Metab. 1993;13:70–79. doi: 10.1038/jcbfm.1993.9. [DOI] [PubMed] [Google Scholar]
- Obrenovitch TP, Urenjak J, Wang M. Nitric oxide formation during cortical spreading depression is critical for rapid subsequent recovery of ionic homeostasis. J Cereb Blood Flow Metab. 2002;22:680–688. doi: 10.1097/00004647-200206000-00006. [DOI] [PubMed] [Google Scholar]
- Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen GJ, Hofker MH, Ferrari MD, Frants RR. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543–552. doi: 10.1016/S0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- Peterlin BL, Rapoport AM, Kurth T. Migraine and obesity: epidemiology, mechanisms, and implications. Headache. 2010;50:631–648. doi: 10.1111/j.1526-4610.2009.01554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, McEwen BS. Introduction for the allostatic load special issue. Physiol Behav. 2012;106:1–4. doi: 10.1016/j.physbeh.2011.12.019. [DOI] [PubMed] [Google Scholar]
- Powers SW, Kashikar-Zuck SM, Allen JR, LeCates SL, Slater SK, Zafar M, Kabbouche MA, O'Brien HL, Shenk CE, Rausch JR, Hershey AD. Cognitive behavioral therapy plus amitriptyline for chronic migraine in children and adolescents: a randomized clinical trial. JAMA. 2013;310:2622–2630. doi: 10.1001/jama.2013.282533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozo-Rosich P, Oshinsky M. Effects of dihydroergotamine (DHE) on central sensitization of neurons in the trigeminal nucleus caudalis. Neurology. 2005;64:D131. [Google Scholar]
- Rodriguez-Raecke R, Niemeier A, Ihle K, Ruether W, May A. Brain gray matter decrease in chronic pain is the consequence and not the cause of pain. J Neurosci. 2009;29:13746–13750. doi: 10.1523/JNEUROSCI.3687-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell MB, Olesen J. A nosographic analysis of the migraine aura in a general population. Brain. 1996;119:355–361. doi: 10.1093/brain/119.2.355. [DOI] [PubMed] [Google Scholar]
- Russo AF. Calcitonin gene-related peptide (CGRP): a new target for migraine. Annu Rev Pharmacol Toxicol. 2015;55:533–552. doi: 10.1146/annurev-pharmtox-010814-124701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachdev A, Marmura MJ. Metabolic syndrome and migraine. Front Neurol. 2012;3:161. doi: 10.3389/fneur.2012.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-del-Río González M. Migraine: ignition of the brain. Rev Neurol. 2013;57:509–514. [PubMed] [Google Scholar]
- Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–1263. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE. Sleep state switching. Neuron. 2010;68:1023–1042. doi: 10.1016/j.neuron.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schock SC, Munyao N, Yakubchyk Y, Sabourin LA, Hakim AM, Ventureyra EC, Thompson CS. Cortical spreading depression releases ATP into the extracellular space and purinergic receptor activation contributes to the induction of ischemic tolerance. Brain Res. 2007;1168:129–138. doi: 10.1016/j.brainres.2007.06.070. [DOI] [PubMed] [Google Scholar]
- Schoenen J, Ambrosini A, Sándor PS, Maertens de Noordhout A. Evoked potentials and transcranial magnetic stimulation in migraine: published data and viewpoint on their pathophysiologic significance. Clin Neurophysiol. 2003;114:955–972. doi: 10.1016/S1388-2457(03)00024-5. [DOI] [PubMed] [Google Scholar]
- Schoonman GG, Evers DJ, Terwindt GM, van Dijk JG, Ferrari MD. The prevalence of premonitory symptoms in migraine: a questionnaire study in 461 patients. Cephalalgia. 2006;26:1209–1213. doi: 10.1111/j.1468-2982.2006.01195.x. [DOI] [PubMed] [Google Scholar]
- Shechter A, Stewart WF, Silberstein SD, Lipton RB. Migraine and autonomic nervous system function: a population-based, case-control study. Neurology. 2002;58:422–427. doi: 10.1212/WNL.58.3.422. [DOI] [PubMed] [Google Scholar]
- Sherman SM. Thalamic relays and cortical functioning. Prog Brain Res. 2005;149:107–126. doi: 10.1016/S0079-6123(05)49009-3. [DOI] [PubMed] [Google Scholar]
- Sherman SM, Guillery RW. On the actions that one nerve cell can have on another: distinguishing “drivers” from “modulators.”. Proc Natl Acad Sci U S A. 1998;95:7121–7126. doi: 10.1073/pnas.95.12.7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberstein SD. Migraine symptoms: results of a survey of self-reported migraineurs. Headache. 1995;35:387–396. doi: 10.1111/j.1526-4610.1995.hed3507387.x. [DOI] [PubMed] [Google Scholar]
- Sluder G. The role of the sphenopalatine ganglion in nasal headaches. N Y State J Med. 1908;27:8–13. [Google Scholar]
- Sprenger T, Borsook D. Migraine changes the brain: neuroimaging makes its mark. Curr Opin Neurol. 2012;25:252–262. doi: 10.1097/WCO.0b013e3283532ca3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankewitz A, May A. Cortical dysbalance in the brain in migraineurs—hyperexcitability as the result of sensitisation? Schmerz. 2008;22(Suppl 1):17–21. doi: 10.1007/s00482-007-0612-x. [DOI] [PubMed] [Google Scholar]
- Steriade M, Llinás RR. The functional states of the thalamus and the associated neuronal interplay. Physiol Rev. 1988;68:649–742. doi: 10.1152/physrev.1988.68.3.649. [DOI] [PubMed] [Google Scholar]
- Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- Stewart WF, Shechter A, Rasmussen BK. Migraine prevalence. A review of population-based studies. Neurology. 1994;44:S17–S23. [PubMed] [Google Scholar]
- Strassman AM, Raymond SA, Burstein R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature. 1996;384:560–564. doi: 10.1038/384560a0. [DOI] [PubMed] [Google Scholar]
- Sugaya E, Takato M, Noda Y. Neuronal and glial activity during spreading depression in cerebral cortex of cat. J Neurophysiol. 1975;38:822–841. doi: 10.1152/jn.1975.38.4.822. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Hardebo JE. The cerebrovascular parasympathetic innervation. Cerebrovasc Brain Metab Rev. 1993;5:33–46. [PubMed] [Google Scholar]
- Tessitore A, Russo A, Esposito F, Giordano A, Taglialatela G, De Micco R, Cirillo M, Conte F, d'Onofrio F, Cirillo S, Tedeschi G. Interictal cortical reorganization in episodic migraine without aura: an event-related fMRI study during parametric trigeminal nociceptive stimulation. Neurol Sci. 2011;32(Suppl 1):S165–S167. doi: 10.1007/s10072-011-0537-0. [DOI] [PubMed] [Google Scholar]
- Tucker DC, Saper CB. Specificity of spinal projections from hypothalamic and brainstem areas which innervate sympathetic preganglionic neurons. Brain Res. 1985;360:159–164. doi: 10.1016/0006-8993(85)91231-4. [DOI] [PubMed] [Google Scholar]
- Uddman R, Edvinsson L, Ekman R, Kingman T, McCulloch J. Innervation of the feline cerebral vasculature by nerve fibers containing calcitonin gene-related peptide: trigeminal origin and co-existence with substance P. Neurosci Lett. 1985;62:131–136. doi: 10.1016/0304-3940(85)90296-4. [DOI] [PubMed] [Google Scholar]
- Valfrè W, Rainero I, Bergui M, Pinessi L. Voxel-based morphometry reveals gray matter abnormalities in migraine. Headache. 2008;48:109–117. doi: 10.1111/j.1526-4610.2007.00723.x. [DOI] [PubMed] [Google Scholar]
- Wahl M, Schilling L, Parsons AA, Kaumann A. Involvement of calcitonin gene-related peptide (CGRP) and nitric oxide (NO) in the pial artery dilatation elicited by cortical spreading depression. Brain Res. 1994;637:204–210. doi: 10.1016/0006-8993(94)91234-3. [DOI] [PubMed] [Google Scholar]
- Waldman SD. Sphenopalatine ganglion block—80 years later. Reg Anesth. 1993;18:274–276. [PubMed] [Google Scholar]
- Wang SJ, Fuh JL, Juang KD, Lu SR. Migraine and suicidal ideation in adolescents aged 13 to 15 years. Neurology. 2009;72:1146–1152. doi: 10.1212/01.wnl.0000345362.91734.b3. [DOI] [PubMed] [Google Scholar]
- Weiller C, May A, Limmroth V, Jüptner M, Kaube H, Schayck RV, Coenen HH, Diener HC. Brain stem activation in spontaneous human migraine attacks. Nat Med. 1995;1:658–660. doi: 10.1038/nm0795-658. [DOI] [PubMed] [Google Scholar]
- Yarnitsky D, Goor-Aryeh I, Bajwa ZH, Ransil BI, Cutrer FM, Sottile A, Burstein R. 2003 Wolff Award: possible parasympathetic contributions to peripheral and central sensitization during migraine. Headache. 2003;43:704–714. doi: 10.1046/j.1526-4610.2003.03127.x. [DOI] [PubMed] [Google Scholar]
- Zhang X, Levy D, Noseda R, Kainz V, Jakubowski M, Burstein R. Activation of meningeal nociceptors by cortical spreading depression: implications to migraine with aura. J Neurosci. 2010;30:8807–8814. doi: 10.1523/JNEUROSCI.0511-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Levy D, Kainz V, Noseda R, Jakubowski M, Burstein R. Activation of central trigeminovascular neurons by cortical spreading depression. Ann Neurol. 2011;69:855–865. doi: 10.1002/ana.22329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Jakubowski M, Buettner C, Kainz V, Gold M, Burstein R. Ezogabine (KCNQ2/3 channel opener) prevents delayed activation of meningeal nociceptors if given before but not after the occurrence of cortical spreading depression. Epilepsy Behav. 2013;28:243–248. doi: 10.1016/j.yebeh.2013.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]