

Abstract
Inflammation is characteristic of most clinical disorders that challenge the neural control of breathing. Since inflammation modulates neuroplasticity, we studied the impact of inflammation caused by prolonged intermittent hypoxia on an important form of respiratory plasticity, acute intermittent hypoxia (three, 5 min hypoxic episodes, 5 min normoxic intervals) induced phrenic long-term facilitation (pLTF). Because chronic intermittent hypoxia elicits neuroinflammation and pLTF is undermined by lipopolysaccharide-induced systemic inflammation, we hypothesized that one night of intermittent hypoxia (IH-1) elicits spinal inflammation, thereby impairing pLTF by a p38 MAP kinase-dependent mechanism. pLTF and spinal inflammation were assessed in anesthetized rats pretreated with IH-1 (2 min hypoxia, 2 min normoxia; 8 h) or sham normoxia and allowed 16 h for recovery. IH-1 (1) transiently increased IL-6 (1.5 ± 0.2-fold; p = 0.02) and inducible nitric oxide synthase (iNOS) (2.4 ± 0.4-fold; p = 0.01) mRNA in cervical spinal homogenates, (2) elicited a sustained increase in IL-1β mRNA (2.4 ± 0.2-fold; p < 0.001) in isolated cervical spinal microglia, and (3) abolished pLTF (−1 ± 5% vs 56 ± 10% in controls; p < 0.001). pLTF was restored after IH-1 by systemic NSAID administration (ketoprofen; 55 ± 9%; p < 0.001) or spinal p38 MAP kinase inhibition (58 ± 2%; p < 0.001). IH-1 increased phosphorylated (activated) p38 MAP kinase immunofluorescence in identified phrenic motoneurons and adjacent microglia. In conclusion, IH-1 elicits spinal inflammation and impairs pLTF by a spinal p38 MAP kinase-dependent mechanism. By targeting inflammation, we may develop strategies to manipulate respiratory motor plasticity for therapeutic advantage when the respiratory control system is compromised (e.g., sleep apnea, apnea of prematurity, spinal injury, or motor neuron disease).
Keywords: inflammation, long-term facilitation, microglia, motor neuron, plasticity, spinal cord
Introduction
Inflammation is prevalent in most clinical disorders challenging ventilatory control, including obstructive sleep apnea (OSA), apnea of prematurity, neurodegenerative disorders, and spinal cord injury. Nevertheless, we know very little concerning interactions between inflammation and any aspect of the neural control of breathing (Huxtable et al., 2011).
Systemic inflammation affects CNS function, including neuroplasticity (Di Filippo et al., 2008; Teeling et al., 2010; Huxtable et al., 2011). However, the impact of inflammation on neuroplasticity is complex, both initiating and undermining distinct forms of neuroplasticity. For example, inflammation initiates plasticity in the spinal dorsal horn, contributing to chronic pain (Woolf and Salter, 2000) via mechanisms involving interactions between spinal microglia and second-order sensory neurons (Coull et al., 2005). Similarly, chronic sustained hypoxia elicits inflammation and sensory plasticity in the carotid body chemoreceptors (Liu et al., 2009), which may contribute to increased carotid body sensitivity during ventilatory acclimatization (Bisgard, 2000). In marked contrast, inflammation undermines other forms of CNS plasticity, including activity-dependent hippocampal synaptic plasticity (Vereker et al., 2000) and spinal instrumental learning (Shaw et al., 2001). Thus, it is of interest to study the impact of inflammation on different forms of respiratory plasticity (Huxtable et al., 2011, 2013; Vinit et al., 2011).
Mild systemic inflammation induced by low-dose lipopolysaccharides elicits spinal inflammation and abolishes phrenic long-term facilitation (pLTF), a form of spinal, respiratory, motor plasticity elicited by acute intermittent hypoxia (AIH; Huxtable et al., 2013). The nonsteroidal anti-inflammatory drug ketoprofen restores pLTF (Huxtable et al., 2013), confirming the role of inflammation. Here, we investigate the impact of a unique form of inflammation induced by one night of intermittent hypoxia in rats (IH-1; 8 h intermittent hypoxia, 16 h recovery). IH-1 mimics aspects of intermittent hypoxia experienced in a single night of OSA, but without preexisting comorbidities (e.g., obesity, hypertension; Gozal and Kheirandish-Gozal, 2008). Although 12 h of intermittent hypoxia elicits cortical and hippocampal inflammation, its impact on spinal inflammation has not been reported (Li et al., 2003, 2004). Here, we test the hypothesis that IH-1 elicits spinal inflammation, thereby undermining AIH-induced pLTF. Furthermore, we test the hypothesis that spinal p38 MAP kinase (MAPK), a key molecule initiating and responding to inflammation (Widmann et al., 1999), plays a key role in the mechanism of pLTF impairment.
This study is the first step toward an understanding of intermittent hypoxia-induced inflammation and its impact on respiratory motor plasticity. Our results are relevant to many clinical disorders associated with chronic intermittent hypoxia (CIH), such as obstructive sleep apnea and apnea of prematurity. On the other hand, ongoing inflammation may undermine the ability to harness “low-dose” intermittent hypoxia therapeutically as a means of restoring motor function in clinical disorders that cause paralysis, such as spinal injury and ALS (Dale et al., 2014).
Materials and Methods
All experiments were approved by the Animal Care and Use Committee at the School of Veterinary Medicine, University of Wisconsin–Madison, and conform to policies outlined in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Experiments were performed on 3–4 month old male Sprague Dawley rats (Harlan colony 211, 300–450 g). Rats were housed under standard conditions, with food and water ad libitum and a 12 h light/dark cycle.
Experimental groups
Rats were placed individually in cylindrical Plexiglas chambers for 8 h (9 A.M. to 5 P.M., the rats' inactive period) with ClearH2O hydrogel to provide nutrition and hydration. Gas flow (4 L/min) and concentrations were regulated by mass flow controllers (Teledyne; Hastings Instruments) and a customized computer program (LabVIEW 2009, Service Pack 1, version 9.0.1; National Instruments). Gas concentrations were continuously monitored (Gemini; CWE Inc.). Rats were exposed to IH-1 (2 min, 10.5% O2; 2 min normoxic intervals; 8 h during the rats' inactive period) or continuous normoxia (Nx; 8 h). Exposures occurred the day before electrophysiology experiments and tissue collection for mRNA or immunohistochemistry (see “Inflammatory gene expression” and “Immunofluorescence”, below). In some cases, rats were sacrificed immediately after exposures for mRNA analysis.
To study the impact of IH-1 on pLTF, rats were assigned to three groups: (1) Nx (n = 6), (2) IH-1 (n = 6), and (3) time controls (four Nx, two IH-1; total, n = 6). In subsequent neurophysiological studies, rats received intraperitoneal injections of the nonsteroidal anti-inflammatory drug ketoprofen [keto; (S)-(+)-ketoprofen; 12.5 mg/kg; Sigma] or vehicle (veh; 50% ethanol and saline, 100 μl/kg) and were divided into three groups: (1) Nx + keto (n = 7), (2) IH-1 + keto (n = 6), and (3) time control + keto (three Nx, three IH-1; total, n = 6). In the final experimental series, rats were instrumented with intrathecal catheters at C4 to deliver a p38 MAP kinase inhibitor (4-[4-(4-fluorophenyl)-5-(4-pyridinyl)-1H-imidazol-2-yl]phenol (SB 202190); 1 mm; p38 inhib; Tocris Bioscience) as described previously (Baker-Herman and Mitchell, 2002; MacFarlane and Mitchell, 2009). Fifteen minutes before AIH, 12 μl (2 μl/30 s) of SB 202190 or vehicle (artificial CSF) were given, and the rats were divided into six groups: (1) Nx + veh (n = 9), (2) IH-1+ veh (n = 6), (3) time control + veh (six Nx, two IH-1; total, n = 8), (4) Nx + p38 inhib (n = 7), (5) IH-1 + p38 inhib (n = 7), and (6) time control + p38 inhib (three Nx, two IH-1; total n = 5).
Inflammatory gene expression
Sample preparation.
Rats were anesthetized with isoflurane and perfused transcardially with ice-cold PBS. Cervical spinal (C3–C6) tissues were removed, homogenized, and used for quantitative PCR (hereafter referred to as “homogenates”). Microglial isolations were performed as described previously (Crain et al., 2009; Nikodemova and Watters, 2012; Huxtable et al., 2013). CD11b+ cells isolated with this method are referred to as “microglia.” The Neural Tissue Dissociation Kit, anti-PE magnetic beads, and MS columns were purchased from Miltenyi Biotec.
Reverse transcription.
RNA was isolated using the TRI-reagent (Sigma), and first-strand cDNA was synthesized from 1 μg of total RNA using M-MLV reverse transcriptase (Invitrogen) and an oligo(dT)/random hexamer cocktail (Promega). The cDNA was then used for quantitative RT-PCR using SYBR Green PCR Master Mix (Applied Biosystems).
Quantitative PCR.
Amplified cDNA was measured by fluorescence in real time using the ABI 7500 Fast Real-Time PCR System (Applied Biosystems) with the following primer sequences: IL-1β, 5′ CTG CAG ATG CAA TGG AAA GA, 5′ TTG CTT CCA AGG CAG ACT TT; IL-6, 5′ GTG GCT AAG GAC CAA GAC CA, 5′ GGT TTG CCG AGT AGA CCT CA; TNF-α, 5′ TCC ATG GCC CAG ACC CTC ACA C, 5′ TCC GCT TGG TGG TTT GCT ACG; iNOS, 5′ AGG GAG TGT TGT TCC AGG TG, 5′ TCT GCA GGA TGT CTT GAA CG; COX-2, 5′ TGT TCC AAC CCA TGT CAA AA, 5′ CGT AGA ATC CAG TCC GGG TA; 18s, 5′ CGG GTG CTC TTA GCT GAG TGT CCC G; 3′ CTC GGG CCT GCT TTG AAC AC.
All primers were designed (using Primer3 software) to span introns where possible; specificity was assessed through the NCBI BLAST. Dissociation curves had a single peak with a Tm consistent with intended amplicon sequences. Primer efficiency was calculated through serial dilutions and a standard curve.
Measurement of pLTF
The pLTF experimental protocol has been described in detail previously (Bach and Mitchell, 1996; Baker-Herman and Mitchell, 2002; Huxtable et al., 2013). In anesthetized, paralyzed, vagotomized, and pump-ventilated rats, integrated phrenic nerve activity was recorded. After stable baseline activity was obtained (>30 min), an arterial blood sample was drawn to assess PO2, PCO2, pH, and base excess (ABL 800; Radiometer). After baseline conditions were established, AIH was administered (three 5 min hypoxic episodes, 9–10.5% O2; 5 min intervals). Blood samples were taken during the first hypoxic episode and 15, 30, 60, and 90 min after AIH. Data were included in analyses only if they complied with the following criteria: (1) baseline and post-AIH PaO2 of >180 mmHg; (2) PaO2 during hypoxic episodes between 35 and 45 mmHg; (3) PaCO2 regulated within 1.5 mmHg of baseline after AIH. Upon completion of the protocols, rats were euthanized with a urethane overdose.
Phrenic motoneuron back-labeling and tissue collection
Separate rats were injected intrapleurally with cholera toxin b subunit bilaterally (25 μg/side; Calbiochem) to retrogradely label phrenic motoneurons (Mantilla et al., 2009; Guenther et al., 2010; Dale-Nagle et al., 2011; Golder et al., 2011; Dale et al., 2012; Lovett-Barr et al., 2012). Three days later, rats were exposed to IH-1 (n = 6) or normoxia (n = 6) and then transcardially perfused (16 h after IH-1) with cold PBS (0.01 m, pH 7.4; Thermo Fisher Scientific) followed by 4% paraformaldehyde (PFA; Thermo Fisher Scientific). After perfusions, spinal cords were immersion fixed in 4% PFA in 0.01 m PBS overnight at 4°C and then saturated in 20% and 30% sucrose at 4°C.
Immunofluorescence
Cervical spinal cords (C3–C6) were sectioned (40 μm, transverse) using a freezing microtome (Leica SM 2000 R). Free-floating sections were washed and nonspecific binding sites blocked (1 h) with 1% bovine serum albumin (BSA; Research Products International). Tissue sections were incubated in the following antibodies (16 h, room temperature, 0.1% BSA): phospho-p38 MAPK (rabbit, 1:250; Cell Signaling Technology), cholera toxin B (goat, 1:5000; Calbiochem), and CD11b (mouse, 1:200; AbD Serotec). Subsequently, tissues were washed and incubated with secondary antibodies (2 h, room temperature): Alexa Fluor 488 donkey anti-rabbit (1:500; Invitrogen), Alexa Fluor 647 donkey anti-goat (1:1000; Invitrogen), and Alexa Fluor 594 donkey anti-mouse (1:1000; Invitrogen). Sections were washed and mounted with antifade solution (ProLong Gold anti-fade reagent; Invitrogen). Controls were run concurrently to ensure specific labeling.
Immunofluorescent images (1024 × 1024 pixels) were viewed with a Nikon C1 laser scanning confocal microscope with lambda strobing in the Nikon EZ-C1 Gold (version 3.80) confocal imaging software (2 μm step increments for z stacks). All image pairs (IH-1 vs Nx) were adjusted identically for contrast and brightness (the same adjustment was made for each image pair) using EZ-C1 FreeViewer Gold software (Nikon).
Data analysis
Gene expression.
Gene expression data were analyzed based on a relative standard curve method, as described previously (Rutledge and Côté, 2003; Huxtable et al., 2013). Statistical significance was determined for each gene by one-way ANOVA with Fisher LSD post hoc tests for individual comparisons (SigmaStat version 11; Systat Software). Differences were considered significant at p < 0.05. Values are expressed as means ± 1 SEM.
Electrophysiology.
Physiological variables and peak amplitude of integrated phrenic nerve activity were averaged for 30 bursts during baseline, hypoxic episodes (short-term hypoxic phrenic response), and 60 and/or 90 min after AIH. Phrenic nerve burst amplitude is expressed as a percentage change from baseline.
Statistical comparisons between short-term hypoxic responses were made at minute 2 of the first hypoxic episode via t tests or one-way ANOVA on Ranks (p38 MAPK data). ANOVA on Ranks was used for the p38 MAPK data since they failed normality/equal variance. Statistical comparisons for post-AIH changes in phrenic burst amplitude were made using two-way repeated measures (RM) ANOVAs with Fisher LSD post hoc tests (SigmaStat version 11; Systat Software). Differences were considered significant at p < 0.05. All values are expressed as means ± 1 SEM.
Results
IH-1 elicits cervical spinal inflammation
In spinal homogenates, IH-1 increased iNOS mRNA 2.4 ± 0.4-fold above normoxic controls (p = 0.010; Fig. 1A). In isolated spinal microglia, IL-1β mRNA was increased (2.4 ± 0.2-fold, p < 0.001) versus normoxic controls (Fig. 1B). No other genes examined (IL-6, TNFα, and COX-2) were significantly changed by IH-1 pretreatment in either homogenates or microglia (p > 0.05; Fig. 1).
Figure 1.

IH-1 increased inflammatory gene expression in the cervical spinal cord. A, Homogenate samples isolated from the cervical spinal cord containing neurons, astrocytes, and microglia showed an initial increase in IL-6 (1.5 ± 0.2-fold change) immediately after 8 h of intermittent hypoxia and an increase in iNOS (2.4 ± 0.4-fold change) mRNA after IH-1 that were significantly greater than those in normoxia control samples. B, In isolated microglia, IL-1β mRNA increased immediately after 8 h IH (2.4 ± 0.2-fold change) and IH-1 (2.4 ± 0.2-fold change) compared to normoxic controls. No other inflammatory genes in the homogenate or microglia samples changed significantly at either time point after IH. **p < 0.01; ***p < 0.001 (significant difference from normoxic controls); #p < 0.05; ##p < 0.01 (significant difference from IH-1, one-way ANOVA, Fisher LSD post-test).
Since the inflammatory response is dynamic, and to better understand the temporal dynamics of the inflammatory response following IH-1, inflammatory gene expression was also examined immediately following 8 h of IH. There was a transient, early increase in IL-6 homogenate mRNA immediately after IH (1.5 ± 0.2-fold) that was greater than IL-6 gene expression the next day (i.e., IH-1; 0.8 ± 0.1-fold; p = 0.017). After IH-1, homogenate IL-6 mRNA was not significantly different from normoxic controls (p = 0.07), demonstrating a return to control levels (Fig. 1A). In microglia, IL-1β gene expression was significantly increased after 8 h of IH (2.4 ± 0.2-fold; p < 0.001) versus normoxic controls and was not significantly different from the increase seen after IH-1 (2.4 ± 0.2-fold; p = 0.753). Homogenate iNOS gene expression (0.8 ± 0.1-fold) was not significantly different from normoxic controls (p = 0.624) immediately after 8 h of IH, but there was a small, significant reduction in iNOS gene expression versus IH-1 (2.4 ± 0.4-fold; p = 0.008), suggesting small, dynamic changes in iNOS gene expression triggered by IH for 8 h.
IH-1 abolishes AIH-induced pLTF
Normoxia-pretreated rats exhibited normal pLTF 60 min after AIH (56 ± 10%, p < 0.001; Fig. 2A). In contrast, pLTF was no longer evident after IH-1 pretreatment (−1 ± 5%, p = 0.866; Fig. 2B), despite no significant (p = 0.967) change in the short-term hypoxic phrenic responses between groups (normoxia, 76 ± 8%; IH-1, 76 ± 11%; Fig. 2D). pLTF was not apparent in time control rats (no AIH) pretreated with IH-1 or normoxia (8 ± 10%, p = 0.320; Fig. 2C). At 60 min, phrenic burst amplitude in Nx + AIH-treated rats was significantly greater than in IH-1 + AIH-treated (p < 0.001) and time control rats (p < 0.001), but there was no significant difference between IH-1 + AIH and time controls (p = 0.336).
Figure 2.

IH-1 significantly reduced AIH-induced pLTF. A–C, Representative integrated phrenic neurograms during AIH protocols for rats receiving normoxia (A), IH-1 (B), and time control (no AIH, C). Development of pLTF is evident as a progressive increase in phrenic nerve amplitude from baseline (black dashed line) over 60 min in normoxic rats, but not in IH-1-treated or time control rats. D, Previous exposure to IH-1 did not alter the short-term hypoxic ventilatory responses (76 ± 11%) versus normoxia-treated (76 ± 8%) rats (t test). E, pLTF was abolished after IH-1 (−1 ± 5%, n = 6) compared to normoxia-treated (56 ± 10%, n = 6) rats. No increase in phrenic nerve amplitude was evident in time control rats (8 ± 10%, n = 6). ***p < 0.001 (significant difference from normoxia, two-way RM ANOVA, Fisher LSD post-test).
Ketoprofen restores pLTF after IH-1
We hypothesized that systemic administration of the NSAID ketoprofen would restore pLTF by reducing systemic inflammation. Ketoprofen (12.5 mg/kg, i.p.) was administered to all groups 3 h before the pLTF protocol. Two groups received AIH: Nx + keto and IH-1 + keto. A third time control group did not receive AIH (time control + keto; three Nx, three IH-1).
Ketoprofen restored pLTF after IH-1 (Fig. 3) but had no significant effect on the short-term hypoxic phrenic response in Nx + keto-treated (93 ± 9%) or IH-1 + keto-treated (116 ± 16%) rats (p = 0.215, t test; Fig. 3D). Nx + keto-treated rats exhibited normal pLTF (34 ± 10%, p < 0.001; Fig. 3A,E) that was significantly greater than time control + keto rats (8 ± 15%, p = 0.003; Fig. 3C). Time controls did not exhibit significant changes in phrenic burst amplitude as expected (Fig. 3E). IH-1 + keto rats exhibited significant AIH-induced pLTF (55 ± 9%, p < 0.001; Fig. 3B), and this effect was not different from Nx + keto (p = 0.221; Fig. 3E). IH-1 + keto rats had significantly greater pLTF than time control + keto rats (p < 0.001; Fig. 3E).
Figure 3.

Systemic treatment with ketoprofen restores pLTF after inflammation induced by IH-1. A–C, Representative integrated phrenic neurograms during AIH protocols for rats receiving normoxia + ketoprofen (nonsteroidal anti-inflammatory, 12.5 mg/kg, i.p.; A), IH-1 + ketoprofen (B), and time control + ketoprofen (no AIH; C). Development of pLTF is evident as a progressive increase in phrenic nerve amplitude from baseline (black dashed line) over 90 min in normoxic + ketoprofen and IH-1 + ketoprofen rats, but not in time control + ketoprofen rats. D, Ketoprofen did not alter short-term hypoxic responses between normoxic (93 ± 9%) and IH-1-treated (116 ± 16%) rats (t test). E, pLTF magnitude was not different between normoxic + ketoprofen (34 ± 10%, n = 7) and IH-1 + ketoprofen-treated rats (55 ± 9%, n = 6), but were significantly greater than time controls + ketoprofen (−6 ± 7%, n = 6). ***p < 0.001 (significant difference from normoxia, two-way RM ANOVA, Fisher LSD post-test), ##p < 0.01 (significant difference from IH-1 + ketoprofen).
Spinal p38 MAPK inhibition restores pLTF after IH-1
We tested the role of spinal p38 MAPK in IH-1 impairment of pLTF since it is activated by multiple proinflammatory molecules and, in turn, triggers additional inflammation. Intrathecal pretreatment with a p38 MAPK inhibitor (SB 202190, 1 mm) (1) did not alter baseline phrenic burst amplitude or other physiological variables (see Table 3) and (2) had no effect on the short-term hypoxic phrenic response (Nx + veh, 123 ± 17%; IH-1 + veh, 100 ± 19%; Nx + p38 inhib, 100 ± 17%; IH-1 + p38 inhib, 108 ± 7%; p = 0.948; Fig. 4D), but (3) restored pLTF in IH-1-pretreated rats (Fig. 4).
Table 3.
Physiological parameters for Sprague Dawley 211 rats during electrophysiological experiments after IH-1, normoxia, or time control and spinal inhibition of p38 MAPK (SB 202190, 1 mM)
| Time | Treatment group | Temperature | PaO2 | PaCO2 | pH | MAP |
|---|---|---|---|---|---|---|
| Baseline | Time control, veh | 37.6 ± 0.2 | 331 ± 9**** | 45 ± 1 | 7.364 ± 0.005 | 124 ± 2 |
| Nx, veh | 37.3 ± 0.1 | 310 ± 12 | 45 ± 1 | 7.355 ± 0.011 | 117 ± 5 | |
| IH-1, veh | 37.3 ± 0.3 | 311 ± 9 | 45 ± 1 | 7.337 ± 0.013 | 128 ± 5 | |
| Nx, p38 | 37.5 ± 0.1 | 328 ± 3 | 44 ± 1 | 7.367 ± 0.007### | 124 ± 6 | |
| IH-1, p38 | 37.2 ± 0.1 | 322 ± 10 | 44 ± 1 | 7.346 ± 0.007 | 125 ± 7 | |
| Time control, p38 | 37.3 ± 0.3 | 312 ± 9.5 | 45 ± 1 | 7.324 ± 0.0183 | 134 ± 3**** | |
| Hx | Time control, veh | 37.6 ± 0.1 | 328 ± 5# | 45 ± 1 | 7.361 ± 0.005 | 123 ± 3# |
| Nx, veh | 37.3 ± 0.1 | 38 ± 1* | 45 ± 1 | 7.341 ± 0.014 | 62 ± 7* | |
| IH-1, veh | 37.2 ± 0.3 | 40 ± 1* | 46 ± 1 | 7.322 ± 0.014## | 71 ± 7* | |
| Nx, p38 | 37.4 ± 0.1 | 35 ± 1* | 44 ± 2 | 7.339 ± 0.012**,## | 63 ± 6* | |
| IH-1, p38 | 37.2 ± 0.1 | 38 ± 1* | 45 ± 1 | 7.332 ± 0.011 | 67 ± 5* | |
| Time control, p38 | 37.2 ± 0.2 | 311 ± 10# | 45 ± 1 | 7.329 ± 0.014 | 134 ± 32 | |
| 60 min | Time control, veh | 37.6 ± 0.1 | 315 ± 10 | 44 ± 1 | 7.381 ± 0.008**,*** | 118 ± 3 |
| Nx, veh | 37.5 ± 0.1 | 297 ± 11 | 45 ± 1 | 7.359 ± 0.009 | 107 ± 5 | |
| IH-1, veh | 37.5 ± 0.2 | 305 ± 11 | 45 ± 1 | 7.339 ± 0.017***,## | 115 ± 5 | |
| Nx, p38 | 37.6 ± 0.1 | 305 ± 5** | 45 ± 1 | 7.362 ± 0.010*** | 119 ± 5 | |
| IH-1, p38 | 37.5 ± 0.1 | 299 ± 8** | 44 ± 1 | 7.348 ± 0.005*** | 114 ± 4** | |
| Time control, p38 | 37.2 ± 0.2 | 308 ± 12 | 45 ± 1 | 7.343 ± 0.014## | 121 ± 5* | |
| 90 min | Time control, veh | 37.6 ± 0.1 | 327 ± 3**** | 44 ± 1 | 7.388 ± 0.007**,*** | 117 ± 6 |
| Nx, veh | 37.5 ± 0.1 | 295 ± 9 | 45 ± 1 | 7.361 ± 0.009*** | 108 ± 4 | |
| IH-1, veh | 37.3 ± 0.2 | 300 ± 10 | 46 ± 2 | 7.341 ± 0.024*** | 113 ± 5 | |
| Nx, p38 | 37.5 ± 0.1 | 308 ± 4**,## | 44 ± 2 | 7.367 ± 0.011*** | 116 ± 6** | |
| IH-1, p38 | 37.5 ± 0.1 | 303 ± 7 | 45 ± 1 | 7.359 ± 0.006*** | 109 ± 4** | |
| Time control, p38 | 37.4 ± 0.2 | 307 ± 11 | 45 ± 1 | 7.376 ± 0.011**,*** | 123 ± 4**** |
*p < 0.001 (difference from all other time points within treatment group);
**p < 0.05 (difference from baseline within treatment group);
***p < 0.05 (difference from Hx within treatment group);
****p < 0.05 difference from Nx, veh within time point);
#p < 0.001 (difference from treatment groups receiving AIH within Hx);
##p < 0.01 (difference from time control, veh within time point);
###p < 0.05 (difference from Nx, p38 and Nx, veh). Hx, Hypoxia; MAP, mean arterial pressure.
Figure 4.

Spinal inhibition of p38 MAPK restored AIH-induced pLTF after IH-1, but did not alter the hypoxic responses or pLTF in normoxic controls. A–C, Representative integrated phrenic neurograms during AIH protocols for rats receiving normoxia + p38 MAPK inhibitor (SB202190, 1 mm, intrathecal; A), IH-1 + p38 MAPK inhibitor (B), and time control + p38 MAPK inhibitor (no AIH, C). Development of pLTF is evident as a progressive increase in phrenic nerve amplitude from baseline (black dashed line) over 90 min in normoxic + p38 MAPK inhibitor and IH-1 + p38 MAPK inhibitor rats, but not in time control + p38 MAPK inhibitor rats. D, Inhibition of p38 MAPK did not alter short-term hypoxic responses between Nx + veh (123 ± 17%), IH-1 + veh (100 ± 19%), Nx + p38 inhib (100 ± 17%), and IH-1 + p38 inhib (108 ± 7%) rats (one-way ANOVA). E, After p38 inhibitor, there was no difference between rats treated with IH-1 or normoxia. Ninety minutes after AIH, pLTF was evident in Nx + vehicle (57 ± 19%), Nx + p38 inhibitor (73 ± 15%), and IH-1 + p38 inhibitor (58 ± 2%), but pLTF in IH-1 + vehicle (11 ± 14%) rats was significantly reduced compared to the aforementioned groups and was not different from time controls + vehicle (13 ± 11%) or time controls + p38 inhibitor (−7 ± 13%). *p < 0.05, **p < 0.01, ***p < 0.001 (significantly different from time control + vehicle); ###p < 0.001 (significantly different from time control + p38 inhibitor); @p < 0.05, @@p < 0.01, @@@p < 0.001 (significantly different from IH-1 + vehicle; two-way RM ANOVA, Fisher LSD post-test).
Spinal p38 MAPK inhibition did not affect pLTF in normoxia-pretreated rats (Fig. 4A,E). Nx + veh- and Nx + p38 inhib-treated rats both exhibited significant pLTF (p < 0.001) 60 min (51 ± 17% and 64 ± 14%, respectively; data not shown) and 90 min after AIH (57 ± 19% and 73 ± 15%, respectively; Fig. 4A,E) and were not different from each other at either time point (60 min, p = 0.382; 90 min, p = 0.266). Spinal p38 MAPK inhibition enhanced pLTF in IH-1-pretreated rats (p < 0.001; Fig. 4B,E); this pLTF was significantly greater than time control + veh (60 min, 20 ± 11%, p = 0.029; 90 min, 13 ± 11%, p = 0.008) and time control + p38 inhib rats (60 min, 2 ± 11%, p = 0.003; 90 min, −8 ± 13%, p < 0.001). With IH-1 + p38 MAPK inhibition, AIH-induced pLTF was greater than in IH-1 + veh at 60 min (21 ± 6%, p = 0.046; data not shown) and 90 min (11 ± 14%, p = 0.018; Fig. 4E) after AIH, but was not greater than in Nx + veh (60 min, p = 0.890; 90 min, p = 0.289) or Nx + p38-pretreated rats (60 min, p = 0.487; 90 min, p = 0.289; Fig. 4E). IH-1 + veh rats did not exhibit pLTF at 60 or 90 min.
Increased phosphorylated p38 MAPK protein in motor neurons and microglia
Since spinal p38 MAPK inhibition restored pLTF after IH-1, we evaluated IH-1 effects on dually phosphorylated (enzymatically activated) p38 MAPK expression in phrenic motor neurons by immunofluorescence. At low magnification (20×), some phospho-p38 MAPK-positive cells were visible in normoxia-treated rats, and some of these were colocalized with cholera toxin B-labeled phrenic motoneurons and CD11b+ cells (used to identify microglia) in the C3–C6 ventral horn (Fig. 5A, top). After IH-1, more phrenic motoneurons and nearby microglia were positive for phospho-p38 MAPK, and staining intensity within cells appeared to increase (Fig. 5A, bottom). This difference between normoxia and IH-1 is emphasized at higher magnification of the phrenic motor nucleus (100×; Fig. 5B, from boxed region in A). These qualitative results suggest that IH-1 increased activated p38 MAPK in both microglia and identified phrenic motoneurons.
Figure 5.
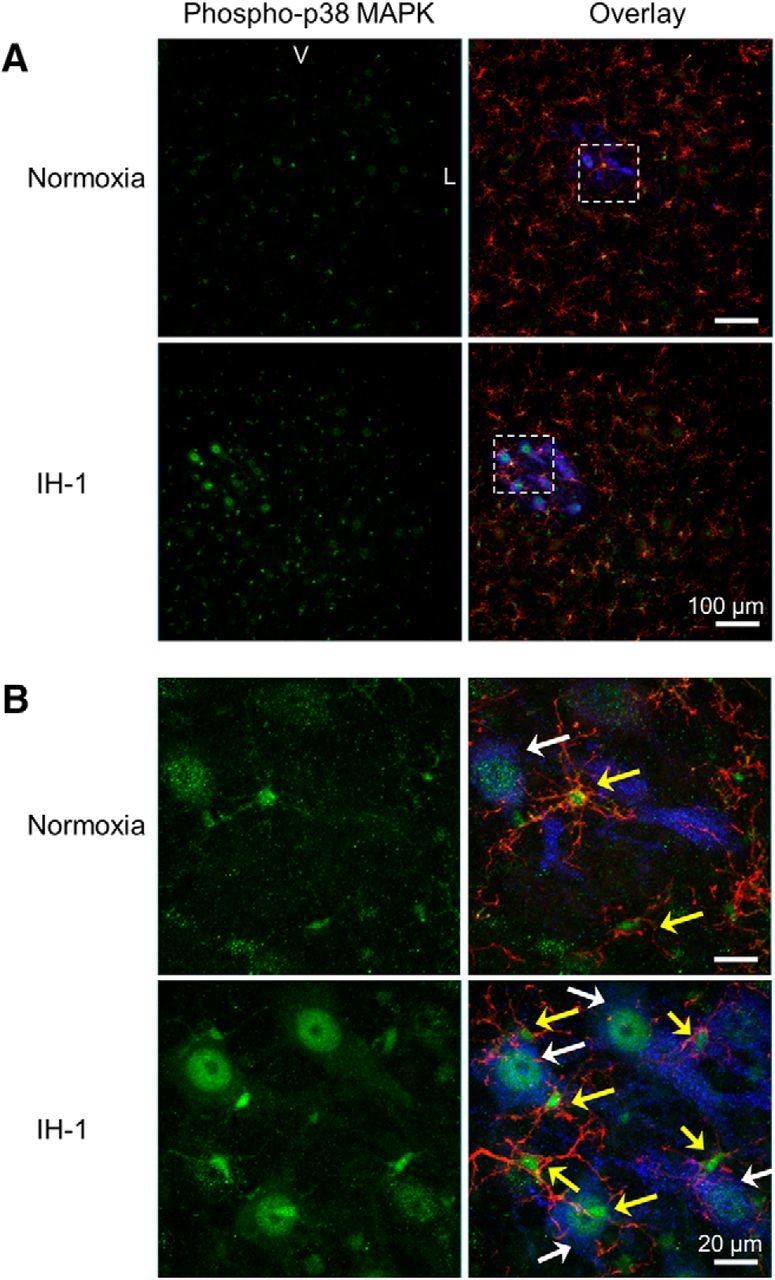
Phospho-p38 MAPK immunofluorescence is prevalent in back-labeled phrenic motoneurons and microglia of the ventral cervical spinal cord after IH-1. A, Confocal images (20×) show representative phospho-p38 MAPK (green) staining in the ventral cervical spinal cord after IH-1 (n = 6), which colocalized with back-labeled phrenic motoneurons (cholera toxin B (CtB); blue) and CD11b (microglia label; red). Minimal staining was evident after the normoxia treatment (n = 6, bottom). B, Higher magnification (100×) from the boxed area in A of the phrenic motor nucleus clearly shows colocalization with phrenic motoneurons and microglia after IH-1 (bottom). Less colocalization is evident after normoxia and is highlighted by white arrows (identifying CtB and phospho-p38 MAPK labeling) and yellow arrows (identifying CD11b and phospho-p38 MAPK labeling; top). V, Ventral; L, lateral.
Physiological variables
In all groups, AIH significantly reduced PaO2 and mean arterial pressure (MAP) during hypoxia (Hx) (Tables 1–3). In time controls pretreated with IH-1 or normoxia, rats had slightly lower pH values at 60 min versus the IH-1 and normoxia groups (Table 1). In rats treated with IH-1,keto + AIH, arterial pH was reduced versus Nx,keto + AIH and time controls (Table 2). Additionally, PaO2 in Nx,keto + AIH rats did not fully recover to baseline levels 60 min after AIH (p = 0.002); it was significantly reduced from IH-1 + keto (p = 0.004) and time control + keto (p = 0.001) 60 min after AIH (Table 2). Since PaO2 was >180 mmHg at all times, we do not believe such minor PaO2 fluctuation affected our results. PaCO2 was also significantly higher at 60 min in IH-1,keto + AIH rats versus baseline (p = 0.038) and during hypoxia (p = 0.002), but was not different from other groups at this same time (Table 2), reflecting higher group variability; nevertheless, it remained within acceptable levels of baseline (see Materials and Methods). Small differences in pH and mean arterial pressure after AIH were evident in some groups (Table 3), but remained within acceptable limits (see Materials and Methods). Similar changes in all treatment groups likely reflect time-dependent changes in this anesthetized experimental preparation.
Table 1.
Physiological variables for Sprague Dawley 211 rats during electrophysiological experiments after IH-1 and normoxia with AIH or time control
| Time | Treatment group | Temperature | PaO2 | PaCO2 | pH | MAP |
|---|---|---|---|---|---|---|
| Baseline | Time control | 37.7 ± 0.1 | 343 ± 12 | 47 ± 1 | 7.336 ± 0.006 | 121 ± 5 |
| IH-1 + AIH | 37.7 ± 0.1 | 328 ± 17 | 45 ± 1 | 7.364 ± 0.008 | 116 ± 7 | |
| Nx + AIH | 37.5 ± 0.2 | 325 ± 20 | 45 ± 1 | 7.371 ± 0.005 | 120 ± 5 | |
| Hx | Time control | 37.6 ± 0.1 | 346 ± 5* | 46 ± 1 | 7.330 ± 0.008* | 118 ± 6* |
| IH-1 + AIH | 37.6 ± 0.1 | 42 ± 2 | 46 ± 1 | 7.350 ± 0.008 | 66 ± 9 | |
| Nx + AIH | 37.3 ± 0.1 | 36 ± 2 | 45 ± 1 | 7.343 ± 0.005 | 64 ± 7 | |
| 60 min | Time control | 37.5 ± 0.1 | 347 ± 3 | 47 ± 1 | 7.343 ± 0.012 | 114 ± 7 |
| IH-1 + AIH | 37.6 ± 0.1 | 347 ± 10 | 45 ± 0.5 | 7.391 ± 0.010 | 112 ± 7 | |
| Nx + AIH | 37.4 ± 0.2 | 308 ± 15** | 45 ± 1 | 7.371 ± 0.005*** | 115 ± 7 |
*p < 0.001 (difference from all other Hx (hypoxia) groups);
**p < 0.05 (difference from time control within 60 min);
***p < 0.01 (difference from time control within 60 min). MAP, Mean arterial pressure.
Table 2.
Physiological variables for Sprague Dawley 211 rats during electrophysiological experiments after IH-1 and normoxia with AIH or time control and systemic treatment with ketoprofen (keto)
| Time | Treatment group | Temperature | PaO2 | PaCO2 | pH | MAP |
|---|---|---|---|---|---|---|
| Baseline | Time control | 37.2 ± 0.1 | 342 ± 5 | 43 ± 2 | 7.380 ± 0.010 | 102 ± 4### |
| Nx, keto + AIH | 37.4 ± 0.1 | 325 ± 13 | 44 ± 1 | 7.376 ± 0.006 | 110 ± 5 | |
| IH-1, keto + AIH | 37.4 ± 0.1 | 340 ± 5 | 43 ± 1 | 7.365 ± 0.009 | 116 ± 3 | |
| Hx | Time control | 37.3 ± 0.1 | 339 ± 4* | 43 ± 2 | 7.381 ± 0.010 | 102 ± 3* |
| Nx, keto + AIH | 37.3 ± 0.1 | 37 ± 2** | 44 ± 1 | 7.367 ± 0.010 | 55 ± 5** | |
| IH-1, keto + AIH | 37.4 ± 0.1 | 37 ± 1** | 42 ± 1 | 7.354 ± 0.010## | 54 ± 3** | |
| 60 min | Time control | 37.3 ± 0.1 | 330 ± 4 | 43 ± 2 | 7.389 ± 0.010 | 100 ± 4 |
| Nx, keto + AIH | 37.4 ± 0.1 | 296 ± 11*,*** | 44 ± 1 | 7.376 ± 0.010 | 104 ± 4 | |
| IH-1, keto + AIH | 37.4 ± 0.2 | 327 ± 8 | 44 ± 1# | 7.376 ± 0.006 | 107 ± 6 |
*p < 0.001 (difference from all other groups within time point);
**p < 0.001 (difference from other time points within group);
***p < 0.01 (difference from baseline within group);
#p < 0.05 (difference from other time points within group);
##p < 0.01 (different from other time points within group);
###p < 0.05 (difference from IH-1, keto + AIH within time point). Hx, Hypoxia; MAP, mean arterial pressure.
Discussion
Although inflammation is prominent in most clinical disorders challenging the control of breathing, little is known concerning how inflammation alters any aspect of ventilatory control. Here, we studied a novel inflammatory stimulus induced by 8 h of intermittent hypoxia during the rat's subjective night (inactive period). Even one “night” of intermittent hypoxia elicits spinal inflammation (demonstrated in microglial gene expression), which subsequently undermines AIH-induced pLTF (demonstrated by reversal after ketoprofen administration). A key molecule in the mechanism whereby IH-1 induced inflammation impairs pLTF is p38 MAPK. Thus, collective evidence demonstrates that a physiologically relevant stimulus (IH-1) impairs respiratory motor plasticity due to spinal inflammation by a p38 MAPK-dependent mechanism. This is the first demonstration that spinal p38 MAPK is a key link between inflammation and pLTF. Thus, p38 MAPK is a molecule of considerable interest in exploring ways of minimizing pathology due to intermittent hypoxia, or in our efforts to harness “low-dose” intermittent hypoxia to treat diverse clinical disorders that impair movement, such as spinal injury and ALS (Dale et al., 2014).
Our results have profound implications concerning the potential impact of even a single night of sleep apnea. In individuals with subclinical OSA, exacerbating factors such as alcohol consumption or acute respiratory infection may tip the balance, transiently increasing the frequency of apneas during sleep. Increased apnea frequency may trigger inflammation, undermining compensatory mechanisms such as pLTF or upper airway LTF, thereby leading to further apneas (i.e., positive feedback loop).
The IH-1 protocol used here differs quantitatively from frequently studied CIH protocols, which typically last for days (Almendros et al., 2014; Navarrete-Opazo and Mitchel, 2014). CIH enhances carotid body hypoxic sensitivity (Peng et al., 2003; Rey et al., 2004; Prabhakar et al., 2007; Pawar et al., 2008), but the extent of carotid body inflammation was not studied. Increased carotid body hypoxic sensitivity could arise from differential effects of inflammation on sensory versus motor plasticity (Huxtable et al., 2011), or because CIH elicits qualitatively/quantitatively different inflammation versus IH-1. The lack of change in the short-term hypoxic response after IH-1 suggests IH-1 does not elicit carotid body inflammation and/or plasticity similar to CIH.
Differences in the quality or quantity of CIH- versus IH-1-induced inflammation may account for differences in the impact of CIH versus IH-1 preconditioning on AIH-induced pLTF. CIH (5 min hypoxic episodes, 5 min intervals, 12 h/d, 7 d during the active period) actually enhances pLTF (Ling et al., 2001) and ventilatory LTF (McGuire et al., 2003, 2004), possibly due to qualitative differences in inflammatory signaling molecules and/or increased expression of trophic factors known to promote respiratory plasticity (Baker-Herman et al., 2004; Dale-Nagle et al., 2011; Dale et al., 2012). After IH-1, the negative effects of inflammation may predominate, undermining respiratory motor plasticity. The presence or absence of respiratory plasticity after prolonged IH may be determined by the duration of IH exposure, diurnal variations, the inflammatory cascade, expression of proplasticity molecules (e.g., growth/trophic factors), and/or other yet to be determined factors (Mateika and Narwani, 2009).
A fundamental understanding of mechanisms undermining AIH-induced motor plasticity is of considerable interest in the treatment of multiple clinical disorders since repetitive (low-dose) acute intermittent hypoxia has been applied therapeutically to restore breathing capacity in rodent models of cervical spinal injury (Lovett-Barr et al., 2012) and ALS (Nichols et al., 2013), and in humans with chronic, incomplete spinal injuries (Tester et al., 2014). Such therapeutic intermittent hypoxia also restores nonrespiratory motor functions, such as leg strength (Trumbower et al., 2012) and walking ability (Lovett-Barr et al., 2012; Hayes et al., 2014) in both rodent models and humans with chronic, incomplete spinal injuries. Since patients with spinal injury and ALS are prone to systemic infections and inflammation, inflammation may undermine the therapeutic efficacy of low-dose IH. Judicious application of anti-inflammatory drugs in association with therapeutic IH may enhance its effectiveness. Here, we have begun to elucidate means of counteracting the impact of inflammation on AIH-induced motor plasticity.
Systemic ketoprofen restores pLTF, demonstrating that inflammation per se undermines pLTF following IH-1. Ketoprofen is a potent anti-inflammatory and analgesic agent used in many species, including humans (Foster et al., 1988) and rats (Foster and Jamali, 1988; Carabaza et al., 1996). Ketoprofen inhibits prostaglandin and leukotriene synthesis (Netter et al., 1985; Walker, 1995; Cabré et al., 1998) by inhibiting the enzymatic activities of both COX-1 and COX-2 peripherally (Gynther et al., 2010; Kokki, 2010), though some work suggests that it can have direct effects centrally (Netter et al., 1985; Mannila et al., 2006) or when modified to increase blood–brain barrier permeability (Gynther et al., 2010). As such, pLTF rescue by ketoprofen most likely results from (1) directly diminishing CNS inflammation; (2) altering systemic inflammation, thereby decreasing transduction of systemic inflammatory signals via the vagus nerve (Hansen et al., 1998); (3) decreasing systemic expression of cytokines that cross the blood–brain barrier (Carson et al., 2006); and/or (4) preventing blood–brain barrier disruption and alteration of the brain's microenvironment (Lim and Pack, 2013). Although systemic NSAIDs do not localize the relevant inflammation, pLTF rescue by ketoprofen confirms that IH-1-induced inflammation undermines pLTF. Targeted inflammatory treatments would be advantageous since chronic NSAID use causes gastrointestinal and cardiovascular problems.
Spinal p38 MAPK inhibition also rescues pLTF, demonstrating that (1) the relevant inflammation is localized within cervical spinal segments encompassing the phrenic motor nucleus, and (2) p38 MAPK is a key link between IH-1-induced inflammation and pLTF suppression. Because inflammation has complex temporal dynamics and involves numerous molecules, we targeted p38 MAPK because it is a convergent, downstream molecule activated by many proinflammatory molecules (Widmann et al., 1999). p38 MAPK also increases posttranslational modification and gene transcription of inflammatory molecules, thereby propagating ongoing inflammation (Kaminska, 2005; Clark et al., 2009). Here, we provide the first demonstration that p38 MAPK is critical for IH-1 pLTF impairment. Consistent with this hypothesis, activated p38 MAPK increases within phrenic motoneurons and adjacent microglia. It is not yet clear whether p38 MAPK orchestrates the relevant inflammation or is a convergent, downstream molecule that directly impairs pLTF. Furthermore, it is not yet clear whether the relevant p38 MAPK activity is within phrenic motoneurons, adjacent microglia, or both. Microglial p38 MAPK plays a key role in neuroplasticity after spinal nerve ligation (Tsuda et al., 2004; Katsura et al., 2006; Ji and Suter, 2007) and spinal cord injury (Hains and Waxman, 2006), and it is associated with other conditions causing chronic pain (Boyle et al., 2006; Cui et al., 2006). Additional research is needed to understand specific p38 MAPK actions following IH-1.
While the p38 MAPK inhibitor used here (SB 202190) is reported to be a selective p38 MAPK inhibitor (Manthey et al., 1998; Lv et al., 2013), other p38 MAPK inhibitors with similar chemical structures (e.g., 4-[5-(4-fluorophenyl)-2-[4-(methylsulfonyl)phenyl]-1H-imidazol-4-yl]pyridine (SB 203580)) inhibit cyclooxygenase enzymatic activity (Borsch-Haubold et al., 1998). Although, no studies are available concerning the actions of SB 202190 on cyclooxygenase activity, we cannot completely rule out cyclooxygenase inhibition as a contributor to pLTF rescue post-IH-1.
In conclusion, we demonstrate a clear link between systemic inflammation initiated by physiologically relevant intermittent hypoxia and impaired respiratory motor plasticity. Even one night (8 h) of intermittent hypoxia abolishes pLTF by inducing cervical spinal inflammation. We demonstrate the role of IH-1 induced inflammation and refine our understanding by demonstrating that spinal inflammation is key, and that p38 MAPK is a likely key molecule linking that inflammation with pLTF.
It is essential to understand the impact of inflammation on the neural control of breathing, including respiratory plasticity. In clinical conditions associated with systemic inflammation, inflammation may undermine respiratory plasticity, potentially explaining at least some differences in carotid body and ventilatory LTF reported in humans following CIH or with OSA (Mateika and Narwani, 2009). Although it is unclear whether plasticity stabilizes or destabilizes breathing, understanding links between inflammation and respiratory plasticity is necessary to determine functional outcomes and appropriate treatments. Understanding and targeting relevant proinflammatory molecules may lead to new therapeutic interventions that restore plasticity in breathing and nonrespiratory motor functions in devastating disorders that compromise ventilatory capacity.
Footnotes
This work was supported by NIH Grant HL111598, the Francis Family Foundation (A.G.H.), an American Physiological Society undergraduate summer research fellowship (T.J.P.), and T32 HL007654 (S.M.S.). We thank B. Wathen for designing the intermittent hypoxia exposure chambers.
The authors declare no competing financial interests.
References
- Almendros I, Wang Y, Gozal D. The polymorphic and contradictory aspects of intermittent hypoxia. Am J Physiol Lung Cell Mol Physiol. 2014;307:L129–L140. doi: 10.1152/ajplung.00089.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respir Physiol. 1996;104:251–260. doi: 10.1016/0034-5687(96)00017-5. [DOI] [PubMed] [Google Scholar]
- Baker-Herman TL, Mitchell GS. Phrenic long-term facilitation requires spinal serotonin receptor activation and protein synthesis. J Neurosci. 2002;22:6239–6246. doi: 10.1523/JNEUROSCI.22-14-06239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat Neurosci. 2004;7:48–55. doi: 10.1038/nn1166. [DOI] [PubMed] [Google Scholar]
- Bisgard GE. Carotid body mechanisms in acclimatization to hypoxia. Respir Physiol. 2000;121:237–246. doi: 10.1016/S0034-5687(00)00131-6. [DOI] [PubMed] [Google Scholar]
- Borsch-Haubold AG, Pasquet S, Watson SP. Direct inhibition of cyclooxygenase-1 and -2 by the kinase inhibitors SB 203580 and PD 98059. SB 203580 also inhibits thromboxane synthase. J Biol Chem. 1998;273:28766–28772. doi: 10.1074/jbc.273.44.28766. [DOI] [PubMed] [Google Scholar]
- Boyle DL, Jones TL, Hammaker D, Svensson CI, Rosengren S, Albani S, Sorkin L, Firestein GS. Regulation of peripheral inflammation by spinal p38 MAP kinase in rats. PLoS Med. 2006;3:e338. doi: 10.1371/journal.pmed.0030338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabré F, Fernández MF, Calvo L, Ferrer X, García ML, Mauleón D. Analgesic, antiinflammatory, and antipyretic effects of S(+)-ketoprofen in vivo. J Clin Pharmacol. 1998;38:3S–10S. doi: 10.1002/j.1552-4604.1998.tb04411.x. [DOI] [PubMed] [Google Scholar]
- Carabaza A, Suesa N, Tost D, Pascual J, Gomez M, Gutierrez M, Ortega E, Montserrat X, Garcia AM, Mis R, Cabre F, Mauleon D, Carganico G. Stereoselective metabolic pathways of ketoprofen in the rat: incorporation into triacylglycerols and enantiomeric inversion. Chirality. 1996;8:163–172. doi: 10.1002/(SICI)1520-636X(1996)8:2<163::AID-CHIR1>3.0.CO%3B2-K. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunol Rev. 2006;213:48–65. doi: 10.1111/j.1600-065X.2006.00441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A, Dean J, Tudor C, Saklatvala J. Post-transcriptional gene regulation by MAP kinases via AU-rich elements. Front Biosci. 2009;14:847–871. doi: 10.2741/3282. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Crain JM, Nikodemova M, Watters JJ. Expression of P2 nucleotide receptors varies with age and sex in murine brain microglia. J Neuroinflammation. 2009;6:24. doi: 10.1186/1742-2094-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Chen Y, Zhi JL, Guo RX, Feng JQ, Chen PX. Activation of p38 mitogen-activated protein kinase in spinal microglia mediates morphine antinociceptive tolerance. Brain Res. 2006;1069:235–243. doi: 10.1016/j.brainres.2005.11.066. [DOI] [PubMed] [Google Scholar]
- Dale EA, Satriotomo I, Mitchell GS. Cervical spinal erythropoietin induces phrenic motor facilitation via extracellular signal-regulated protein kinase and Akt signaling. J Neurosci. 2012;32:5973–5983. doi: 10.1523/JNEUROSCI.3873-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale EA, Ben Mabrouk F, Mitchell GS. Unexpected benefits of intermittent hypoxia: enhanced respiratory and nonrespiratory motor function. Physiology. 2014;29:39–48. doi: 10.1152/physiol.00012.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale-Nagle EA, Satriotomo I, Mitchell GS. Spinal vascular endothelial growth factor induces phrenic motor facilitation via extracellular signal-regulated kinase and Akt signaling. J Neurosci. 2011;31:7682–7690. doi: 10.1523/JNEUROSCI.0239-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Filippo M, Sarchielli P, Picconi B, Calabresi P. Neuroinflammation and synaptic plasticity: theoretical basis for a novel, immune-centred, therapeutic approach to neurological disorders. Trends Pharmacol Sci. 2008;29:402–412. doi: 10.1016/j.tips.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Foster RT, Jamali F. Stereoselective pharmacokinetics of ketoprofen in the rat. Influence of route of administration. Drug Metab Dispos. 1988;16:623–626. [PubMed] [Google Scholar]
- Foster RT, Jamali F, Russell AS, Alballa SR. Pharmacokinetics of ketoprofen enantiomers in healthy subjects following single and multiple doses. J Pharm Sci. 1988;77:70–73. doi: 10.1002/jps.2600770113. [DOI] [PubMed] [Google Scholar]
- Golder FJ, Fuller DD, Lovett-Barr MR, Vinit S, Resnick DK, Mitchell GS. Breathing patterns after mid-cervical spinal contusion in rats. Exp Neurol. 2011;231:97–103. doi: 10.1016/j.expneurol.2011.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozal D, Kheirandish-Gozal L. Cardiovascular morbidity in obstructive sleep apnea: oxidative stress, inflammation, and much more. Am J Respir Crit Care Med. 2008;177:369–375. doi: 10.1164/rccm.200608-1190PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther CH, Vinit S, Windelborn JA, Behan M, Mitchell GS. Atypical protein kinase C expression in phrenic motor neurons of the rat. Neuroscience. 2010;169:787–793. doi: 10.1016/j.neuroscience.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gynther M, Jalkanen A, Lehtonen M, Forsberg M, Laine K, Ropponen J, Leppänen J, Knuuti J, Rautio J. Brain uptake of ketoprofen-lysine prodrug in rats. Int J Pharm. 2010;399:121–128. doi: 10.1016/j.ijpharm.2010.08.019. [DOI] [PubMed] [Google Scholar]
- Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci. 2006;26:4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen MK, Taishi P, Chen Z, Krueger JM. Vagotomy blocks the induction of interleukin-1beta (IL-1beta) mRNA in the brain of rats in response to systemic IL-1beta. J Neurosci. 1998;18:2247–2253. doi: 10.1523/JNEUROSCI.18-06-02247.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes HB, Jayaraman A, Herrmann M, Mitchell GS, Rymer WZ, Trumbower RD. Daily intermittent hypoxia enhances walking after chronic spinal cord injury: a randomized trial. Neurology. 2014;82:104–113. doi: 10.1212/01.WNL.0000437416.34298.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxtable AG, Vinit S, Windelborn JA, Crader SM, Guenther CH, Watters JJ, Mitchell GS. Systemic inflammation impairs respiratory chemoreflexes and plasticity. Respir Physiol Neurobiol. 2011;178:482–489. doi: 10.1016/j.resp.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxtable AG, Smith SM, Vinit S, Watters JJ, Mitchell GS. Systemic LPS induces spinal inflammatory gene expression and impairs phrenic long-term facilitation following acute intermittent hypoxia. J Appl Physiol. 2013;114:879–887. doi: 10.1152/japplphysiol.01347.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Suter MR. p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain. 2007;3:33. doi: 10.1186/1744-8069-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminska B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy–from molecular mechanisms to therapeutic benefits. Biochim Biophys Acta. 2005;1754:253–262. doi: 10.1016/j.bbapap.2005.08.017. [DOI] [PubMed] [Google Scholar]
- Katsura H, Obata K, Mizushima T, Sakurai J, Kobayashi K, Yamanaka H, Dai Y, Fukuoka T, Sakagami M, Noguchi K. Activation of Src-family kinases in spinal microglia contributes to mechanical hypersensitivity after nerve injury. J Neurosci. 2006;26:8680–8690. doi: 10.1523/JNEUROSCI.1771-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokki H. Ketoprofen pharmacokinetics, efficacy, and tolerability in pediatric patients. Paediatr Drugs. 2010;12:313–329. doi: 10.2165/11534910-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Li RC, Row BW, Gozal E, Kheirandish L, Fan Q, Brittian KR, Guo SZ, Sachleben LR, Jr, Gozal D. Cyclooxygenase 2 and intermittent hypoxia-induced spatial deficits in the rat. Am J Respir Crit Care Med. 2003;168:469–475. doi: 10.1164/rccm.200211-1264OC. [DOI] [PubMed] [Google Scholar]
- Li RC, Row BW, Kheirandish L, Brittian KR, Gozal E, Guo SZ, Sachleben LR, Jr, Gozal D. Nitric oxide synthase and intermittent hypoxia-induced spatial learning deficits in the rat. Neurobiol Dis. 2004;17:44–53. doi: 10.1016/j.nbd.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Lim DC, Pack AI. Obstructive sleep apnea and cognitive impairment: Addressing the blood-brain barrier. Sleep Med Rev. 2013;18:35–48. doi: 10.1016/j.smrv.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L, Fuller DD, Bach KB, Kinkead R, Olson EB, Jr, Mitchell GS. Chronic intermittent hypoxia elicits serotonin-dependent plasticity in the central neural control of breathing. J Neurosci. 2001;21:5381–5388. doi: 10.1523/JNEUROSCI.21-14-05381.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, He L, Stensaas L, Dinger B, Fidone S. Adaptation to chronic hypoxia involves immune cell invasion and increased expression of inflammatory cytokines in rat carotid body. Am J Physiol Lung Cell Mol Physiol. 2009;296:L158–L166. doi: 10.1152/ajplung.90383.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett-Barr MR, Satriotomo I, Muir GD, Wilkerson JE, Hoffman MS, Vinit S, Mitchell GS. Repetitive intermittent hypoxia induces respiratory and somatic motor recovery after chronic cervical spinal injury. J Neurosci. 2012;32:3591–3600. doi: 10.1523/JNEUROSCI.2908-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv H, Li S, Zhang J, Liang W, Mu X, Jiang Y. In Vitro Effects of SB202190 on Echinococcus granulosus. Korean J Parasitol. 2013;51:255–258. doi: 10.3347/kjp.2013.51.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane PM, Mitchell GS. Episodic spinal serotonin receptor activation elicits long-lasting phrenic motor facilitation by an NADPH oxidase-dependent mechanism. J Physiol. 2009;587:5469–5481. doi: 10.1113/jphysiol.2009.176982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannila A, Kokki H, Heikkinen M, Laisalmi M, Lehtonen M, Louhisto HL, Järvinen T, Savolainen J. Cerebrospinal fluid distribution of ketoprofen after intravenous administration in young children. Clin pharmacokinet. 2006;45:737–743. doi: 10.2165/00003088-200645070-00008. [DOI] [PubMed] [Google Scholar]
- Manthey CL, Wang SW, Kinney SD, Yao Z. SB202190, a selective inhibitor of p38 mitogen-activated protein kinase, is a powerful regulator of LPS-induced mRNAs in monocytes. J Leukoc Biol. 1998;64:409–417. doi: 10.1002/jlb.64.3.409. [DOI] [PubMed] [Google Scholar]
- Mantilla CB, Zhan WZ, Sieck GC. Retrograde labeling of phrenic motoneurons by intrapleural injection. J Neurosci Methods. 2009;182:244–249. doi: 10.1016/j.jneumeth.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateika JH, Narwani G. Intermittent hypoxia and respiratory plasticity in humans and other animals: does exposure to intermittent hypoxia promote or mitigate sleep apnoea? Exp Physiol. 2009;94:279–296. doi: 10.1113/expphysiol.2008.045153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire M, Zhang Y, White DP, Ling L. Chronic intermittent hypoxia enhances ventilatory long-term facilitation in awake rats. J Appl Physiol. 2003;95:1499–1508. doi: 10.1152/japplphysiol.00044.2003. [DOI] [PubMed] [Google Scholar]
- McGuire M, Zhang Y, White DP, Ling L. Serotonin receptor subtypes required for ventilatory long-term facilitation and its enhancement after chronic intermittent hypoxia in awake rats. Am J Physiol Regul Integr Comp Physiol. 2004;286:R334–R341. doi: 10.1152/ajpregu.00463.2003. [DOI] [PubMed] [Google Scholar]
- Navarrete-Opazo A, Mitchel GS. Therapeutic potential of intermittent hypoxia: A matter of dose. Am J Physiol Regul Integr Comp Physiol. 2014;307:R1181–R1197. doi: 10.1152/ajpregu.00208.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netter P, Lapicque F, Bannwarth B, Tamisier JN, Thomas P, Royer RJ. Diffusion of intramuscular ketoprofen into the cerebrospinal fluid. Eur J Clin Pharmacol. 1985;29:319–321. doi: 10.1007/BF00544088. [DOI] [PubMed] [Google Scholar]
- Nichols NL, Gowing G, Satriotomo I, Nashold LJ, Dale EA, Suzuki M, Avalos P, Mulcrone PL, McHugh J, Svendsen CN, Mitchell GS. Intermittent hypoxia and stem cell implants preserve breathing capacity in a rodent model of amyotrophic lateral sclerosis. Am J Respir Crit Care Med. 2013;187:535–542. doi: 10.1164/rccm.201206-1072OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikodemova M, Watters JJ. Efficient isolation of live microglia with preserved phenotypes from adult mouse brain. J Neuroinflammation. 2012;9:147. doi: 10.1186/1742-2094-9-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawar A, Peng YJ, Jacono FJ, Prabhakar NR. Comparative analysis of neonatal and adult rat carotid body responses to chronic intermittent hypoxia. J Appl Physiol. 2008;104:1287–1294. doi: 10.1152/japplphysiol.00644.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc Natl Acad Sci U S A. 2003;100:10073–10078. doi: 10.1073/pnas.1734109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR, Peng YJ, Kumar GK, Pawar A. Altered carotid body function by intermittent hypoxia in neonates and adults: relevance to recurrent apneas. Respir Physiol Neurobiol. 2007;157:148–153. doi: 10.1016/j.resp.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Rey S, Del Rio R, Alcayaga J, Iturriaga R. Chronic intermittent hypoxia enhances cat chemosensory and ventilatory responses to hypoxia. J Physiol. 2004;560:577–586. doi: 10.1113/jphysiol.2004.072033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge RG, Côté C. Mathematics of quantitative kinetic PCR and the application of standard curves. Nucleic Acids Res. 2003;31:e93. doi: 10.1093/nar/gng093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw KN, Commins S, O'Mara SM. Lipopolysaccharide causes deficits in spatial learning in the watermaze but not in BDNF expression in the rat dentate gyrus. Behav Brain Res. 2001;124:47–54. doi: 10.1016/S0166-4328(01)00232-7. [DOI] [PubMed] [Google Scholar]
- Teeling JL, Cunningham C, Newman TA, Perry VH. The effect of non-steroidal anti-inflammatory agents on behavioural changes and cytokine production following systemic inflammation: implications for a role of COX-1. Brain Behav Immun. 2010;24:409–419. doi: 10.1016/j.bbi.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tester NJ, Fuller DD, Fromm JS, Spiess MR, Behrman AL, Mateika JH. Long-term facilitation of ventilation in humans with chronic spinal cord injury. Am J Respir Crit Care Med. 2014;189:57–65. doi: 10.1164/rccm.201305-0848OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumbower RD, Jayaraman A, Mitchell GS, Rymer WZ. Exposure to acute intermittent hypoxia augments somatic motor function in humans with incomplete spinal cord injury. Neurorehabil Neural Repair. 2012;26:163–172. doi: 10.1177/1545968311412055. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Mizokoshi A, Shigemoto-Mogami Y, Koizumi S, Inoue K. Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia. 2004;45:89–95. doi: 10.1002/glia.10308. [DOI] [PubMed] [Google Scholar]
- Vereker E, Campbell V, Roche E, McEntee E, Lynch MA. Lipopolysaccharide inhibits long term potentiation in the rat dentate gyrus by activating caspase-1. J Biol Chem. 2000;275:26252–26258. doi: 10.1074/jbc.M002226200. [DOI] [PubMed] [Google Scholar]
- Vinit S, Windelborn JA, Mitchell GS. Lipopolysaccharide attenuates phrenic long-term facilitation following acute intermittent hypoxia. Respir Physiol Neurobiol. 2011;176:130–135. doi: 10.1016/j.resp.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JS. NSAID: an update on their analgesic effects. Clin Exp Pharmacol Physiol. 1995;22:855–860. doi: 10.1111/j.1440-1681.1995.tb01950.x. [DOI] [PubMed] [Google Scholar]
- Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]