

Abstract
The development of effective and simple methods of vaccine preparation is desired for the prophylaxis and treatment of a variety of infectious diseases and cancers. We have created novel polyion complex (PIC) nanoparticles (NPs) composed of amphiphilic anionic biodegradable poly(γ-glutamic acid) (γ-PGA) and cationic polymers as a vaccine adjuvant. PIC NPs can be prepared by mixing γ-PGA-graft-l-phenylalanine ethylester (γ-PGA-Phe) polymer with cationic polymer in phosphate-buffered saline. We examined the efficacy of PIC NPs for antigen delivery and immunostimulatory activity in vitro and in vivo. PIC NPs enhanced the uptake of ovalbumin (OVA) by dendritic cells (DCs) and subsequently induced DC maturation. The immunization of mice with OVA-carrying PIC NPs induced potent and antigen-specific cellular and humoral immunity. Since PIC NPs can be created with water-soluble anionic γ-PGA-Phe and a cationic polymer by simple mixing in the absence of any organic solvents, PIC NPs may have potential as a novel candidate for an effective antigen carrier and vaccine adjuvant.
INTRODUCTION
The induction of potent adaptive immunity is essential for an effective vaccine. Although using only a part of a pathogen, such as a purified protein component, is generally much safer than using the whole pathogen, such components can induce a modest humoral immune response but little or no cell-mediated immune response (1, 2). Removal or eradication of intracellular pathogens by the cell-mediated immune response is highly desired for the next generation of vaccines (3, 4). Several lines of evidence suggest that an appropriate adjuvant can strongly enhance the immunogenicity of antigens (5–7). Such adjuvants must be first recognized by the innate immune cells in order to induce adaptive immunity. Alum-based adjuvants (alum) that are approved for clinical use in humans can increase humoral immune responses without unacceptable side effects. However, they elicit cell-mediated immune responses only poorly (1, 5). Therefore, ideal adjuvants are expected to effectively induce not only humoral but also cell-mediated immune responses.
Stimulation of the antigen-presenting cells (APCs) is considered to be important for the induction of adaptive immune responses (8, 9). Dendritic cells (DCs) are the most potent APCs that connect innate and adaptive immune responses (10). The interaction between antigens and DCs triggers the maturation of DCs in peripheral tissue. Immature DCs (iDCs) develop to mature DCs (mDCs) by interaction with various antigens, leading to the upregulation of costimulatory molecules and major histocompatibility complexes (MHCs). Subsequently, DCs migrate into the secondary lymphoid organs, present the processed antigen peptides to naive T cells, and induce adaptive immune responses. Thus, targeting selected antigens to DCs and subsequent stimulation of DCs constitute a pivotal strategy for establishing effective vaccine development.
Particle-based adjuvants made from certain biomaterials have also been explored as an antigen carrier in vaccine studies (4). Particles carrying antigens are capable of inducing potent immune responses via efficient antigen delivery to the APCs. In previous studies, we created biodegradable amphiphilic nanoparticles (NPs) composed of poly(γ-glutamic acid)-graft-l-phenylalanine ethylester (γ-PGA-Phe) (11–13). The NPs proved to have great potential as a vaccine adjuvant. However, there is an obstacle to developing this classical type of NPs as a vaccine adjuvant for clinical use, because they are likely contaminated with a small amount of an organic solvent, such as dimethyl sulfoxide (DMSO). Solubilization of hydrophobic γ-PGA with an organic solvent is inevitable during the production process of γ-PGA-Phe NPs. To circumvent such contamination in producing γ-PGA-Phe NPs, we have successfully created novel polyion complex (PIC) NPs composed of water-soluble anionic γ-PGA-Phe and a cationic polymer by simple mixing without using any organic solvents (14). The PIC NPs showed high stability in phosphate-buffered saline (PBS) and other buffers with a wide range of pH values. The stability of PIC NPs depended on the γ-PGA-Phe/cationic polymer ratio and hydrophobicity of γ-PGA. The hydrophobic interaction between γ-PGA-Phe and a cationic polymer is important for the stability of NPs as a drug delivery carrier. γ-PGA is a naturally occurring biodegradable and nontoxic polyamide produced from certain strains of Bacillus (15). The cationic polymers poly(ε-lysine) (ε-PL) and protamine are also naturally occurring polymers and have been used as food additives due to their antimicrobial activities (16, 17). In this study, we show that antigen-carrying PIC NPs are capable of inducing potent and antigen-specific cellular and humoral immunity in mice.
MATERIALS AND METHODS
Mice.
Female C57BL/6 (H-2Kb) mice (6 to 8 weeks of age) were purchased from Charles River (Yokohama, Japan). Experiments in mice were approved by Kagoshima University and carried out in accordance with its guideline for animal experimentation.
PIC nanoparticles.
The biodegradable anionic polymer γ-PGA (molecular weight, 3.8 × 105) and cationic polymer ε-PL (molecular weight, 4.7 × 103) were kindly provided by Meiji Seika Co. Ltd. (Tokyo, Japan) and Chisso Corporation (Tokyo, Japan). The cationic protein protamine (molecular weight, 4.3 × 103) was purchased from Sigma (St. Louis, MO). The amphiphilic graft copolymers consisting of γ-PGA-Phe were synthesized as previously described (14, 18). PIC NPs composed of γ-PGA-Phe and ε-PL [PIC (ε-PL) NPs] or protamine [PIC (protamine) NPs] were prepared by a simple mixing method (Fig. 1A). γ-PGA-Phe in PBS (10 mg/ml) was added to the cationic polymer in PBS (2 mg/ml) at the same volume to yield a translucent solution. To prepare ovalbumin (OVA)-carrying PIC NPs (OVA-PIC NPs), OVA or Alexa Fluor 488-labeled OVA (1 mg/ml) mixed with ε-PL or protamine (2 mg/ml) was added into the γ-PGA-Phe solution (10 mg/ml) at the same volume (Fig. 1B). γ-PGA-Phe with a 40% Phe-grafting degree was used to prepare PIC NPs.
FIG 1.

(A) Chemical structures of Phe-grafted γ-PGA (γ-PGA-Phe) and ε-PL. (B) Schematic representation of antigen-carrying PIC NP formation. PIC NPs were formed and stabilized by hydrophobic interaction of γ-PGA-Phe and electrostatic interaction between γ-PGA and cationic polymers.
Characterization of PIC NPs.
The particle size and distribution of PIC NPs (a mixed solution of γ-PGA-Phe and ε-PL or protamine) in PBS were measured without filtering by a dynamic light scattering (DLS) method using a Zetasizer nano ZS (Malvern Instruments, United Kingdom). The surface charge of the PIC NPs was determined by zeta potential measurements using a Zetasizer nano ZS. Alexa Fluor 488-labeled OVA was used to measure the OVA-loading content in OVA-PIC NPs. The mixed solution of Alexa Fluor 488-labeled OVA, ε-PL or protamine, and γ-PGA-Phe was centrifuged at 14,000 × g for 15 min. After centrifugation, the fluorescence intensity of the supernatant was measured by using a fluorescence microplate reader (Fluoroskan Ascent FL; Thermo Fisher Scientific, Inc., USA). The amount of OVA loaded on/into PIC NPs was calculated as the loaded OVA weight (μg) per total NP weight.
Preparation of DCs.
Bone marrow-derived DCs were generated according to a previously described method (19). Briefly, femurs and tibias were removed from mice and cleaned of the surrounding muscle tissues. Both ends of the bones were cut with scissors, and the marrow was flushed with PBS using a syringe with a 26-gauge needle. The suspension was passed through a 50-μm cell strainer. Bone marrow cells (2.5 × 106 cells/10 ml) were suspended in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 100 units/ml penicillin G, 100 μg/ml streptomycin, and 20 ng/ml recombinant mouse granulocyte-macrophage colony-stimulating factor (PeproTech, Rocky Hill, NJ) and placed into a bacteriological petri dish. Fresh culture medium was added to the dish on day 3. On day 7, nonadherent or loosely adherent cells were harvested and used as iDCs. More than 80% of the cells expressed CD11c, as determined by flow cytometry (FACSCalibur; BD Biosciences, San Jose, CA).
Uptake of OVA-PIC nanoparticles by DCs.
iDCs (1 × 106 cells) were incubated with PBS, Alexa Fluor 488-labeled OVA (10 μg/ml), or Alexa Fluor 488-labeled OVA in PIC NPs (10 μg/ml Alexa Fluor 488-OVA,100 μg/ml γ-PGA-Phe, and 20 μg/ml ε-PL) for 1 h at 37°C. After incubation, the cells were washed with PBS. The cell-associated fluorescence was measured by flow cytometry, and data were analyzed using FlowJo (Tree Star, Inc., Ashland, OR).
Analysis of DCs for CD40 expression.
iDCs were incubated in a 48-well plate (1 × 106 cells per well) with PIC NPs (100 μg/ml γ-PGA-Phe and 20 μg/ml ε-PL) for 24 h at 37°C. iDCs treated with PBS or lipopolysaccharide (LPS; 1 μg/ml) (Sigma) were also incubated as the negative and positive controls, respectively. The cells were washed with PBS, stained with a fluorescein isothiocyanate (FITC)-conjugated anti-CD40 monoclonal antibody (MAb) (BD Bioscience) for 30 min at 4°C, and subjected to flow cytometry.
Immunization of mice with OVA-PIC NPs.
C57BL/6 mice were anesthetized by an intraperitoneal injection of sodium pentobarbital and immunized with PBS, OVA alone (100 μg), OVA-PIC NPs (1 mg of γ-PGA-Phe, 200 μg of ε-PL, and 100 μg of OVA), or OVA plus alum (Sigma) (100 μg of OVA and 1 mg of alum) through a subcutaneous route on days 0 and 7. In another experiment, C57BL/6 mice were immunized with PBS, OVA-PIC (ε-PL) NPs (1,000 or 100 μg of γ-PGA-Phe, 200 or 20 μg of ε-PL, and 100 or 10 μg of OVA), or OVA-PIC (protamine) NPs (1,000 or 100 μg of γ-PGA-Phe, 200 or 20 μg of protamine, and 100 or 10 μg of OVA) through a subcutaneous route on days 0 and 7. On day 14 after the final immunization, spleen cells were harvested.
Pentamer staining.
Pentameric H-2Kb complexes folded with the immunodominant CD8+ T-cell peptide OVA257–264 (SIINFEKL) were purchased from Proimmune (Oxford, United Kingdom). Spleen cells were stained with the allophycocyanin-labeled pentamer and an FITC-conjugated anti-CD8 MAb (Proimmune) to detect OVA-specific CD8+ T cells. The cells were washed with PBS containing 0.1% sodium azide and 0.1% bovine serum albumin (BSA) and fixed with PBS containing 2.5% formaldehyde (Wako, Osaka, Japan). The cell-associated fluorescence was determined by flow cytometry.
ELISPOT.
Gamma interferon (IFN-γ)-producing cells were determined by using an enzyme-linked immunospot (ELISPOT) kit for mouse IFN-γ (BD Biosciences). Spleen cells (1 × 106 cells/ml) were stimulated with the OVA257–264 peptide at 10 μg/ml in a 96-well ELISPOT plate. The plate was incubated for 24 h at 37°C, washed, and further incubated with a biotin-conjugated detection antibody for 2 h at room temperature. After washing, the plate was incubated with streptavidin-conjugated horseradish peroxidase and incubated for 1 h. The plate was washed again and incubated with the substrate for 15 min. The colorimetric reaction was terminated by washing with water. After drying the plate, the number of spots was counted microscopically.
Intracellular cytokine staining.
Spleen cells isolated from immunized mice were stimulated with the OVA257–264 peptide at 10 μg/ml for 10 h. The protein transport inhibitor BD GolgiPlug (1:1,000; BD Biosciences) was added to accumulate intracellular cytokines. The cells were washed, incubated for 10 min at 4°C with a purified anti-mouse CD16/32 antibody to block Fc receptors, and stained with a phycoerythrin (PE)-conjugated anti-CD8 MAb (BD Biosciences) for 30 min at 4°C. The cells were permeabilized with Cytofix/Cytoperm Plus (BD Biosciences) and stained with an FITC-conjugated anti-IFN-γ MAb or an FITC-conjugated anti-tumor necrosis factor alpha (TNF-α) MAb (all from BD Biosciences) for 30 min at 4°C. The cells were washed with PBS, and cell-associated fluorescence was determined by flow cytometry.
Measurement of serum antibodies.
Serum samples were examined for their anti-OVA antibody titers by enzyme-linked immunosorbent assay (ELISA). Briefly, a flat-bottomed 96-well plate was coated with OVA suspended in carbonate-bicarbonate buffer (10 μg/ml) and subjected to overnight incubation at 4°C. The plate was washed with PBS containing Tween 20 (T-PBS) and incubated with blocking buffer (1% BSA and 1% skim milk in T-PBS) for 2 h at room temperature. The serum samples, serially diluted with blocking buffer, were added to each well. After incubation for 2 h at room temperature, the plate was incubated with an alkaline phosphatase-conjugated goat anti-mouse IgG antibody (Southern Biotechnology). The plate was developed by adding the substrate to each well and incubated for 10 min. The colorimetric reaction was terminated by adding a stop solution (2 M H2SO4), and the absorbance of each well at 450 nm was measured by a microplate reader. Endpoint titers were determined by the x axis intercept of dilution curves.
Statistical analysis.
The statistical significance of the results was determined by the Student paired t test. P values below 0.05 were considered to be significant.
RESULTS
Preparation of PIC NPs and OVA-PIC NPs.
PIC NPs composed of γ-PGA-Phe and a cationic protein were prepared by a simple mixing method. In this study, two kinds of cationic proteins, ε-PL and protamine, were used to form PIC NPs. The sizes of PIC (ε-PL) NPs and PIC (protamine) NPs were measured by DLS. These NPs showed a monodispersed size distribution with a mean diameter ranging from 200 to 300 nm. The mean diameters (±standard deviations) of the PIC (ε-PL) NPs and PIC (protamine) NPs used in this study were 240 ± 16 nm (polydispersity index [PDI] of 0.21) and 221 ± 19 nm (PDI of 0.15), respectively.
The OVA-PIC NPs obtained were investigated for their protein loading capability. The loading efficiency was found to be in the range of 20 to 25%, and the amounts of OVA loaded per NP weight were almost identical, irrespective of the cationic protein used (ε-PL or protamine). In this study, OVA-PIC NPs with an OVA loading of 20 μg per milligram of NPs were used for these experiments. The sizes of the OVA-PIC (ε-PL) NPs and OVA-PIC (protamine) NPs were 251 ± 35 nm (PDI = 0.29) and 229 ± 23 nm (PDI = 0.19), respectively. There was no significant difference in particle sizes between PIC NPs and OVA-PIC NPs. PIC NPs and OVA-PIC NPs had a highly negative zeta potential (about −25 mV). PIC NPs were obtained by the addition of excess amounts of γ-PGA-Phe to cationic proteins (nonstoichiometric mixing ratios), suggesting that the negative charge of the NP surface is due to the existence of carboxyl groups of γ-PGA. OVA-PIC NPs were stable under physiological conditions, exhibiting no aggregation, precipitation, or dissociation for a long period of time. After 1 week, PIC NPs released less than 5% of the total OVA into PBS at 37°C.
Uptake of antigen-carrying PIC NPs and DC maturation.
When iDCs were incubated with Alexa Fluor 488-labeled OVA or Alexa Fluor 488-labeled OVA-PIC (ε-PL) NPs and analyzed for their antigen uptake by flow cytometry, Alexa Fluor 488-labeled OVA-PIC (ε-PL) NPs were more efficiently taken up by iDCs than Alexa Fluor 488-labeled OVA alone (Fig. 2A), suggesting that PIC NPs function as an effective antigen carrier. To determine whether the uptake of PIC NPs induces the maturation of iDCs, iDCs were incubated with PIC (ε-PL) NPs and analyzed for their expression of CD40 as a maturation marker of DCs. CD40 is an important costimulatory molecule of DCs, which link innate and adaptive immunity (10). As appropriate negative and positive controls, iDCs were also incubated with PBS or LPS, respectively. Stimulation of iDCs with LPS markedly enhanced the expression of CD40 (Fig. 2B). Modest enhancement of CD40 expression was observed in iDCs stimulated with PIC (ε-PL) NPs compared with the CD40 expression of PBS-stimulated iDCs.
FIG 2.

Uptake of PIC (ε-PL) NPs by DCs and their maturation. (A) iDCs were incubated in the absence (PBS) or presence of Alexa Fluor 488-labeled OVA (10 μg/ml) or Alexa Fluor 488-labeled OVA (10 μg/ml) in PIC NPs (OVA-PIC) for 1 h at 37°C. After incubation, cell-associated fluorescence was assessed by flow cytometry. (B) iDCs were incubated in the absence (PBS) or presence of LPS (1 μg/ml) or PIC NPs (100 μg/ml γ-PGA-Phe and 20 μg/ml ε-PL). After incubation for 24 h, the cells were collected, stained with an anti-CD40–FITC MAb, and analyzed by flow cytometry. Representative results of three separate experiments are shown.
Induction of antigen-specific adaptive immunity.
Vaccines against intracellular pathogens are required to induce CD8+ T cells, while those targeting extracellular pathogens need to produce antibodies (2). DCs can present the uptake antigens, along with the MHC class I molecule, to CD8+ T cells through the antigen cross-presentation pathway (20). Therefore, the cellular immune responses in mice after immunization with antigen-carrying PIC (ε-PL) NPs were examined. The mice were immunized with PBS, OVA alone, OVA-carrying PIC (ε-PL) NPs, or OVA plus alum twice at an interval of 7 days. Spleen cells were stained with pentameric H-2Kb complexes folded with the OVA257–264 epitope peptide. OVA-specific CD8+ T cells were not observed in the spleen cells obtained from the mice immunized with PBS (control) or OVA alone (Fig. 3). OVA-carrying PIC (ε-PL) NPs could induce more OVA-specific CD8+ T cells than OVA plus alum could.
FIG 3.
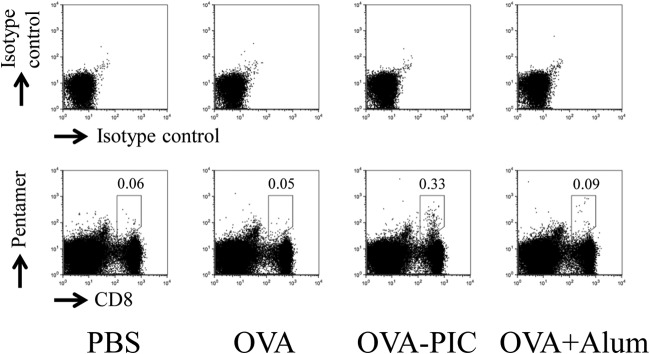
Antigen-specific CD8+ T cells induced by OVA-PIC (ε-PL) NPs. Mice (n = 3) were subcutaneously immunized twice with PBS, OVA (100 μg), OVA-PIC NPs (OVA-PIC) (1 mg of γ-PGA-Phe, 200 μg ε-PL, and 100 μg of OVA), or OVA plus alum (100 μg of OVA and 1 mg of alum). Spleen cells were isolated on day 14 after the final immunization. The cells were stained with an anti-CD8 MAb and the H-2Kb/OVA257–264 pentamer. The number in each panel indicates the percentage of CD8+ pentamer+ T cells. A representative result for one mouse in each group is shown.
In the next experiment, the functional ability of antigen-specific T cells following stimulation with the OVA257–264 epitope peptide was determined by IFN-γ ELISPOT. IFN-γ-producing cells were identified following in vitro stimulation with the OVA257–264 epitope peptide. As shown by the results in Fig. 4A, OVA-specific IFN-γ-producing cells were not observed in the spleen when mice were immunized with PBS or OVA alone. In contrast, a number of IFN-γ-producing cells were observed in mice immunized with OVA-carrying PIC (ε-PL) NPs. The ability of OVA-PIC (ε-PL) NPs to induce IFN-γ-producing cells was significantly higher than that observed for OVA alone or OVA plus alum. To confirm that the cytokine-producing cells were CD8+ T cells, intracellular cytokine staining of the cells was conducted. The spleen cells obtained from the mice immunized with OVA-carrying PIC (ε-PL) NPs contained more IFN-γ- and TNF-α-producing CD8+ T cells than those obtained from the mice immunized with OVA plus alum (Fig. 4B). Cytokine-producing CD8+ T cells were not observed in the spleen cells obtained from the mice immunized with PBS or OVA.
FIG 4.
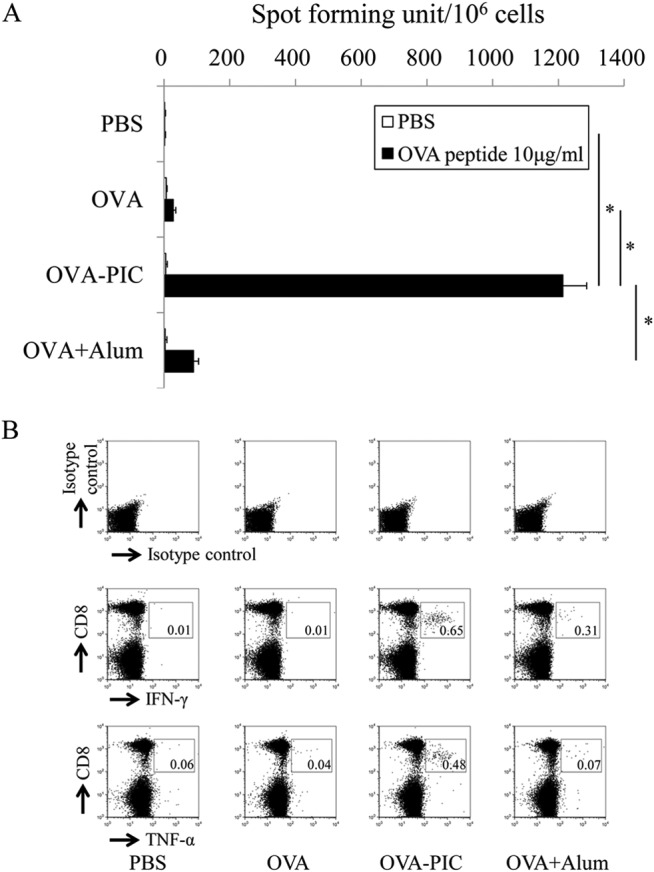
Functional analysis of antigen-specific cytokine-producing T cells induced by OVA-PIC (ε-PL) NPs. Mice (n = 3) were subcutaneously immunized twice with PBS, OVA, OVA-PIC NPs (OVA-PIC), or OVA plus alum. Spleen cells were isolated on day 14 after the final immunization. (A) Spleen cells were stimulated with no peptide (PBS) or the OVA257–264 CTL epitope peptide (10 μg/ml) and evaluated for their IFN-γ production by ELISPOT. Data are expressed as the number of IFN-γ-positive spots per million cells and represent the mean result ± standard deviation (SD) for each group. Statistical analysis was carried out among the 4 groups stimulated with the OVA257–264 epitope peptide (10 μg/ml) (*, P < 0.05). (B) Spleen cells were stimulated with the OVA257–264 peptide and examined for their production of IFN-γ and TNF-α by intracellular cytokine staining. The number in each panel indicates the percentage of cytokine-positive CD8+ T cells.
OVA-specific serum antibody levels were measured by ELISA. Mice immunized with OVA, OVA-carrying PIC (ε-PL) NPs, and OVA plus alum showed significantly higher levels of OVA-specific IgG antibody than those immunized with PBS, although the antibody levels did not differ significantly among the three groups (Fig. 5).
FIG 5.
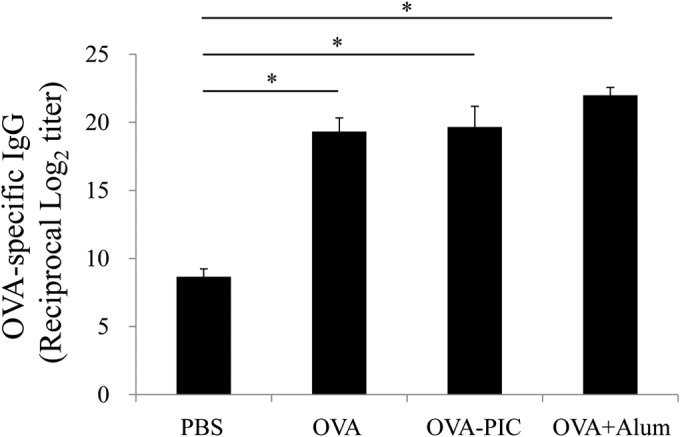
Induction of antigen-specific IgG antibody responses in mice immunized with OVA-PIC (ε-PL) NPs. Mice (n = 3) were subcutaneously immunized twice with PBS, OVA, OVA-PIC NPs (OVA-PIC), or OVA plus alum. Sera were collected on day 14 after the final immunization. The levels of OVA-specific IgG antibody were measured by ELISA. Data represent the means ± SD of the endpoint titers for groups of three mice. Statistical analysis was carried out among the results from 4 groups (*, P < 0.05).
Improvement of antigen-specific adaptive immunity by different PIC formulations.
Biodegradable γ-PGA-Phe-based materials have great potential as vaccine adjuvants with sufficient safety profiles (11–13, 21). PIC NPs are prepared as amphiphilic-graft copolymers of γ-PGA-Phe and a cationic protein in PBS. Therefore, different biodegradable cationic polymers were evaluated for the induction of antigen-specific immune responses in mice. ε-PL and protamine were selected for the cationic component of PIC NPs. ε-PL is characterized as a cationic, water-soluble, biodegradable, and nontoxic polymer (16). Protamine is also a cationic and naturally occurring substance, derived from fish (17).
When the CD8+ T-cell response to the H-2Kb-restricted OVA257–264 epitope was examined by the pentamer-staining assay, immunization with protamine-based OVA-PIC NPs was found to induce a much higher level of the antigen-specific CD8+ T-cell response than immunization with the same amount of ε-PL-based OVA-PIC NPs (Fig. 6). The functional ability of antigen-specific CD8+ T cells was assessed by IFN-γ ELISPOT after stimulation with the epitope peptide. The number of antigen-specific IFN-γ-producing cells was much greater in the mice immunized with OVA-PIC (protamine) NPs than in those immunized with OVA-PIC (ε-PL) NPs (Fig. 7A). Similar to the ELISPOT results, immunization with protamine-based OVA-PIC NPs could induce the cytokine-producing CD8+ T cells more potently than immunization with ε-PL-based OVA-PIC NPs (Fig. 7B). In addition, the mice immunized with protamine- and ε-PL-based OVA-PIC NPs showed equally high levels of induction of antigen-specific IgG antibody (Fig. 8).
FIG 6.
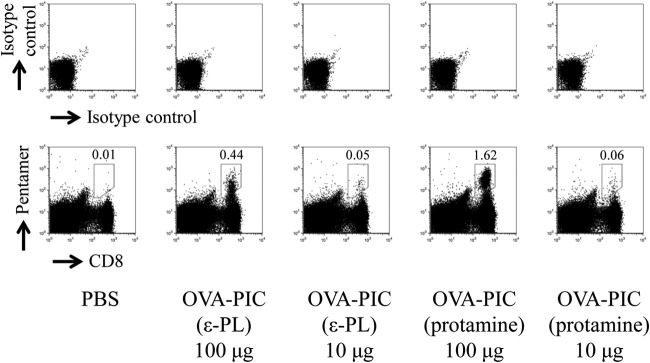
Effects of different cationic proteins on PIC NPs for induction of antigen-specific CD8+ T cells. Mice (n = 3) were subcutaneously immunized twice with ε-PL-based OVA-PIC NPs [OVA-PIC (ε-PL) 100 μg (1 mg of γ-PGA-Phe, 200 μg of ε-PL, and 100 μg of OVA) or OVA-PIC (ε-PL) 10 μg (100 μg of γ-PGA-Phe, 20 μg of ε-PL, and 10 μg of OVA)] or protamine-based OVA-PIC NPs [OVA-PIC (protamine) 100 μg (1 mg of γ-PGA-Phe, 200 μg of protamine, and 100 μg of OVA) or OVA-PIC (protamine) 10 μg (100 μg of γ-PGA-Phe, 20 μg of protamine, and 10 μg of OVA)]. Spleen cells were isolated on day 14 after the final immunization. The cells were stained with an anti-CD8 MAb and the H-2Kb/OVA257–264 pentamer. The number in each panel indicates the percentage of CD8+ pentamer+ T cells. A representative result for one mouse in each group is shown.
FIG 7.
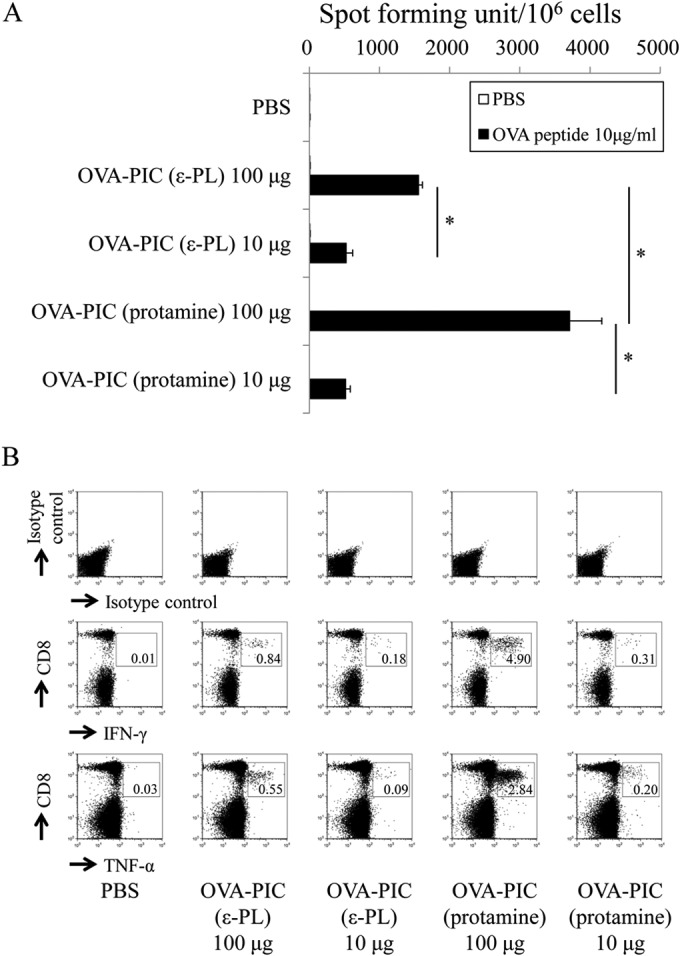
Functional analysis of antigen-specific cytokine-producing T cells induced by OVA-PIC NPs containing different cationic proteins. Mice (n = 3) were subcutaneously immunized twice with PBS, OVA-PIC (ε-PL) 100 μg (1 mg of γ-PGA-Phe, 200 μg of ε-PL, and 100 μg of OVA), OVA-PIC (ε-PL) 10 μg (100 μg of γ-PGA-Phe, 20 μg of ε-PL, and 10 μg of OVA), OVA-PIC (protamine) 100 μg (1 mg of γ-PGA-Phe, 200 μg of protamine, and 100 μg of OVA), or OVA-PIC (protamine) 10 μg (100 μg of γ-PGA-Phe, 20 μg of protamine, and 10 μg of OVA). Spleen cells were isolated on day 14 after the final immunization. (A) Spleen lymphocytes were stimulated with no peptide (PBS) or the OVA257–264 CTL epitope peptide (10 μg/ml) and evaluated for their IFN-γ production by ELISPOT. Data are expressed as the numbers of IFN-γ-positive spots per million cells. Data represent the mean result ± SD for each group. Statistical analysis was carried out among the 4 groups stimulated with the OVA257–264 epitope peptide (10 μg/ml) (*, P < 0.05). (B) Spleen lymphocytes were stimulated with the OVA257–264 peptide and examined for their production of IFN-γ and TNF-α by intracellular cytokine staining. The number in each panel indicates the percentage of cytokine-positive CD8+ T cells.
FIG 8.
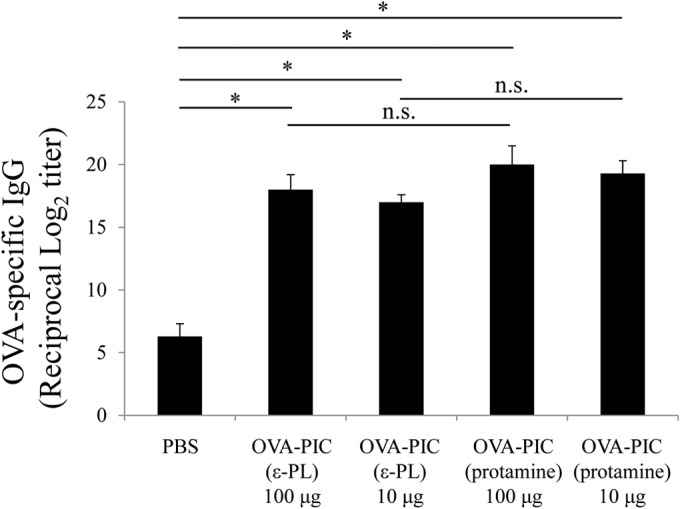
Induction of antigen-specific IgG antibody in mice immunized with OVA-PIC NPs containing different cationic proteins. Mice (n = 3) were subcutaneously immunized twice with PBS, OVA-PIC (ε-PL) 100 μg (1 mg of γ-PGA-Phe, 200 μg of ε-PL, and 100 μg of OVA), OVA-PIC (ε-PL) 10 μg (100 μg of γ-PGA-Phe, 20 μg of ε-PL, and 10 μg of OVA), OVA-PIC (protamine) 100 μg (1 mg of γ-PGA-Phe, 200 μg of protamine, and 100 μg of OVA), or OVA-PIC (protamine) 10 μg (100 μg of γ-PGA-Phe, 20 μg of protamine, and 10 μg of OVA). Sera were collected on day 14 after the final immunization. The levels of OVA-specific IgG antibody were measured by ELISA. Data represent the means ± SD of the endpoint titers for groups of three mice. Statistical analysis was carried out among the results for the 5 groups (*, P < 0.05). n.s., not statistically significant.
DISCUSSION
Antigen uptake by APCs and their subsequent maturation are key steps in initiating and regulating adaptive immunity (1, 2). The interaction between antigens and iDCs appears to lead to the maturation of DCs and enhance their antigen-presenting capacity, costimulatory molecule expression, and cytokine production (9, 22). The immunogenicity of particulate materials seems to be negligible, since they are inert in stimulating innate immunity (4). In some cases, such as for poly(I·C), monophosphoryl lipid A (MPLA), and imidazoquinolines, particulate materials themselves have immunostimulatory activity to improve the immunogenicity of vaccines (23–27). However, it is sometimes difficult to control the formulation of these materials. PIC NPs can be prepared by simple mixing methods, and PIC NP-treated DCs induce DC maturation.
The innate immune system can sense microbes through pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), that are expressed in various cells, including DCs (1, 28, 29). In addition to TLRs, other types of PRRs, including the C-type lectin-like receptors and cytosolic Nod-like receptors, also sense a broad range of microbial stimuli, and the cytosolic RIG-I-like receptors interact with viral nucleic acids (30–32). Several studies have recently demonstrated that hydrophobicity of particulate polymeric materials and antigens plays an important role in stimulating the innate immune system (33–35). In addition, the hydrophobicity of such materials influences endosomal membrane disruption (36). PIC NPs are formed from γ-PGA-Phe copolymers and a cationic protein through their hydrophobic interactions, suggesting that they also stimulate the innate immune system. In our previous studies, it was demonstrated that γ-PGA-Phe NPs possess adjuvant activity and can stimulate DCs via TLR4 on the cell surface (19). Thus, it is expected that PIC NPs also exert their adjuvant activity by the same mechanism as γ-PGA-Phe NPs.
DCs reside in the periphery, where they are situated to recognize and capture antigens. Upon receiving an activating stimulus, DCs migrate to lymphoid organs, where the processed peptides derived from captured antigens are presented to CD4+ T cells in the context of MHC class II molecules (8). Furthermore, DCs directly present the captured antigens, along with the MHC class I molecule, to CD8+ T cells through the antigen cross-presenting pathway (20). It was reported that escape of antigens from the endosomes into the cytoplasm is crucial for initiating the cross-presenting pathway (37). Since OVA-PIC NPs strongly induced OVA-specific cellular immune responses, it is assumed that the combinations of PIC NPs with other antigens can effectively induce antigen-specific CD8+ T cell responses. Furthermore, strong antigen-specific antibody responses were also observed in mice after immunization with OVA-PIC NPs, suggesting that PIC NPs are capable of inducing both cell-mediated and humoral immune responses in vivo.
Protamine-based OVA-PIC NPs induced OVA-specific cellular immune responses more strongly than did ε-PL-based OVA-PIC NPs (Fig. 6 and 7). Although PIC NPs are capable of inducing both cell-mediated and humoral immune responses irrespective of the cationic polymer used for NP formation, the induction of immune responses appears to depend on the kind of cationic polymers. The lower capacity of OVA-PIC (ε-PL) NPs may be attributed to the lower stability of ε-PL in NPs compared with that of protamine. In fact, PIC (ε-PL) NPs did not maintain a stable particle form in PBS containing BSA for a long period of time compared to the length of time for which PIC (protamine) NPs were stable (data not shown). Thus, protamine seems to be more suited to being a component of PIC NPs.
The degradation profiles of PIC NPs composed of γ-PGA-Phe and ε-PL or protamine remain to be clarified. In a previous study, we studied hydrolytic and enzymatic degradation of NPs composed of γ-PGA-Phe (38). The γ-PGA-Phe NPs were degraded with different degradation patterns by various enzymes, such as pronase E, cathepsin B, lipase, and γ-GTP. The enzymatic degradation of the γ-PGA-Phe NPs occurred via the hydrolysis of γ-PGA as the main chain and Phe as the side chain. The size of the NPs increased during the initial degradation period and decreased gradually when the degradation time was extended, and finally, the NPs disappeared completely. These results suggest that the hydrophobic interactions of Phe attached to γ-PGA were significantly reduced with increasing time. Further degradation probably occurred upon the dissociation of γ-PGA-Phe from NPs. Since PIC NPs are also composed of γ-PGA-Phe, it is assumed that PIC NPs are degraded by a mechanism similar to that of γ-PGA-Phe NPs. Further studies are in progress to determine the biodegradation of PIC NPs in vitro and in vivo.
In conclusion, the novel antigen-carrying PIC NPs, which can be easily prepared by mixing anionic γ-PGA-Phe polymer, cationic polymer, and antigen in PBS, strongly induce DC activation in vitro and elicit significant antigen-specific adaptive immune responses in vivo. Therefore, PIC NPs may have great potential as a novel vaccine adjuvant and should be further pursued for their clinical applications.
ACKNOWLEDGMENTS
This work was supported by Core Research for Evolutional Science and Technology from the Japan Science and Technology Agency (JST).
All authors are inventors of PIC NPs and have submitted their patent application.
REFERENCES
- 1.Pulendran B, Ahmed R. 2011. Immunological mechanisms of vaccination. Nat Immunol 12:509–517. doi: 10.1038/ni.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reed SG, Orr MT, Fox CB. 2013. Key roles of adjuvants in modern vaccines. Nat Med 19:1597–1608. doi: 10.1038/nm.3409. [DOI] [PubMed] [Google Scholar]
- 3.Reed SG, Bertholet S, Coler RN, Friede M. 2009. New horizons in adjuvants for vaccine development. Trends Immunol 30:23–32. doi: 10.1016/j.it.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Sahdev P, Ochyl LJ, Moon JJ. 2014. Biomaterials for nanoparticle vaccine delivery systems. Pharm Res 31:2563–2582. doi: 10.1007/s11095-014-1419-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Gregorio E, Caproni E, Ulmer JB. 2013. Vaccine adjuvants: mode of action. Front Immunol 31:214. doi: 10.3389/fimmu.2013.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenthal JA, Chen L, Baker JL, Putnam D, DeLisa MP. 2014. Pathogen-like particles: biomimetic vaccine carriers engineered at the nanoscale. Curr Opin Biotechnol 7:51–58. doi: 10.1016/j.copbio.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 7.Smith DM, Simon JK, Baker JR Jr. 2013. Applications of nanotechnology for immunology. Nat Rev Immunol 13:592–605. doi: 10.1038/nri3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohn L, Delamarre L. 2014. Dendritic cell-targeted vaccines. Front Immunol 30:255. doi: 10.3389/fimmu.2014.00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steinman RM. 2008. Dendritic cells in vivo: a key target for a new vaccine science. Immunity 29:319–324. doi: 10.1016/j.immuni.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 10.Steinman RM. 2012. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol 30:1–22. doi: 10.1146/annurev-immunol-100311-102839. [DOI] [PubMed] [Google Scholar]
- 11.Uto T, Wang X, Sato K, Haraguchi M, Akagi T, Akashi M, Baba M. 2007. Targeting of antigen to dendritic cells with poly(γ-glutamic acid) nanoparticles induces antigen-specific humoral and cellular immunity. J Immunol 178:2979–2986. doi: 10.4049/jimmunol.178.5.2979. [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Uto T, Akagi T, Akashi M, Baba M. 2007. Induction of potent CD8+ T-cell responses by novel biodegradable nanoparticles carrying human immunodeficiency virus type 1 gp120. J Virol 81:10009–10016. doi: 10.1128/JVI.00489-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akagi T, Wang X, Uto T, Baba M, Akashi M. 2007. Protein direct delivery to dendritic cells using nanoparticles based on amphiphilic poly(amino acid) derivatives. Biomaterials 28:3427–3436. doi: 10.1016/j.biomaterials.2007.04.023. [DOI] [PubMed] [Google Scholar]
- 14.Akagi T, Watanabe K, Kim H, Akashi M. 2010. Stabilization of polyion complex nanoparticles composed of poly(amino acid) using hydrophobic interactions. Langmuir 26:2406–2413. doi: 10.1021/la902868g. [DOI] [PubMed] [Google Scholar]
- 15.Shih IL, Van YT. 2001. The production of poly-(gamma-glutamic acid) from microorganisms and its various applications. Bioresour Technol 79:207–225. doi: 10.1016/S0960-8524(01)00074-8. [DOI] [PubMed] [Google Scholar]
- 16.Saimura M, Takehara M, Mizukami S, Kataoka K, Hirohara H. 2008. Biosynthesis of nearly monodispersed poly(epsilon-L-lysine) in Streptomyces species. Biotechnol Lett 30:377–385. doi: 10.1007/s10529-007-9563-7. [DOI] [PubMed] [Google Scholar]
- 17.Sorgi FL, Bhattacharya S, Huang L. 1997. Protamine sulfate enhances lipid-mediated gene transfer. Gene Ther 4:961–968. doi: 10.1038/sj.gt.3300484. [DOI] [PubMed] [Google Scholar]
- 18.Akagi T, Kaneko T, Kida T, Akashi M. 2005. Preparation and characterization of biodegradable nanoparticles based on poly(gamma-glutamic acid) with l-phenylalanine as a protein carrier. J Control Release 108:226–236. doi: 10.1016/j.jconrel.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 19.Uto T, Akagi T, Yoshinaga K, Toyama M, Akashi M, Baba M. 2011. The induction of innate and adaptive immunity by biodegradable poly(γ-glutamic acid) nanoparticles via a TLR4 and MyD88 signaling pathway. Biomaterials 32:5206–5212. doi: 10.1016/j.biomaterials.2011.03.052. [DOI] [PubMed] [Google Scholar]
- 20.Guermonprez P, Saveanu L, Kleijmeer M, Davoust J, Van Endert P, Amigorena S. 2003. ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature 425:397–402. doi: 10.1038/nature01911. [DOI] [PubMed] [Google Scholar]
- 21.Uto T, Wang X, Akagi T, Zenkyu R, Akashi M, Baba M. 2009. Improvement of adaptive immunity by antigen-carrying biodegradable nanoparticles. Biochem Biophys Res Commun 379:600–604. doi: 10.1016/j.bbrc.2008.12.122. [DOI] [PubMed] [Google Scholar]
- 22.Sato K, Fujita S. 2007. Dendritic cells: nature and classification. Allergol Int 56:183–191. doi: 10.2332/allergolint.R-06-139. [DOI] [PubMed] [Google Scholar]
- 23.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 24.Trumpfheller C, Caskey M, Nchinda G, Longhi MP, Mizenina O, Huang Y, Schlesinger SJ, Colonna M, Steinmam RM. 2008. The microbial mimic poly IC induces durable and protective CD4+ T cell immunity together with a dendritic cell targeted vaccine. Proc Natl Acad Sci U S A 105:2574–2579. doi: 10.1073/pnas.0711976105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guy B. 2007. The perfect mix: recent progress in adjuvant research. Nat Rev Microbiol 5:505–517. doi: 10.1038/nrmicro1681. [DOI] [PubMed] [Google Scholar]
- 26.Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC. 2007. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science 316:1628–1632. doi: 10.1126/science.1138963. [DOI] [PubMed] [Google Scholar]
- 27.Vasilakos JP, Tomai MA. 2013. The use of Toll-like receptor 7/8 agonists as vaccine adjuvants. Expert Rev Vaccines 12:809–819. doi: 10.1586/14760584.2013.811208. [DOI] [PubMed] [Google Scholar]
- 28.Iwasaki A, Medzhitov R. 2010. Regulation of adaptive immunity by the innate immune system. Science 327:291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 30.Geijtenbeek TB, Gringhuis SI. 2009. Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol 9:465–479. doi: 10.1038/nri2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ting JP, Duncan JA, Lei Y. 2010. How the noninflammasome NLRs function in the innate immune system. Science 327:286–290. doi: 10.1126/science.1184004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilkins C, Gale M Jr. 2010. Recognition of viruses by cytoplasmic sensors. Curr Opin Immunol 22:41–47. doi: 10.1016/j.coi.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shima F, Akagi T, Uto T, Akashi M. 2013. Manipulating the antigen-specific immune response by the hydrophobicity of amphiphilic poly(γ-glutamic acid) nanoparticles. Biomaterials 34:9709–9716. doi: 10.1016/j.biomaterials.2013.08.064. [DOI] [PubMed] [Google Scholar]
- 34.Kreuter J, Liehl E, Berg U, Soliva M, Speiser PP. 1988. Influence of hydrophobicity on the adjuvant effect of particulate polymeric adjuvants. Vaccine 6:253–256. doi: 10.1016/0264-410X(88)90220-4. [DOI] [PubMed] [Google Scholar]
- 35.Seong SY, Matzinger P. 2004. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol 4:469–478. doi: 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]
- 36.Murthy N, Robichaud JR, Tirrell DA, Stayton PS, Hoffman AS. 1999. The design and synthesis of polymers for eukaryotic membrane disruption. J Control Release 61:137–143. doi: 10.1016/S0168-3659(99)00114-5. [DOI] [PubMed] [Google Scholar]
- 37.Shen H, Ackerman AL, Cody V, Giodini A, Hinson ER, Cresswell P, Edelson RL, Saltzman WM, Hanlon DJ. 2006. Enhanced and prolonged cross-presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles. Immunology 117:78–88. doi: 10.1111/j.1365-2567.2005.02268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Akagi T, Higashi M, Kaneko T, Kida T, Akashi M. 2006. Hydrolytic and enzymatic degradation of nanoparticles based on amphiphilic poly(gamma-glutamic acid)-graft-L-phenylalanine copolymers. Biomacromolecules 7:297–303. doi: 10.1021/bm050657i. [DOI] [PubMed] [Google Scholar]