

Abstract
Synaptic plasticity deficits are increasingly recognized as causing the memory impairments which define Alzheimer's disease (AD). In AD mouse models, evidence of abnormal synaptic function is present before the onset of cognitive deficits, and presents as increased synaptic depression revealed only when synaptic homeostasis is challenged, such as with suppression of ryanodine receptor (RyR)-evoked calcium signaling. Otherwise, at early disease stages, the synaptic physiology phenotype appears normal. This suggests compensatory mechanisms are recruited to maintain a functionally normal net output of the hippocampal circuit. A candidate calcium-regulated synaptic modulator is nitric oxide (NO), which acts presynaptically to boost vesicle release and glutamatergic transmission. Here we tested whether there is a feedforward cycle between the increased RyR calcium release seen in presymptomatic AD mice and aberrant NO signaling which augments synaptic plasticity. Using a combination of electrophysiological approaches, two-photon calcium imaging, and protein biochemistry in hippocampal tissue from presymptomatic 3xTg-AD and NonTg mice, we show that blocking NO synthesis results in markedly augmented synaptic depression mediated through presynaptic mechanisms in 3xTg-AD mice. Additionally, blocking NO reduces the augmented synaptically evoked dendritic calcium release mediated by enhanced RyR calcium release. This is accompanied by increased nNOS levels in the AD mice and is reversed upon normalization of RyR-evoked calcium release with chronic dantrolene treatment. Thus, recruitment of NO is serving a compensatory role to boost synaptic transmission and plasticity during early AD stages. However, NO's dual role in neuroprotection and neurodegeneration may convert to maladaptive functions as the disease progresses.
Keywords: calcium, homeostasis, nitric oxide, ryanodine receptor, synaptic depression, synaptic plasticity
Introduction
Synaptic plasticity deficits are increasingly recognized as causing memory loss in AD (Selkoe, 2002; Knobloch and Mansuy, 2008; Nistico et al., 2012). However, the early mechanisms driving synaptic pathophysiology are poorly understood. Short and long-term plasticity are calcium-dependent processes, and subject to additional modifications through second messengers, such as nitric oxide (NO). This gaseous neuromodulator is generated by NO synthase (NOS) through NMDAR-mediated calcium entry (Boehning and Snyder, 2003; Garthwaite, 2008). NO can either facilitate or suppress plasticity through biphasic effects on NMDAR and AMPAR insertion, presynaptic vesicle regulation, S-nitrosylation of synaptic proteins, and cyclic GMP (cGMP) generation (Reyes-Harde et al., 1999; Stanton et al., 2005; Ratnayaka et al., 2012; Selvakumar et al., 2013).
NO signaling is associated with neurodegenerative diseases through formation of reactive nitrogen species and cGMP signaling cascades (Nakamura et al., 2013; Zhao et al., 2015). Yet, NO also has neuroprotective capacities, as shown in AD mouse models where it appears to be particularly important in reducing cell loss and tau pathology (Colton et al., 2006, 2008; Wilcock et al., 2008). In AD models, NO can be altered through several mechanisms. For example, the NMDAR-mediated calcium entry which activates NOS is augmented by abnormal ryanodine receptor (RyR)-evoked calcium-induced calcium release (Goussakov et al., 2010). NOS protein levels are also increased in AD mouse models and human AD brains (Luth et al., 2002; Müller et al., 2011; Shilling et al., 2014), as are RyR levels (Chakroborty et al., 2009, 2012a; Bruno et al., 2012). In presymptomatic AD mice, these conditions, which amplify NO, occur alongside exaggerated hippocampal synaptic depression; these deficits are revealed when homeostasis is challenged, such as suppressing RyR-calcium release. Otherwise the hippocampal network, and cognitive performance, appear normal but clearly are not (Oddo et al., 2003; Chakroborty et al., 2009; Chakroborty and Stutzmann, 2011). The undetected deficits in synaptic plasticity suggest recruitment of compensatory mechanisms. Here we examine whether increased NOS is sustaining synaptic potentiation and demonstrate a NO-mediated increase in synaptically evoked calcium responses and neurotransmitter release which counters maladaptive synaptic depression in AD neurons.
Materials and Methods
Experimental animals.
Six to 8-week-old 3xTg-AD and NonTg (nontransgenic) mice, of both genders, were used. These experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee at Rosalind Franklin University. The 3xTg-AD (APPswe, TauP301L, and PS1M164VKI) mice used were described previously (Oddo et al., 2003). NonTg mice are on the same background strain (129/C57BL6) as the 3xTg-AD mice.
Hippocampal slice preparation.
Mice were deeply anesthetized, decapitated, and brains removed into sucrose cutting solution (in mm: 200 sucrose, 1.5 KCl, 0.5 CaCl2, 4.0 MgCl2, 1.0 KH2PO4, 25 NaHCO3, 10 Na-ascorbate, and 20 dextrose, equilibrated with 95% O2/5% CO2). Transverse hippocampal slices (300 μm for patch recordings; 400 μm for field recordings) were prepared as described previously (Chakroborty et al., 2012a) and maintained in standard aCSF (in mm: 130 NaCl, 2.5 KCl, 2.0 CaCl2, 1.2 MgSO4, 1.25 KH2PO4, 25 NaHCO3 and 10 dextrose) at 30°C for 1 h before use. For experiments with l-NAME (Nω-Nitro-l-arginine methyl ester hydrochloride; NOS inhibitor), slices were incubated in 200 μm l-NAME for 2 h before use. For experiments with SNAP (S-nitroso-N-Acetyl-d, l-penicillamine; NO donor), slices were washed with 100 μm SNAP for 40 min after baseline responses were recorded.
Extracellular field potential recording.
Hippocampal brain slices (400 μm) were placed in an interface chamber (Harvard Apparatus) and perfused at 1.5 ml/min with aCSF equilibrated with 95% O2/5% CO2 at room temperature (27°C). Data were acquired at 10 kHz using pClamp 9.2 software with a DigiData 1322 A-D converter and AxoClamp 2B amplifier. Synaptic responses were evoked by stimulating the Schaffer collateral/commissural pathway with a bipolar stimulating electrode. Field EPSPs (fEPSPs) were recorded in the stratum radiatum of the CA1 subfield of the hippocampus using recording microelectrodes (2–4 MΩ) filled with aCSF. Input/output (I/O) curves were generated using stimulus intensities from 0–225 μA in steps of 25 μA. Baseline stimulus intensity was determined from I/O curves as the stimulus intensity evoking a 30% or 70% of maximal response for LTP or LTD experiments, respectively (data not shown). Baseline fEPSPs were recorded at 0.05 Hz for 20 min before and for 60 min after induction of LTP or LTD. LTP was induced at baseline intensity by a high-frequency stimulus (HFS) consisting of two 100 Hz trains 10 s apart. LTD was induced at baseline intensity by a low-frequency stimulus (LFS) consisting of 900 pulses at 1 Hz.
Whole-cell patch-clamp recording.
Hippocampal brain slices (300 μm) were placed in a perfusion chamber mounted on a movable stage assembly on an upright microscope (BX50WI; Olympus Optical) and superfused at 2 ml/min with aCSF equilibrated with 95% O2/5% CO2 at room temperature (27°C). Patch pipettes (4–6 MΩ) pulled from borosilicate glass tubing were filled with intracellular solution (in mm: 135 K-gluconate, 2 MgCl2, 4 Na-ATP, 0.4 Na-GTP, 10 Na-phosphocreatine and 10 HEPES, pH adjusted to 7.3 with KOH) and 100 μm bis-fura-2. Hippocampal CA1 pyramidal neurons were identified visually via infrared differential interference contrast optics, and electrophysiologically by their passive membrane properties and spike frequency accommodation. Membrane potentials and evoked responses were obtained in current-clamp mode using pClamp 10 software acquired at 10 kHz with a Digidata 1322 A-D converter and MultiClamp 700B amplifier. Spontaneous postsynaptic potentials were recorded for 1 min. Synaptic responses were evoked by stimulating the Schaffer collateral/commissural pathway with a monopolar stimulating electrode. I/O curves were generated using stimulus intensities from 0–500 pA in steps of 50 pA. Baseline stimulus intensity was determined from I/O curves as the stimulus intensity evoking a 70% of maximal response (data not shown). LTD was induced at baseline intensity by a low-frequency stimulus (LFS) consisting of 900 pulses at 1 Hz. Paired-pulse facilitation (PPF) was assessed before and after LFS using interpulse intervals of 25, 50, and 200 ms. Five successive responses were recorded for each interpulse interval at 0.05 Hz. Access resistance was continually monitored, and cells were used for recording only if the access resistance was maintained <10 MΩ.
Calcium imaging.
Imaging of fluorescent calcium signals was performed in acute hippocampal brain slices using a custom-made video-rate multiphoton imaging system. Individual CA1 pyramidal neurons were filled with the calcium indicator bis-fura-2 via a patch pipette and calcium responses were imaged from dendrites and dendritic spines. Laser excitation was provided by 80 MHz trains of ultra-short (100 fs) pulses at 780 nm from a titanium/sapphire laser (Mai Tai Broadband, Spectra-Physics). The laser beam was scanned by paired galvanometers to provide a full-frame scan rate of 30 Hz. The scanned beam was focused onto the tissue through an Olympus 40× water-immersion objective (NA 0.8). Emitted fluorescence light was detected by a wide-field photomultiplier (R5929, Hamamatsu) and captured by frame-grabber software VideoSavant 5.0 (IO Industries). Imaging was synced with electrophysiological protocols through Digidata 1322 A-D board controlled by pClamp 10 software. Calcium responses were generated by 30 Hz trains (1.5 s) before, 1 min after and 20 min after LFS. 30 Hz trains were administered at baseline stimulus intensity. Images were analyzed offline with MetaMorph software. Results are expressed as inverse ratios so that increases in calcium concentration correspond to increasing ratios. The percentage change is calculated as [(F0/ΔF) − 1] × 100, where F0 is the average resting fluorescence at baseline and ΔF is the decrease in fluorescence reflecting calcium release.
Immunoblot analysis.
Methodological details were described previously by Chakroborty et al., 2012b. Hippocampal tissue was homogenized in tissue protein extraction reagents and total protein was separated by SDS-PAGE on 3–8% Tris-acetate NuPAGE gradient gels. Protein was transferred onto polyvinylidene difluoride membranes at 30 V for 2 h. Rabbit anti-NOS and anti-β-actin primary antibodies (Abcam) were diluted 1:1000 in 2.5% nonfat milk and applied for 72 h at 4°C. HRP-conjugated secondary antibodies were applied for 1 h at room temperature (27°C). Images were acquired using VersaDoc Imaging System and quantified with ImageJ software.
Data analysis and statistics.
Evoked responses were analyzed offline using Clampfit 10 and Origin Pro8 software. For PPF recordings, EPSP amplitudes (mV) were expressed as the ratio of the second EPSP over the first EPSP. For LTP and LTD recordings, fEPSP slopes recorded 0–2, 15–20, and 50–60 min after HFS or LFS were averaged and expressed as a percentage of the average slope from pre-HFS or pre-LFS baseline recordings. Minianalysis (v6.0.9, Synaptosoft) was used to measure spontaneous EPSP events with minimal amplitude of 0.2 mV and minimal area of 3 mV·ms. Baseline was determined from a 1 ms average immediately before each event using “complex peak detection” in Minianalysis. Data were expressed as ±SEM and assessed for significance using paired t tests, or two-way ANOVAs with Scheffe's post hoc analysis, where n denotes number of slices examined in extracellular field experiments and number of neurons in whole-cell patch-clamp experiments. For all experiments, n = 6–8 for each group. Five to six mice were used for each set of experiments.
Results
Divergent roles of NO in modulating synaptic plasticity
Augmented calcium signals in 3xTg-AD mice may be aberrantly increasing NO to sustain normal synaptic function in AD mice. We tested this hypothesis by measuring hippocampal synaptic plasticity in aCSF (control) and l-NAME (NOS inhibitor) perfused slices from NonTg and 3xTg-AD mice (Fig. 1). Hippocampal synaptic plasticity occurs in multiple phases that involve short-term and long-term changes at the synapse. For example, short-term plasticity (PTP and STD) involve alterations in presynaptic neurotransmitter release properties (Fioravante and Regehr, 2011). LTP and LTD can be divided into a transient early phase that is mediated by modification of pre-existing proteins (E-LTP and E-LTD), and a more persistent late phase that requires gene transcription and new protein synthesis (L-LTP and L-LTD; Davies et al., 1989; Frey et al., 1993). We chose to compare short-term plasticity, as well as early and late phases of long-term plasticity to query which phases were particularly vulnerable to NO modification, which in turn provides further insight regarding the underlying mechanisms driving homeostasis and pathology in AD. First, we measured post-tetanic potentiation (PTP), a calcium-dependent form of short-term presynaptic plasticity (Zucker and Regehr, 2002). PTP was similar between NonTg and 3xTg-AD mice in control conditions, and blocking NOS abolished PTP in both groups (F(3,27) = 72.6, p < 0.01).
Figure 1.
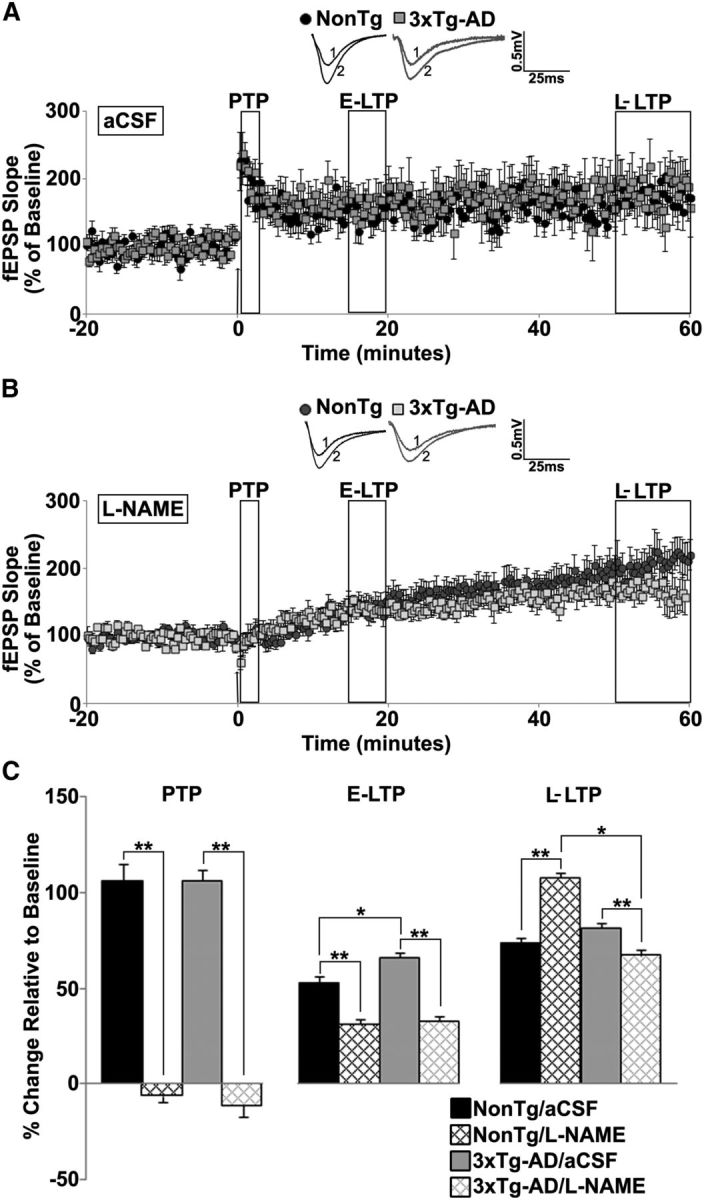
NO maintains long term potentiation in young 3xTg-AD mice. A, B, Graph shows averaged time course of LTP from NonTg and 3xTg-AD mice in aCSF (A) and l-NAME (B). Insets, Baseline traces before (1) and after (2) HFS from NonTg (black) and 3xTg-AD (gray) mice. C, Bar graph shows percentage change in post-HFS response relative to baseline 0–2 min after HFS (PTP), 15–20 min after HFS (E-LTP), and 50–60 min after HFS (L-LTP). The arrow denotes the time at which the HFS (2 × 100 Hz, 10 s apart) was administered; *p < 0.05 indicates significantly different from NonTg; **p < 0.05 indicates significantly different from control aCSF-treated slices.
Early-LTP (E-LTP, 15–20 min post-HFS) was slightly higher in 3xTg-AD compared with NonTg mice under control conditions. Application of l-NAME decreased E-LTP similarly in NonTg and 3xTg-AD hippocampi (F(3,27) = 29.51, p < 0.01). Late-LTP (L-LTP), measured 50–60 min post-HFS, was similar in both mouse strains in control conditions (Fig. 1A–C). However, l-NAME increases L-LTP in NonTg mice, whereas it decreased L-LTP in 3xTg-AD mice (F(3,27) = 74.64, p < 0.01).
Presymptomatic AD mice can exhibit abnormal synaptic depression when homeostasis is challenged (Chakroborty et al., 2009; Palop and Mucke, 2010a,b; Chakroborty and Stutzmann, 2011; Chakroborty et al., 2012a). Thus, we investigated whether NO-augmented plasticity could compensate for aberrant synaptic depression in AD mice (Fig. 2A–D). Short-term depression (STD), a form of presynaptic short-term plasticity (Zucker and Regehr, 2002; Fioravante and Regehr, 2011), was measured for 2 min post-LFS and was similar in NonTg and 3xTg-AD hippocampi in control conditions. However, l-NAME blocked STD in NonTg hippocampus, yet increased STD in 3xTg-AD (F(3,21) = 34.36, p < 0.01). In the presence of the NO donor SNAP, there was significant potentiation in NonTg hippocampus, whereas STD was completely abolished in 3xTg-AD (F(3,23) = 146.46, p < 0.01).Thus, NO exerts vastly different regulatory effects on presynaptic STD mechanisms between NonTg (NO is permissive for depression) and 3xTg-AD (NO may augment vesicle release to constrain LTD; Fig. 3).
Figure 2.
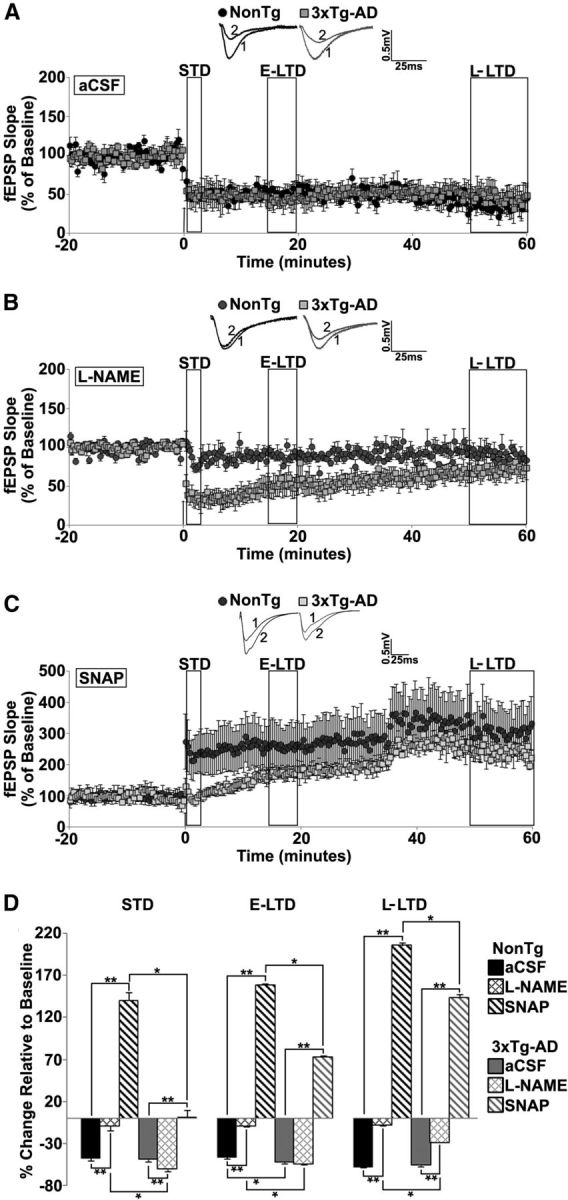
NO maintains long term depression in young 3xTg-AD mice. A–C, Graph shows averaged time course of LTD from NonTg and 3xTg-AD mice in aCSF (A), l-NAME (B), and SNAP (C). Insets, Representative baseline traces before (1) and after (2) LFS from NonTg (black) and 3xTg-AD (gray) mice. D, Bar graph shows percentage change in post-LFS responses relative to baseline 0–2 min after LFS (STD), 15–20 min after LFS (E-LTD), and 50–60 min after LFS (L-LTD). The arrow denotes the time at which the LFS (900 pulses at 1 Hz) was administered; *p < 0.05 indicates significantly different from NonTg; **p < 0.05 indicates significantly different from control aCSF-treated slices.
Figure 3.

NOS inhibition normalizes neurotransmitter release in 3xTg-AD neurons. A–C, Representative traces of postsynaptic spontaneous potentials recorded from NonTg (black) and 3xTg-AD (gray) CA1 neurons in aCSF (A), l-NAME (B), and SNAP (C). D, E, Bar graphs show averaged frequency (D) and amplitude (E) of spontaneous events from NonTg and 3xTg-AD neurons in aCSF, l-NAME and SNAP before (baseline) and 20 min after LFS (E-LTD); *p < 0.05 indicates significantly different from NonTg; **p < 0.05 indicates significantly different from control aCSF-treated neurons.
E-LTD (15–20 min post-LFS), was moderately higher in 3xTg-AD mice than NonTg mice in control conditions. However, l-NAME treatment revealed divergent effects between the mouse strains: in NonTg mice, E-LTD amplitude is markedly reduced, but l-NAME did not alter E-LTD in 3xTg-AD hippocampus (F(3,21) = 257.75, p < 0.01). SNAP treatment resulted in a reversal in the polarity of plasticity in both NonTg and 3xTg-AD hippocampus, though potentiation was significantly larger in NonTg mice than in 3xTg-AD (F(3,23) = 751.77, p < 0.01). L-LTD, measured 50–60 min post-LFS, was similar in both strains in control conditions, however, different responses are revealed when NO synthesis is blocked. Here, L-LTD is nearly abolished in NonTg mice but only partially reduced in 3xTg-AD mice (F(3,21) = 79.54, p < 0.01). With SNAP, 50–60 min post-LFS, the reversal in polarity of plasticity persisted in both strains, with potentiation being considerably greater in NonTg than 3xTg-AD mice (F(3,23) = 130.92, p < 0.01). The increased potentiation in NonTg hippocampus after SNAP treatment is likely driven by activity-dependent neurotransmitter vesicle release; whereas the blunted potentiation observed in 3xTg-AD mice may reflect reduced vesicle store content due to the high baseline activity (Fig. 3).
Inhibition of NO signaling normalizes neurotransmitter release and presynaptic plasticity to NonTg levels
To explore differences in presynaptic effects mediated by NO, we measured spontaneous neurotransmitter vesicle release and paired-pulse facilitation before and 20 min after LFS. We first measured spontaneous glutamatergic EPSPs (sEPSPs) to determine whether NO alters release properties in CA1 neurons (Fig. 3A–E). In control aCSF, pre-LFS, the sEPSP frequency was significantly greater in 3xTg-AD than in NonTg neurons. l-NAME had no significant effects on sEPSP properties in NonTg neurons; yet, in 3xTg-AD neurons, l-NAME reduced the frequency of sEPSPs to NonTg levels (F(3,28) = 5.73, p < 0.05). SNAP significantly increased the pre-LFS frequency of sEPSPs in both NonTg, as well as 3xTg-AD, compared with control aCSF (F(3,20) = 8.82, p < 0.01), though frequency of sEPSPs in 3xTg-AD neurons were significantly greater than in NonTg neurons (F(3,20) = 4.65, p < 0.05). This supports a facilitatory presynaptic role for NO in spontaneous neurotransmitter release in 3xTg-AD neurons. LFS does not change the frequency of sEPSPs in either NonTg or 3xTg-AD neurons. There were no differences in sEPSP amplitude between NonTg and 3xTg-AD neurons regardless of condition. Thus, NO retrogradely increases presynaptic vesicle release in 3xTg-AD mice.
An additional short-term presynaptic assay is PPF (Zucker and Regehr, 2002). Paired-pulse ratios were measured across a range of interpulse intervals (25–200 ms) under conditions as described above (Fig. 4). In NonTg mice, l-NAME or LFS did not affect PPF ratios. SNAP decreased pre-LFS PPF ratios at 25 ms (t(1,6) = 4.18, p < 0.01) and 50 ms (t(1,6) = 4.98, p < 0.01) intervals and at the 25 ms interval after LFS (t(1,6) = 3.83, p < 0.01) indicating a role of NO in presynaptic vesicle release. However, in 3xTg-AD neurons, PPF ratios decreased at 25 ms (t(1,8) = 2.95, p < 0.01) and 50 ms (t(1,8) = 2.94, p < 0.01) intervals after LFS in 3xTg-AD neurons, indicative of increased calcium and vesicle release probability. Notably, blocking NO synthesis increased PPF ratios to pre-LFS levels in 3xTg-AD neurons and were not different from NonTg. Similar to NonTg mice, SNAP decreased pre-LFS PPF ratios in 3xTg-AD neurons at the 25 ms (t(1,6) = 1.74, p < 0.05) and 50 ms (t(1,6) = 2.4, p < 0.05) intervals. There were no changes in PPF with a 200 ms interval in either group.
Figure 4.

NO increases probability of neurotransmitter release. A, Representative PPF traces at 50 ms interpulse interval from NonTg (black) and 3xTg-AD (gray) neurons. B, PPF measured at indicated interstimulus intervals. Bar graph shows paired-pulse ratio for NonTg and 3xTg-AD neurons in aCSF, l-NAME and SNAP before (baseline) and 20 min after LFS (E-LTD); *p < 0.05 indicates significantly different from NonTg; **p < 0.05 indicates significantly different from control aCSF-treated neurons; ***p < 0.05 indicates significantly different from baseline.
Presynaptic calcium is a fundamental variable increasing neurotransmitter vesicle release probability; l-NAME occludes this process in the 3xTg-AD mice only, whereas SNAP promotes this process in 3xTg-AD mice, suggesting that NO exerts a unique facilitatory role on glutamatergic vesicle release and thus increases transmitter tone in the synaptic cleft. The calcium dynamics underlying the increased release probability may also contribute to the increased E-LTD in the AD mice, and increased synaptic depression upon sustained stimulation due to depleted vesicles in the readily releasable or reserve pools within presynaptic terminals.
Blocking NO synthesis decreases synaptically evoked calcium responses in 3xTg-AD neurons
Synaptic plasticity polarity (potentiation or depression) is reflective of spatial and temporal calcium patterning (Cummings et al., 1996; Fitzjohn and Collingridge, 2002; Raymond and Redman, 2006); this patterning can be shaped by NO-mediated nitrosylation of calcium channels (Kakizawa et al., 2012, 2013). To examine interactions among NO signaling, calcium responses, and plasticity, we measured synaptically evoked (30 Hz for 1.5 s) calcium responses in CA1 dendrites from NonTg and 3xTg-AD mice before and after administering a LFS (Fig. 5). Evoked dendritic calcium responses were imaged at three time points: baseline (1 min pre-LFS), STD (1 min post-LFS), and E-LTD (20 min post-LFS). LTD (50–60 min post-LFS) was not measured due to washout of intracellular messengers through the patch pipette. Under control conditions, calcium responses were 3- to 4-fold greater in 3xTg-AD CA1 dendrites relative to NonTg dendrites at each time point. LFS did not alter NonTg calcium responses; however there was an increasing trend in 3xTg-AD neurons (p = 0.08). We next incubated hippocampal slices with l-NAME to investigate the role of NO in synaptically evoked calcium signals. In NonTg dendrites, blocking NO synthesis did not alter synaptically evoked calcium responses. However, l-NAME markedly reduced calcium responses in 3xTg-AD CA1 dendrites (Fig. 5; baseline: F(3,28) = 16.85, p < 0.01; STD: F(3,28) = 20.08, p < 001; E-LTD: F(3,28) = 8.17, p < 0.01). Administration of LFS did not alter calcium responses to 30 Hz trains within either group. We next treated slices with NO donor SNAP to further investigate the role of NO in synaptically evoked calcium responses. SNAP did not affect synaptically evoked calcium responses in NonTg or 3xTg-AD dendrites compared with control aCSF, although responses were still 3- to 4-fold greater in 3xTg-AD neurons at each time point. The similar magnitude of evoked calcium responses in 3xTg-AD neurons in SNAP and in control aCSF may indicate a “ceiling effect” for the S-nitrosylation of the RyRs whereby increasing NO levels does not further increase evoked calcium responses. This observation is supported by our data that demonstrate a reduction in evoked calcium responses in 3xTg-AD neurons treated with the NOS inhibitor l-NAME.
Figure 5.
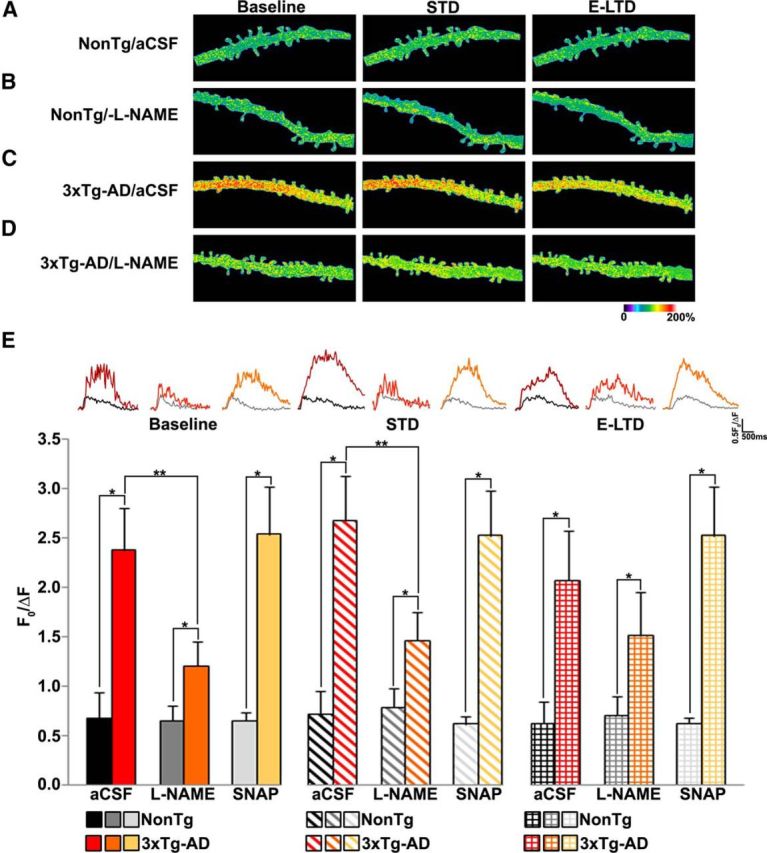
NOS inhibition decreases synaptically evoked calcium responses in 3xTg-AD CA1 neurons. A, Pseudocolored images of dendritic calcium response from a NonTg neuron in aCSF to 30 Hz stimulus before LFS (baseline), 1 min after LFS (STD), and 20 min after LFS (E-LTD). B–D, Same as in A, but from l-NAME-treated NonTg neuron (B), a 3xTg-AD neuron in aCSF (C), and 3xTg-AD neuron treated with l-NAME (D). E, Bar graphs comparing averaged maximal calcium changes between NonTg and 3xTg-AD neurons with and without l-NAME or SNAP after 30 Hz stimulus at baseline, STD and E-LTD time-points. Inset, Calcium traces from NonTg/aCSF (black), NonTg/l-NAME (gray), NonTg/SNAP (light gray), 3xTg-AD/aCSF (red), 3xTg-AD/l-NAME (orange), and 3xTg-AD/SNAP (yellow) neurons; *p < 0.05 indicates significantly different from NonTg neurons; **p < 0.05 indicates significantly different from control aCSF-treated neurons.
Stabilizing RyR-calcium restores nNOS levels in 3xTg-AD hippocampus
ER-mediated calcium release can activate transcription factors such as pCREB and drive neuronal NOS (nNOS) expression (Ziviani et al., 2011). Because the AD mice have increased ER-calcium release, we next measured hippocampal nNOS protein expression in young 3xTg-AD mice and found increased levels similar to those previously described (Muller et al., 2011; Shilling et al., 2014; Fig. 6). We then asked whether increased nNOS is associated with increased RyR-calcium signaling in AD mice by treating mice with dantrolene (10 mg/kg, i.p.; 4 weeks), which normalizes RyR calcium signaling in AD mice (Chakroborty et al., 2012b; Oules et al., 2012; Peng et al., 2012). Notably, restoring RyR-calcium levels prevented the increased nNOS expression in 3xTg-AD mice (F(3,23) = 5.95, p < 0.05). Importantly, this treatment did not affect NonTg nNOS levels, which argues against off-target effects and supports the hypothesis that increased RyR-evoked calcium signaling drives the increased nNOS expression in 3xTg-AD mice.
Figure 6.

Increased nNOS protein expression in 3xTg-AD mice. A, Western blot showing hippocampal nNOS protein levels in saline- or dantrolene-treated NonTg and 3xTg-AD mice. B, Bar graph shows nNOS protein levels relative to β-actin controls; *p < 0.05 indicates significantly different from NonTg; **p < 0.05 indicates significantly different from dantrolene-treated mice.
Discussion
Memory loss is the most salient feature of AD and is likely mediated by defects in synaptic functionality. Notably, early dysfunctions in calcium-regulated synaptic plasticity are observed before detectable memory impairments, yet the net output of the hippocampal network remains functionally intact. This functional disconnect raises interesting questions regarding the mechanisms by which plasticity homeostasis is maintained at the hippocampal circuit level despite profound Ca2+ signaling abnormalities at the cellular level. Here, we demonstrate a mechanism for synaptic homeostasis in presymptomatic 3xTg-AD mice via a facilitatory interaction between NO and RyR-evoked calcium release. This NO-mediated augmentation of presynaptic functions may initially compensate for synaptic depression associated with AD and maintain the plasticity required for memory encoding, yet over time the cumulative effects of NO and S-nitrosylation may convert to pathogenic cascades and accelerate AD pathology (Fig. 7).
Figure 7.
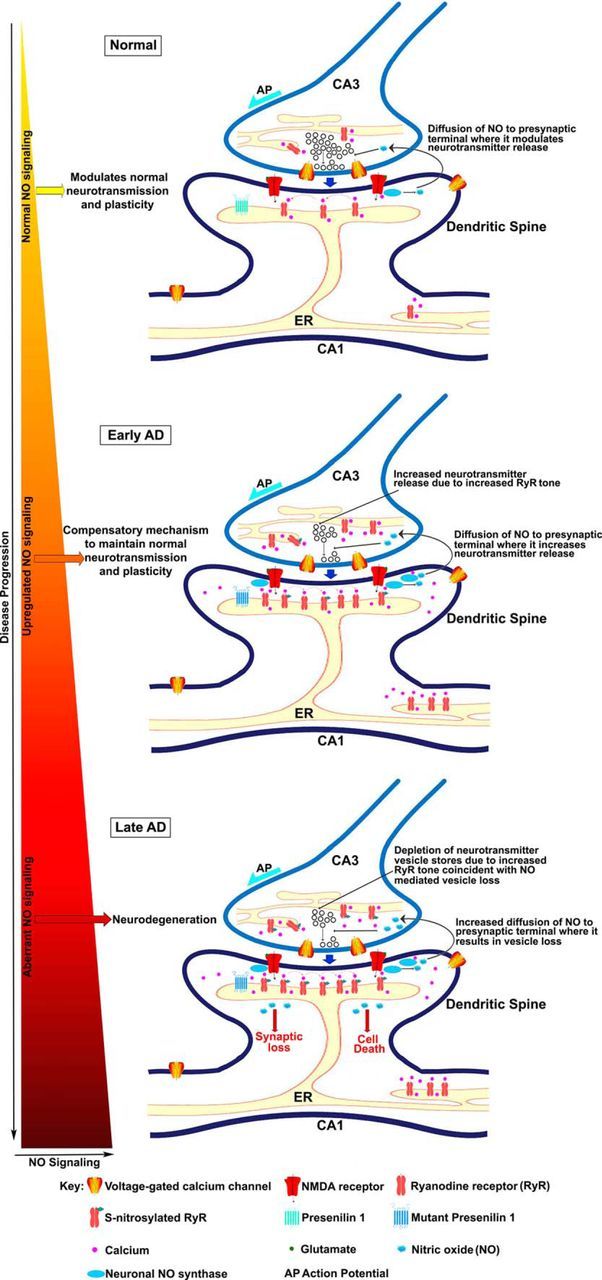
Proposed role of NO signaling in early synaptic homeostasis and later neurodegeneration at the CA3–CA1 synapse in AD. Normal: NO generated by nNOS in the postsynaptic CA1 terminal diffuses to the presynaptic CA3 terminal and modulates neurotransmitter release. Early AD: increased RyR expression significantly increases CICR (calcium-induced calcium release). At the presynaptic terminal, RyR-evoked CICR can facilitate spontaneous neurotransmitter release. Increased RyR-evoked calcium also drives increased nNOS production, which in turn, generates increased levels of NO that, diffuses to the presynaptic terminal. Increased CICR and NO signaling mediate increased release of neurotransmitter vesicles to maintain normal levels of plasticity. Late AD: sustained increases in RyR-evoked calcium signaling and NO signaling can deplete vesicles from the readily releasable pool, as well as the reserve pool and cause metabolic, oxidative and nitrosative stress, mitochondrial and ER dysfunction, and ultimately loss of synapses and neuronal death. Further, NO can aberrantly S-nitrosylate several proteins that eventually also contribute to neurodegeneration.
NO maintains functional plasticity in 3xTg-AD mice
In our studies, the primary site of NO regulation in the 3xTg-AD mice is in presynaptic terminals where it increases evoked and spontaneous vesicle release, as indicated by PPF assays and spontaneous vesicle release properties. Here, NO can augment transmitter release through cGMP signaling and S-nitrosylation of synaptic proteins which enhances presynaptic syntaxin binding with VAMP and SNAP25 (Meffert et al., 1994; Palmer et al., 2008). Additionally, NO influences the magnitude of vesicular release by converting reserve pool vesicles to readily releasable pool vesicles (Ratnayaka et al., 2012). NO can also increase RyR channel open probability through S-nitrosylation (Kakizawa et al., 2012). These reciprocal interactions between increased RyR-calcium signaling and upregulated nNOS expression in AD neurons can sustain increased NO generation and increase presynaptic gain. Postsynaptically, the neuroprotective feature of NO curbs excessive NMDAR-mediated calcium influx and excitotoxicity through S-nitrosylation of the NR2A subunit (Lipton et al., 1993; Kim et al., 1999). Concurrently, S-nitrosylation of caspase 3, 8, and 9 reduces apoptosis (Zhao et al., 2015).
Thus, in healthy neurons, there is a self-limiting cycle of synaptic activity, NO production, and presynaptic and postsynaptic excitability. However, in 3xTg-AD brains, an entirely different baseline of calcium signaling, presynaptic activity, and NOS levels are present, and NO may have very different functions under these circumstances.
Sustained NO synthesis and AD pathology
The increased nNOS levels and NO activity in AD brains may initially serve a neuroprotective role as suggested by the selectively spared NOS-positive neurons in AD (Hyman et al., 1992; Law et al., 2001). However, sustained increases in NO has deleterious effects, including oxidative stress, protein misfolding, mitochondrial stress and fragmentation, loss of synaptic function, and apoptosis (Andersen, 2004; Sayre et al., 2008; Steinert et al., 2010).
In AD, sustained NO and S-nitrosylation upregulation can tap into several neurodegenerative cascades, including increased RyR channel activity and calcium release, thus establishing a pathogenic feedforward cycle accelerating synaptic dysfunction, amyloid aggregation, and tau phosphorylation (Stutzmann, 2007; Bezprozvanny and Mattson, 2008). Other aberrantly S-nitrosylated proteins in AD brains include protein disulfide isomerase which regulates protein folding and ER stress (Uehara et al., 2006; Hatahet and Ruddock, 2009; Roussel et al., 2013), Drp1(dynamin-related protein) a GTPase regulating mitochondrial fission and synaptic function (Li et al., 2004), and cdk5 (cyclin-dependent kinase) which is vital to cell survival, neuronal differentiation, and synaptic plasticity, and when nitrosylated contributes to Aβ-induced spine loss (Qu et al., 2011; Cheung and Ip, 2012).
Therapeutic opportunities
The incidence of AD will increase dramatically over the next few decades and more effective therapies need to be developed. Because NO-mediated homeostatic signaling prevents excess synaptic depression by facilitating neurotransmitter release, strengthening these compensative mechanisms could provide novel therapies to maintain synaptic function in AD. These proposed mechanisms are in addition to those already ascribed to NO neuroprotection in AD mouse models (Colton et al., 2006, 2008; Wilcock et al., 2008). Further elucidation of the bimodal role of NO signaling during AD progression can facilitate potential therapeutic strategies for AD drug discovery.
Footnotes
This work was supported by NIH R01-AG030205 (G.E.S.) and the Alzheimer's Drug Discovery Foundation (G.E.S.).
The authors declare no competing financial interests.
References
- Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nat Med. 2004;10:S18–S25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008;31:454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehning D, Snyder SH. Novel neural modulators. Annu Rev Neurosci. 2003;26:105–131. doi: 10.1146/annurev.neuro.26.041002.131047. [DOI] [PubMed] [Google Scholar]
- Bruno AM, Huang JY, Bennett DA, Marr RA, Hastings ML, Stutzmann GE. Altered ryanodine receptor expression in mild cognitive impairment and Alzheimer's disease. Neurobiol Aging. 2012;33:1001.e1–1001.e16. doi: 10.1016/j.neurobiolaging.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakroborty S, Stutzmann GE. Early calcium dysregulation in Alzheimer's disease: setting the stage for synaptic dysfunction. Sci China Life Sci. 2011;54:752–762. doi: 10.1007/s11427-011-4205-7. [DOI] [PubMed] [Google Scholar]
- Chakroborty S, Goussakov I, Miller MB, Stutzmann GE. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J Neurosci. 2009;29:9458–9470. doi: 10.1523/JNEUROSCI.2047-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakroborty S, Kim J, Schneider C, Jacobson C, Molgó J, Stutzmann GE. Early presynaptic and postsynaptic calcium signaling abnormalities mask underlying synaptic depression in presymptomatic Alzheimer's disease mice. J Neurosci. 2012a;32:8341–8353. doi: 10.1523/JNEUROSCI.0936-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakroborty S, Briggs C, Miller MB, Goussakov I, Schneider C, Kim J, Wicks J, Richardson JC, Conklin V, Cameransi BG, Stutzmann GE. Stabilizing ER Ca2+ channel function as an early preventative strategy for Alzheimer's disease. PloS One. 2012b;7:e52056. doi: 10.1371/journal.pone.0052056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung ZH, Ip NY. Cdk5: a multifaceted kinase in neurodegenerative diseases. Trends Cell Biol. 2012;22:169–175. doi: 10.1016/j.tcb.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Colton CA, Vitek MP, Wink DA, Xu Q, Cantillana V, Previti ML, Van Nostrand WE, Weinberg B, Dawson H. NO synthase 2 (NOS2) deletion promotes multiple pathologies in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:12867–12872. doi: 10.1073/pnas.0601075103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton CA, Wilcock DM, Wink DA, Davis J, Van Nostrand WE, Vitek MP. The effects of NOS2 gene deletion on mice expressing mutated human AbetaPP. J Alzheimers Dis. 2008;15:571–587. doi: 10.3233/jad-2008-15405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings JA, Mulkey RM, Nicoll RA, Malenka RC. Ca2+ signaling requirements for long-term depression in the hippocampus. Neuron. 1996;16:825–833. doi: 10.1016/S0896-6273(00)80102-6. [DOI] [PubMed] [Google Scholar]
- Davies SN, Lester RA, Reymann KG, Collingridge GL. Temporally distinct pre- and post-synaptic mechanisms maintain long-term potentiation. Nature. 1989;338:500–503. doi: 10.1038/338500a0. [DOI] [PubMed] [Google Scholar]
- Fioravante D, Regehr WG. Short-term forms of presynaptic plasticity. Curr Opin Neurobiol. 2011;21:269–274. doi: 10.1016/j.conb.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzjohn SM, Collingridge GL. Calcium stores and synaptic plasticity. Cell Calcium. 2002;32:405–411. doi: 10.1016/S0143416002001999. [DOI] [PubMed] [Google Scholar]
- Frey U, Huang YY, Kandel ER. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- Garthwaite J. Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci. 2008;27:2783–2802. doi: 10.1111/j.1460-9568.2008.06285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goussakov I, Miller MB, Stutzmann GE. NMDA-mediated Ca(2+) influx drives aberrant ryanodine receptor activation in dendrites of young Alzheimer's disease mice. J Neurosci. 2010;30:12128–12137. doi: 10.1523/JNEUROSCI.2474-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatahet F, Ruddock LW. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal. 2009;11:2807–2850. doi: 10.1089/ars.2009.2466. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Marzloff K, Wenniger JJ, Dawson TM, Bredt DS, Snyder SH. Relative sparing of nitric oxide synthase-containing neurons in the hippocampal formation in Alzheimer's disease. Ann Neurol. 1992;32:818–820. doi: 10.1002/ana.410320618. [DOI] [PubMed] [Google Scholar]
- Kakizawa S, Yamazawa T, Chen Y, Ito A, Murayama T, Oyamada H, Kurebayashi N, Sato O, Watanabe M, Mori N, Oguchi K, Sakurai T, Takeshima H, Saito N, Iino M. Nitric oxide-induced calcium release via ryanodine receptors regulates neuronal function. EMBO J. 2012;31:417–428. doi: 10.1038/emboj.2011.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakizawa S, Yamazawa T, Iino M. Nitric oxide-induced calcium release: activation of type 1 ryanodine receptor by endogenous nitric oxide. Channels. 2013;7:1–5. doi: 10.4161/chan.22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WK, Choi YB, Rayudu PV, Das P, Asaad W, Arnelle DR, Stamler JS, Lipton SA. Attenuation of NMDA receptor activity and neurotoxicity by nitroxyl anion, NO. Neuron. 1999;24:461–469. doi: 10.1016/S0896-6273(00)80859-4. [DOI] [PubMed] [Google Scholar]
- Knobloch M, Mansuy IM. Dendritic spine loss and synaptic alterations in Alzheimer's disease. Mol Neurobiol. 2008;37:73–82. doi: 10.1007/s12035-008-8018-z. [DOI] [PubMed] [Google Scholar]
- Law A, Gauthier S, Quirion R. Say NO to Alzheimer's disease: the putative links between nitric oxide and dementia of the Alzheimer's type. Brain Res Brain Res Rev. 2001;35:73–96. doi: 10.1016/S0165-0173(00)00051-5. [DOI] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Choi YB, Pan ZH, Lei SZ, Chen HS, Sucher NJ, Loscalzo J, Singel DJ, Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- Luth HJ, Munch G, Arendt T. Aberrant expression of NOS isoforms in Alzheimer's disease is structurally related to nitrotyrosine formation. Brain Res. 2002;953:135–143. doi: 10.1016/S0006-8993(02)03280-8. [DOI] [PubMed] [Google Scholar]
- Meffert MK, Haley JE, Schuman EM, Schulman H, Madison DV. Inhibition of hippocampal heme oxygenase, nitric oxide synthase, and long-term potentiation by metalloporphyrins. Neuron. 1994;13:1225–1233. doi: 10.1016/0896-6273(94)90060-4. [DOI] [PubMed] [Google Scholar]
- Müller M, Cardenas C, Mei L, Cheung KH, Foskett JK. Constitutive cAMP response element binding protein (CREB) activation by Alzheimer's disease presenilin-driven inositol trisphosphate receptor (InsP3R) Ca2+ signaling. Proc Natl Acad Sci U S A. 2011;108:13293–13298. doi: 10.1073/pnas.1109297108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Tu S, Akhtar MW, Sunico CR, Okamoto S, Lipton SA. Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron. 2013;78:596–614. doi: 10.1016/j.neuron.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisticò R, Cavallucci V, Piccinin S, Macrì S, Pignatelli M, Mehdawy B, Blandini F, Laviola G, Lauro D, Mercuri NB, D'Amelio M. Insulin receptor beta-subunit haploinsufficiency impairs hippocampal late-phase LTP and recognition memory. Neuromolecular Med. 2012;14:262–269. doi: 10.1007/s12017-012-8184-z. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/S0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Oulès B, Del Prete D, Greco B, Zhang X, Lauritzen I, Sevalle J, Moreno S, Paterlini-Bréchot P, Trebak M, Checler F, Benfenati F, Chami M. Ryanodine receptor blockade reduces amyloid-beta load and memory impairments in Tg2576 mouse model of Alzheimer disease. J Neurosci. 2012;32:11820–11834. doi: 10.1523/JNEUROSCI.0875-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer ZJ, Duncan RR, Johnson JR, Lian LY, Mello LV, Booth D, Barclay JW, Graham ME, Burgoyne RD, Prior IA, Morgan A. S-nitrosylation of syntaxin 1 at Cys(145) is a regulatory switch controlling Munc18–1 binding. Biochem J. 2008;413:479–491. doi: 10.1042/BJ20080069. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Synaptic depression and aberrant excitatory network activity in Alzheimer's disease: two faces of the same coin? Neuromolecular Med. 2010a;12:48–55. doi: 10.1007/s12017-009-8097-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Amyloid-β-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci. 2010b;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Liang G, Inan S, Wu Z, Joseph DJ, Meng Q, Peng Y, Eckenhoff MF, Wei H. Dantrolene ameliorates cognitive decline and neuropathology in Alzheimer triple transgenic mice. Neurosci Lett. 2012;516:274–279. doi: 10.1016/j.neulet.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu J, Nakamura T, Cao G, Holland EA, McKercher SR, Lipton SA. S-nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by beta-amyloid peptide. Proc Natl Acad Sci U S A. 2011;108:14330–14335. doi: 10.1073/pnas.1105172108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnayaka A, Marra V, Bush D, Burden JJ, Branco T, Staras K. Recruitment of resting vesicles into recycling pools supports NMDA receptor-dependent synaptic potentiation in cultured hippocampal neurons. J Physiol. 2012;590:1585–1597. doi: 10.1113/jphysiol.2011.226688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond CR, Redman SJ. Spatial segregation of neuronal calcium signals encodes different forms of LTP in rat hippocampus. J Physiol. 2006;570:97–111. doi: 10.1113/jphysiol.2005.098947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Harde M, Empson R, Potter BV, Galione A, Stanton PK. Evidence of a role for cyclic ADP-ribose in long-term synaptic depression in hippocampus. Proc Natl Acad Sci U S A. 1999;96:4061–4066. doi: 10.1073/pnas.96.7.4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel BD, Kruppa AJ, Miranda E, Crowther DC, Lomas DA, Marciniak SJ. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 2013;12:105–118. doi: 10.1016/S1474-4422(12)70238-7. [DOI] [PubMed] [Google Scholar]
- Sayre LM, Perry G, Smith MA. Oxidative stress and neurotoxicity. Chem Res Toxicol. 2008;21:172–188. doi: 10.1021/tx700210j. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Selvakumar B, Jenkins MA, Hussain NK, Huganir RL, Traynelis SF, Snyder SH. S-nitrosylation of AMPA receptor GluA1 regulates phosphorylation, single-channel conductance, and endocytosis. Proc Natl Acad Sci U S A. 2013;110:1077–1082. doi: 10.1073/pnas.1221295110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilling D, Muller M, Takano H, Mak DO, Abel T, Coulter DA, Foskett JK. Suppression of InsP3 receptor-mediated Ca2+ signaling alleviates mutant presenilin-linked familial Alzheimer's disease pathogenesis. J Neurosci. 2014;34:6910–6923. doi: 10.1523/JNEUROSCI.5441-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton PK, Winterer J, Zhang XL, Müller W. Imaging LTP of presynaptic release of FM1-43 from the rapidly recycling vesicle pool of Schaffer collateral-CA1 synapses in rat hippocampal slices. Eur J Neurosci. 2005;22:2451–2461. doi: 10.1111/j.1460-9568.2005.04437.x. [DOI] [PubMed] [Google Scholar]
- Steinert JR, Chernova T, Forsythe ID. Nitric oxide signaling in brain function, dysfunction, and dementia. Neuroscientist. 2010;16:435–452. doi: 10.1177/1073858410366481. [DOI] [PubMed] [Google Scholar]
- Stutzmann GE. The pathogenesis of Alzheimers disease is it a lifelong “calciumopathy”? Neuroscientist. 2007;13:546–559. doi: 10.1177/1073858407299730. [DOI] [PubMed] [Google Scholar]
- Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Lewis MR, Van Nostrand WE, Davis J, Previti ML, Gharkholonarehe N, Vitek MP, Colton CA. Progression of amyloid pathology to Alzheimer's disease pathology in an amyloid precursor protein transgenic mouse model by removal of nitric oxide synthase 2. J Neurosci. 2008;28:1537–1545. doi: 10.1523/JNEUROSCI.5066-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao QF, Yu JT, Tan L. S-Nitrosylation in Alzheimer's disease. Mol Neurobiol. 2015;51:268–280. doi: 10.1007/s12035-014-8672-2. [DOI] [PubMed] [Google Scholar]
- Ziviani E, Lippi G, Bano D, Munarriz E, Guiducci S, Zoli M, Young KW, Nicotera P. Ryanodine receptor-2 upregulation and nicotine-mediated plasticity. EMBO J. 2011;30:194–204. doi: 10.1038/emboj.2010.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]