

Abstract
The anthrax protective antigen (PA) is the central component of the three-part anthrax toxin, and it is the primary immunogenic component in the approved AVA anthrax vaccine and the “next-generation” recombinant PA (rPA) anthrax vaccines. Animal models have indicated that PA-specific antibodies (AB) are sufficient to protect against infection with Bacillus anthracis. In this study, we investigated the PA domain specificity, affinity, mechanisms of neutralization, and synergistic effects of PA-specific antibodies from a single donor following vaccination with the rPA vaccine. Antibody-secreting cells were isolated 7 days after the donor received a boost vaccination, and 34 fully human monoclonal antibodies (hMAb) were identified. Clones 8H6, 4A3, and 22F1 were able to neutralize lethal toxin (LeTx) both in vitro and in vivo. Clone 8H6 neutralized LeTx by preventing furin cleavage of PA in a dose-dependent manner. Clone 4A3 enhanced degradation of nicked PA, thereby interfering with PA oligomerization. The mechanism of 22F1 is still unclear. A fourth clone, 2A6, that was protective only in vitro was found to be neutralizing in vivo in combination with a toxin-enhancing antibody, 8A7, which binds to domain 3 of PA and PA oligomers. These results provide novel insights into the antibody response elicited by the rPA vaccine and may be useful for PA-based vaccine and immunotherapeutic cocktail design.
INTRODUCTION
Bacillus anthracis, the causative agent of anthrax, has long been considered a serious public health threat, particularly after the bioterrorism attacks that occurred after 11 September 2001. B. anthracis secretes a tripartite toxin that consists of protective antigen (PA; 83 kDa), lethal factor (LF; 85 kDa), and edema factor (EF; 89 kDa). LF is a Zn2+-dependent metalloprotease (1) that combines with PA to form lethal toxin (LeTx), which causes death when injected into animals intravenously. EF is a Ca2+-dependent adenylyl cyclase enzyme (2) that combines with PA to form edema toxin (EdTx), which causes edema at the site of inoculation. PA by itself is not known to have pathogenic effects but is considered the central component of the anthrax toxin. It primarily functions as a vehicle mediating the cellular uptake of EF and LF. PA binds to cell surface receptors (CMG-2/TEM-8) on target host cells (3, 4). Following binding, PA is cleaved by furin-like protease produced by the target cells to PA20, a 20-kDa peptide that is released into the surroundings. The remaining fragment, PA63 (also known as nicked/activated PA63), remains bound to the receptor and forms a heptamer or octamer that can translocate as many as three molecules of EF or LF from the cell surface into the cytosol via endocytosis, leading to the biological effects of EF and LF (5).
Vaccination against PA is sufficient to elicit immune protection. PA is the primary immunogenic component in the anthrax vaccine adsorbed (AVA) formulation that is currently licensed in the United States for human use. Although effective, AVA requires a much longer time (6 months) to produce protective immunity than that required for any effective postexposure anthrax prophylaxis, and the exact antigenic composition of the vaccine remains unknown and varies between batches (6, 7). The only immunogenic component in the “next-generation” anthrax vaccine is recombinant PA (rPA). The rPA vaccine can potentially elicit a faster protective immune response to be effective in postexposure cases and be produced in more homogenous standard formulations (7). Clinical trials have demonstrated the safety and immunogenicity of the rPA vaccine (8, 9). Vaccination with rPA is known to elicit a polyclonal antibody (Ab) response. Most of the studies that have been conducted characterize the antibody responses induced by the AVA vaccine in humans (10). To date, there have not been very many studies to characterize the human antibody responses to the rPA vaccine.
The conventional treatment following potential exposure to aerosolized B. anthracis spores was antibiotics combined with PA-based anthrax vaccine. While antibiotics are effective in killing bacteria, they are unable to clear toxin components from the bloodstream (11). Passive immunization with monoclonal antibodies (MAbs) against toxin components has been shown to be highly protective in postexposure cases, particularly when combined with antibiotics (12, 13). Characterizing the protective rPA vaccine-induced antibody response in humans may identify naturally occurring neutralizing antibodies that could be used therapeutically.
Most of the monoclonal antibodies neutralizing PA-mediated toxicity have been produced in murine sources (14, 15), although some protective monoclonal antibodies against PA have been developed in chimpanzees, transgenic mice, and humans (16–18). The first Food and Drug Administration-approved monoclonal antibody against PA is fully human (19). Identification and characterization of novel protective anti-PA antibodies could contribute to the development of additional therapeutics for treating anthrax in patients.
In this study, we assessed the neutralizing potential of a large panel of monoclonal antibodies generated from an individual donor immunized with the rPA vaccine. We further characterized the domain specificities, affinities, mechanisms of neutralization, and potential synergistic effects of the protective human monoclonal antibodies (hMAbs). Overall, our results provide novel insights into the donor's antibody responses to the rPA vaccine and information about the design of PA-based vaccines and immunotherapeutic cocktails of hMAbs.
MATERIALS AND METHODS
Recombinant anthrax toxins.
Recombinant protective antigen (rPA) and lethal factor (rLF) were expressed in Escherichia coli and purified as described previously (20, 21). The four domains of PA—domain 1 (residues 1 to 258), domain 2 (residues 258 to 487), domain 3 (residues 488 to 595), and domain 4 (residues 596 to 735)—were expressed as previously described (22, 23). To obtain PA63, PA83 was treated with trypsin at a final trypsin/PA weight ratio of 1:1,000 for 30 min at room temperature. The reaction was stopped by adding a 10-fold excess of soybean trypsin inhibitor (Sigma), and PA63 was purified using a HiTrap Q HP column (GE Healthcare Life Sciences).
Human subjects.
After informed consent was obtained, a male volunteer was immunized with rPA vaccine (an investigational anthrax vaccine developed by Beijing Institute of Biotechnology, which is composed of purified rPA absorbed by aluminum hydroxide adjuvant) and received his second rPA vaccination 28 days later. Peripheral blood (35 ml) was drawn 7 days after the second (boost) vaccination (day 35). Peripheral blood mononuclear cells (PBMCs) were isolated from the blood by a standard Ficoll-Hypaque (Dakewe Biotech) density gradient method (24). The presence of PA-specific antibody-secreting cells (ASCs) was confirmed by linked immunosorbent spot assay (ELISpot) as described previously (25). Briefly, filter plates with hydrophilic mixed cellulose ester (MCE) membrane (Millipore) were coated with 10 μg/ml of PA or anti-human IgG antibody (BD Pharmingen). After washing and blocking, 1 × 106 cells from PBMC suspensions were added to each well. Anti-human IgG conjugated to horseradish peroxidase (HRP) antibody (Sigma) was then added after appropriate washing. The individual ASCs were visualized by addition of the 3-amino-9-ethylcarbazole substrate solution (Dakewe Biotech), and the spots were counted with a BioReader 4000 (Bio-Sys GmbH). The presence of toxin-neutralizing antibodies in the blood serum was confirmed by a cell viability assay (26).
Fluorescent cell sorting and single-cell PCR.
PBMCs were stained with anti-CD3-fluorescein isothiocyanate (FITC), anti-CD20-FITC, anti-CD19-phycoerythrin (PE)-Alexa 610, and anti-CD27-PE (Molecular Probes) and anti-CD38-PE-Cy5 (Beckman Coulter). ASCs were defined as CD19+ CD3− CD20− CD27high CD38high. Single ASCs were sorted using a MoFlo XDP cell sorter (Beckman Coulter) into 96-well plates. PCR amplification of paired VH and VL genes was performed as described previously (24), with the following modifications. Briefly, single cells were sorted into 96-well plates containing 20 μl of RNase-free water (Qiagen) and 0.25 μl of RNasin (Promega). Double-stranded cDNA was produced using the OneStep reverse transcription-PCR (RT-PCR) kit (Qiagen). The nested PCR was carried out using TransStart Taq DNA polymerase (TransGen Biotech). The PCR products of positive clones were purified and sequenced by Sangon Biotech. Sequences were analyzed using IMGT/V-QUEST (http://www.imgt.org/IMGT_vquest/share/textes/).
Expression of recombinant hMAbs from linear expression cassettes.
The cytomegalovirus (CMV) promoter, Ig leader sequence, VH/VL gene, Ig constant egion (IgG1), and poly(A) sequence fragments were assembled by overlapping PCR into full-length Ig heavy- and light-chain linear expression cassettes as previously described (27). The purified PCR products of paired full-length Ig H and L genes were cotransfected into a HEK293T (human embryonic kidney cell line; ATCC CRL-11268) cell expression system using TurboFect transfection reagent (Thermo Scientific). The culture supernatants were collected by aspiration and stored at −20°C until further use.
Enzyme-linked immunosorbent assay (ELISA).
Nunc plates (Thermo Scientific) were coated overnight with 2 μg/ml of PA, PA domain 1, PA domain 2, PA domain 3, or PA domain 4. After a washing with phosphate-buffered saline (PBS) plus 0.1% Tween 20, serial dilutions of culture supernatants of HEK293T cells starting at the desired dilution were added to each well and incubated for 1 h at 37°C. HRP-conjugated anti-human IgG antibody (Sigma) and then 3,3′,5,5′-tetramethylbenzidine (TMB) substrate (Merck KGaA) were added after appropriate washing. The reaction was stopped with 2 M H2SO4. The optical density (OD) was detected at 450 nm.
BLI.
The kinetics of binding of the hMAbs to PA was studied using a ForteBio Octet instrument (Pall Life Science) based on biolayer interferometry (BLI) measurements by following the manufacturer's instructions. Briefly, anti-hIgG Fc capture Biosensors (Pall Life Science) were loaded with hMAbs at 20 μg/ml diluted in HEPES buffer (GE Healthcare Life Sciences). After capture, a 1-min wash in loading buffer removed excess unbound hMAbs to establish a new baseline signal. The sensors were dipped into wells containing PA at several diluted concentrations of from 300 to 50 nM in loading buffer. The sensors were then dipped into wells containing loading buffer to measure the (off-rate) dissociation rate. Equilibrium dissociation constant (KD) values were calculated as the ratio of off-rate values to on-rate values. Data analysis was performed using ForteBio Data Analysis software package 7.0.
Cell viability assay.
J774A.1 cells (murine macrophage cell line; ATCC TIB-67) were seeded in 96-well plates (Costar) at a density of 3 × 104/well and cultured overnight at 37°C. Twofold serial dilutions of hMAbs were preincubated with LeTx (0.1 μg/ml of PA and 0.1 μg/ml of LF) for 1 h at 37°C. The cell culture medium was replaced with the hMAb-toxin mixture and incubated with the cells for 1.5 h or 4 h at 37°C. 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT; Invitrogen) was added to each well to a final concentration of 1 mg/ml, and the cells were incubated at 37°C for an additional 4 h. The hMAb-toxin mixture was replaced with the extraction buffer (0.5% [wt/vol] SDS, 25 mM HCl in 90% isopropyl alcohol). The OD of each well was measured at 570 nm, and the percent viability was calculated as (ODmeasured − ODdead control)/(ODlive control − ODdead control). The live and dead cell controls were treated with media only or a 10-fold excess of LeTx, respectively. The experiments were performed in duplicate; at least three independent experiments were performed.
Animal experiments.
Male Fischer 344 rats (Vital River Laboratories) weighing 160 to 180 g were injected intravenously with a mixture of antibody and LeTx (10 μg of PA and 10 μg of LF per rat) at the desired molar ratios in 200 μl of PBS, or with LeTx only. Animals were observed for signs of malaise and mortality over 48 h. All animal experiments were performed according to protocols approved by the Animal Care and Use Committee of the Beijing Institute of Biotechnology.
Inhibition of furin cleavage.
PA83 (36 pmol, 3 μg) was incubated with 0.25 μl of furin (New England BioLabs) in the absence or presence of antibody (33 pmol, 5 μg) for 1 h at 37°C. The reaction was performed in 20 mM Tris-HCl (pH 8.5) to preserve monomeric PA63 after furin cleavage. Samples were resolved on a 7.5% nonreducing SDS-PAGE gel (Bio-Rad Laboratories) and visualized using Coomassie brilliant blue (Amresco).
Inhibition of oligomerization.
PA63 (48 pmol, 3 μg) was incubated with antibody (33 pmol, 5 μg) for 30 min at room temperature. Oligomerization was induced using 50 mM morpholineethanesulfonic acid (MES) buffer (pH 5.5). Samples were resolved on a 4% to 20% nonreducing SDS-PAGE gel (Bio-Rad Laboratories) and visualized using Coomassie brilliant blue (Amresco). Subsequently, Western blot assay was performed with a rabbit anti-PA polyclonal antibodies (previously prepared in our laboratory) and a goat anti-rabbit HRP-conjugated IgG antibody (Santa Cruz Biotechnology).
Statistical analysis.
Data from the in vitro assays were analyzed using the Student t test. Animal survival curves were generated and analyzed using the log rank test. All statistical analyses were performed using GraphPad Prism 5. A P value of <0.05 was considered statistically significant.
RESULTS
Human antibody response to immunization with the rPA vaccine.
A male donor was immunized twice with the next-generation rPA vaccine. A blood sample was collected 7 days after the second (boost) vaccination (day 35). The frequencies of total IgG+ and PA-specific IgG+ ASCs were determined as a percentage of total PBMCs using a standard ELISpot (25). The number of total IgG+ spots increased from 421 per million at day 28 to 647.5 per million at day 35 (Fig. 1A). Similarly, the number of PA-specific spots increased from 3.5 per million at day 28 to 154 spots per million at day 35 (Fig. 1A). The percentage of PA-specific IgG+ ASCs as a fraction of total IgG+ ASCs increased from 0.8% to 23.8% 7 days after the boost vaccination. The protective immune response after vaccination was confirmed using the MTT assay as previously described (22). Prior to vaccination, the serum isolated from the vaccinated patient did not mitigate LeTx-induced cell death. However, at day 28 the 50% inhibitory concentration (IC50) in the serum against LeTx was 87.4 and increased at day 35 to 514.1 (Fig. 1B). Taken together, the results indicated that PA-specific and neutralizing polyclonal Ab responses were effectively induced by the boost rPA vaccination.
FIG 1.
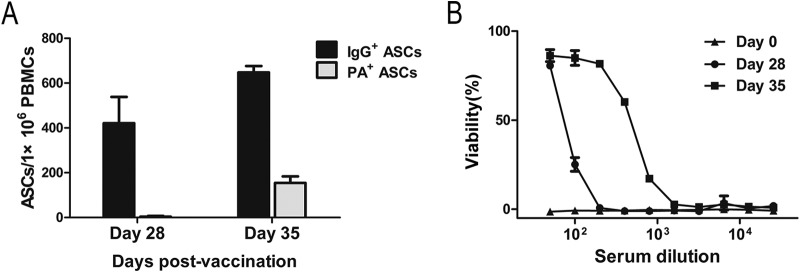
Characterization of the serum antibody response to recombinant anthrax protective antigen (rPA) vaccination. (A) ELISpot was used to determine the number of PA-specific and total IgG+ ASCs per million PBMCs at day 28 and day 35 postvaccination. Error bars indicate the standard deviations of duplicates. (B) The ability of the donor serum, collected at day 0, day 28, and day 35, to protect J774A.1 cells from LeTx-mediated cell death was assessed using the MTT assay. Data are means ± SDs from at least three independent experiments, each conducted in duplicate.
Rapid generation and characterization of PA-specific hMAbs following rPA vaccination.
Seven days after the donor received the boost vaccination, ASCs defined as CD19+ CD3− CD20− CD27high CD38high were sorted from total PBMCs as single cells using fluorescent cell sorting. A total of 529 pairs of VH and VL genes were amplified from single ASCs. To rapidly express the MAb genes, linear expression cassettes (27) were produced by PCR. Full-length Ig H and L genes were cotransfected into a HEK293T cell expression system. IgG concentrations of approximately 1 μg/ml were obtained from the transfected culture supernatants. The recovery was sufficient for screening clones in ELISAs and cell viability assays. ELISA was used to screen 529 hMAbs produced from single ASCs for reactivity to PA. Of the hMAbs screened, 34 clones (6.4%) were PA specific (Table 1). Domain specificity of the PA-specific hMAbs was determined by ELISA. Of the 34 PA-specific clones, 14 hMAbs did not clearly bind to any of the PA domains, and only one hMAb bound to domain 2. Surprisingly, 12 of the 34 hMAbs specifically bound to domain 4 of PA (Table 1).
TABLE 1.
Characteristics of the 34 PA-specific human monoclonal antibodies generated in this study
| Functiona | Clone | VHb | DHc | JHd | HCDR3e | VLf | JLg | Specificityh |
|---|---|---|---|---|---|---|---|---|
| Protective | 2A6 | HV4-31 | HD2-15 | HJ4 | ATGDGGVAGTFDF | KV3-15 | KJ4 | Whole PA |
| 4A3 | HV4-39 | HD3-9 | HJ6 | ASLPLVYDSLTGHYFQYYGLNV | LV1-40 | LJ2 | Whole PA | |
| 8H6 | HV4-39 | HD6-13 | HJ4 | ASGYSTSFDY | KV1-12 | KJ2 | Whole PA | |
| 22F1 | HV4-31 | HD4-17 | HJ5 | ARASLRGIL | KV3-11 | KJ4 | PA63 | |
| Enhancing | 1H2 | HV3-30 | HD1-26 | HJ4 | AKDGREALLAS | LV3-25 | LJ2 | D1 |
| 1H5 | HV3-23 | HD4-23 | HJ5 | AKGGGNSGWFDT | KV4-1 | KJ1 | D4 | |
| 1D10 | HV3-74 | HD3-10 | HJ4 | AGDPGYYGSGSRAGFDS | KV3-11 | KJ5 | Whole PA | |
| 2A2 | HV4-61 | HD3-3 | HJ6 | ATGKYYDFWTGPKFGMDV | KV1-9 | KJ4 | D3 | |
| 3C4 | HV1-3 | HD2-8 | HJ4 | ARQGGPSRTGGYYFDY | KV4-1 | KJ4 | D4 | |
| 8A7 | HV3-23 | HD3-10 | HJ1 | AKFSGHYGSGSFHLPEYFQH | KV1-5 | KJ1 | D3 | |
| 9F11 | HV3-48 | HD5-18 | HJ4 | ARDRDTNIWYPDGFDS | KV1-5 | KJ1 | D3 | |
| 11G6 | HV3-23 | HD4-23 | HJ5 | ARGGGNSGWFDP | KV4-1 | KJ1 | D4 | |
| 11B10 | HV3-23 | HD4-23 | HJ5 | AKGGGNSGWFDS | KV4-1 | KJ1 | D4 | |
| 11E12 | HV3-23 | HD3-10 | HJ6 | AKVRFGDDGMDV | KV1-5 | KJ2 | D1 | |
| 12B8 | HV3-23 | HD4-23 | HJ5 | AKGGGNSGWFDS | KV4-1 | KJ1 | D4 | |
| 12C9 | HV4-39 | HD4-17 | HJ4 | ARMLGYGDYHCDY | KV1-12 | KJ4 | Whole PA | |
| 14A4 | HV3-23 | HD4-23 | HJ5 | AKGGGNSGWFDP | KV4-1 | KJ1 | D4 | |
| 15F8 | HV5-10-1 | HD3-3 | HJ4 | TRHSDFWSGYHDY | KV1-5 | KJ1 | Whole PA | |
| 15H10 | HV3-7 | HD1-26 | HJ4 | ARGATWTDF | KV3-15 | KJ1 | Whole PA | |
| 18C11 | HV3-30 | HD5-18 | HJ2 | AKEFGHDHWYFDF | KV3-15 | KJ4 | Whole PA | |
| 19D2 | HV3-23 | HD4-23 | HJ5 | ARGGGNSGWFDS | KV4-1 | KJ1 | D4 | |
| 19H4 | HV3-23 | HD6-19 | HJ2 | AKRQGAALHKWHFDR | KV4-1 | KJ1 | Whole PA | |
| 19F8 | HV4-59 | HD5-12 | HJ4 | ARGGDGDYLQF | KV1-NL1 | KJ2 | Whole PA | |
| Indifferent | 2G6 | HV3-23 | HD3-10 | HJ3 | AKDVSMIREFDAFDI | LV4-69 | LJ3 | D4 |
| 3D6 | HV5-51 | HD3-3 | HJ4 | ARTNVGVPGFDF | LV6-57 | LJ3 | Whole PA | |
| 3C8 | HV3-53 | HD5-18 | HJ6 | ASLVDTTMGYYGVDV | KV1-33 | KJ2 | Whole PA | |
| 3A11 | HV3-30 | HD6-6 | HJ4 | AKLYTSSTSLDF | LV3-25 | LJ2 | D4 | |
| 7D5 | HV3-23 | HD4-23 | HJ5 | AKGGGNSGWFDP | KV4-1 | KJ2 | D4 | |
| 8D4 | HV3-30 | HD5-18 | HJ6 | AKDLRERYTYGYWVSLYYDGMDV | LV1-44 | LJ3 | Whole PA | |
| 11D6 | HV3-30 | HD6-19 | HJ4 | ARISLASSSGWFDY | KV3-11 | KJ4 | Whole PA | |
| 11F11 | HV3-23 | HD4-23 | HJ5 | AKGGGNSGWFDS | KV4-1 | KJ1 | D4 | |
| 12H3 | HV3-23 | HD3-10 | HJ3 | AKDVSMIREFDAFDI | LV4-69 | LJ3 | D4 | |
| 15H8 | HV1-2 | HD5-18 | HJ6 | ASHLRMEV | KV1-27 | KJ1 | D3 | |
| 16D2 | HV3-30 | HD3-10 | HJ4 | AKDQLYFGSQSPGHY | KV4-1 | KJ4 | D2 |
Function categorized based on the cell viability assays.
H-chain V-region gene usage.
H-chain D-region gene usage.
H-chain J-region gene usage.
Amino acid sequence of the heavy-chain third complementarity-determining region.
L-chain V-region gene usage.
L-chain J-region gene usage.
PA domain specificity of the hMAbs based on subunit ELISA.
To determine the neutralizing capabilities of the PA-specific hMAbs, a cell viability assay was performed using J774A.1 cells. The 34 hMAbs were categorized as protective (11.8%), toxin enhancing (55.9%), or toxin indifferent (32.3%) depending on their ability to prevent cell death after a 1.5-h incubation with the cells (Fig. 2A; Table 1). The viabilities of cells treated with LeTx only were used as references to determine hMAbs to be protective, indifferent, or enhancing (28). We selected five of the clones for in-depth characterization (Table 2).
FIG 2.
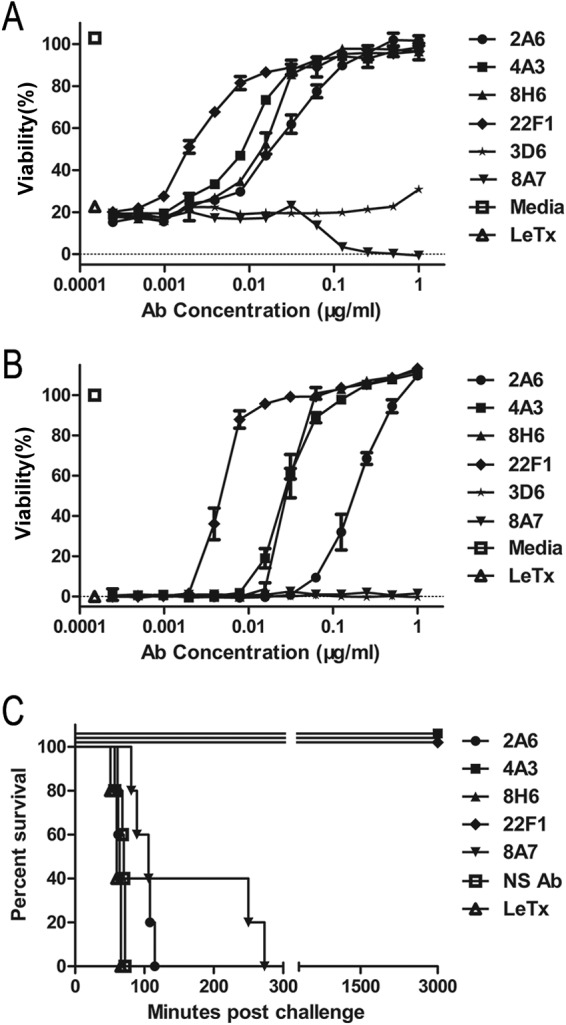
Protective efficacy of PA-specific human monoclonal antibodies (hMAbs) in vitro and in vivo. Serial 2-fold dilutions of the indicated hMAbs mixed with LeTx were incubated with J774A.1 cells for 1.5 h (A) or 4 h (B). The viability of the J774A.1 cells was determined using the MTT assay. Data are means ± SDs from at least three independent experiments, each conducted in duplicate. (C) Fischer 344 rats (n = 5/group) were challenged with LeTx (10 μg of PA and 10 μg of LF per rat) to determine whether the hMAbs provided in vivo protection (the molar ratio of antibodies to PA was 1:1). NS Ab, nonspecific antibody.
TABLE 2.
Human monoclonal antibody clones characterized in detail
| Clone | KD (nM)a | In vitro IC50 (nM)b | In vivo survival rate (%)c | Domain specificityd | Functione |
|---|---|---|---|---|---|
| 2A6 | 3.05 ± 1.36 | 1.33 | 0 | Whole PA | Unknown |
| 4A3 | 179 ± 54.4 | 0.19 | 100 | Whole PA | Interference with oligomerization |
| 8H6 | 0.636 ± 0.838 | 0.2 | 100 | Whole PA | Inhibition of furin cleavage |
| 22F1 | Very low affinity | 0.03 | 100 | PA63 | Unknown |
| 8A7 | 0.869 ± 2.48 | Domain 3 | Binding to oligomers |
Determined by the Fortebio Octet system.
Determined using cell viability assays.
Determined using animal experiments.
Based on subunit ELISA.
Based on furin cleavage and oligomerization assays.
Among the protective hMAbs in a 4-h incubation, 22F1 (IC50 = 0.0047 μg/ml, 0.03 nM) conferred the highest level of protection and 4A3 (IC50 = 0.029 μg/ml, 0.19 nM) and 8H6 (IC50 = 0.03 μg/ml, 0.2 nM) showed similar neutralization capacities, while the remaining hMAb, 2A6 (IC50 = 0.2 μg/ml, 1.33 nM), required a much higher concentration to achieve measurable protection (Fig. 2B). Clone 8A7 is a representative toxin-enhancing antibody that augmented LeTx toxicity in vitro (Fig. 2A). Fischer 344 rats (n = 5) were challenged with LeTx to evaluate the in vivo neutralization potential of the hMAbs, as previously described (29). Consistent with the in vitro results, 22F1 showed the highest level of protection and resulted in 100% survival even when mixed with PA at the molar ratio of 1:3 (data not shown). However, 2A6 was not able to offer any protection in vivo. Similarly, 8A7 provided no in vivo protection (Fig. 2C).
The binding affinities of all of the hMAbs were determined using the Fortebio Octet system. Interestingly, 22F1, which had the highest neutralizing capacity both in vitro and in vivo, had an extremely low binding affinity for PA83 but bound to PA63 with moderate affinity (KD = 4.21 nM). The four protective antibodies did not clearly bind to a specific domain of PA.
Protective hMAb 8H6 inhibited furin cleavage of PA.
In vivo, after PA83 binds to its cell surface receptors, PA20 is proteolytically removed by furin or furin-like proteases. In vitro, PA63 and PA20 remain associated with each other after furin cleavage, but the two fragments can be dissociated by SDS-PAGE. To determine whether any of the hMAbs inhibited the furin cleavage of PA, the antibodies were preincubated with PA83 and then furin was added. Furin cleavage was inhibited by 8H6 but none of the other hMAbs (Fig. 3A). 8H6 inhibited furin-mediated PA83 cleavage in a dose-dependent manner; 16.5 pmol (2.5 μg) of antibody completely inhibited cleavage of 36 pmol (3 μg) of PA83 (Fig. 3B). 8H6 provided an excellent level of protection both in vitro and in vivo and had a very high binding affinity with PA83, yet it did not show clear domain specificity.
FIG 3.
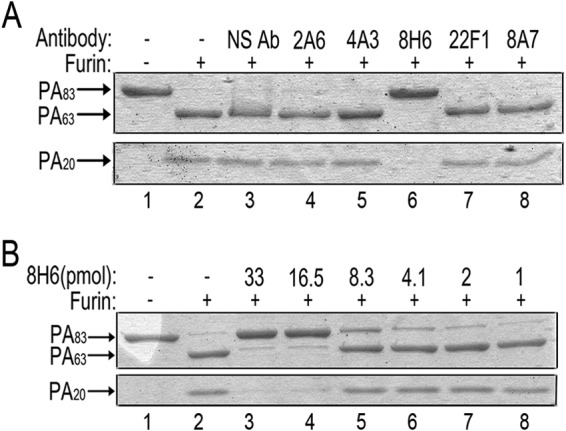
Clone 8H6 neutralized PA by inhibiting furin cleavage. (A) PA was incubated with furin in the presence or absence of each hMAb. Furin cleavage was determined by the absence of the full-length PA83 band and the presence of the PA63 and PA20 bands. NS Ab, nonspecific antibody. (B) 8H6 inhibited furin cleavage in a dose-dependent manner. In vitro proteolysis of 36 pmol of PA83 was completely inhibited by 16.5 pmol of antibody. Results shown were obtained by nonreducing SDS-PAGE.
Protective hMAb 4A3 enhanced the degradation of nicked PA.
Following the furin-mediated cleavage of PA83, the remaining PA63 fragment oligomerizes, facilitating the binding of LF/EF. In vitro, PA63 remains associated with PA20 after cleavage, preventing the oligomerization of PA63. However, the electrostatic interactions between PA63 and PA20 can be disrupted by anion-exchange chromatography, and low pH (pH 5.5 to 6.0) can favor the oligomerization of PA63. To determine whether any of the hMAbs inhibited PA oligomerization, each antibody was preincubated with purified PA63 before the pH was reduced to 5.5. Then the samples were resolved by nonreducing SDS-PAGE. No oligomers were observed in the presence of 4A3, indicating that 4A3 inhibited oligomerization. Interestingly, the presence of 4A3 also resulted in the appearance of two distinct bands (approximately 17 kDa and 30 kDa), several faint bands between 63 kDa and 15 kDa, and a weaker PA63 band (Fig. 4A, lanes 7 and 8). To confirm that these bands were PA fragments, the membranes were probed with rabbit anti-PA polyclonal antibodies (Fig. 4B). Taken together, the results suggested that 4A3 enhanced the degradation of PA63, thereby possibly interfering with PA oligomerization and providing protection in vitro and in vivo. While 4A3 bound to PA83 with low affinity, it bound to PA63 with high affinity (KD = 0.198 nM), suggesting that 4A3 specially interacted with PA63 after PA20 was cleaved off.
FIG 4.
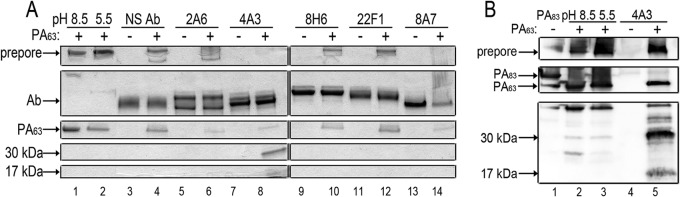
Clone 4A3 inhibited PA63 oligomerization and clone 8A7 bound PA63 oligomers. (A) PA63 was allowed to oligomerize in the presence (+) or absence (−) of each hMAb. Samples where antibody was present were preincubated to allow binding. Samples were resolved by nonreducing SDS-PAGE gel. NS Ab, nonspecific antibody. (B) Clone 4A3 enhanced the degradation of PA63 and thereby interfered with PA oligomerization. Samples were subjected to nonreducing SDS-PAGE and Western blotting using rabbit anti-PA polyclonal antibodies.
Toxin-enhancing hMAb 8A7 bound PA63 oligomers and synergized with hMAb 2A6 to provide better protection.
We also assessed the effect of clone 8A7 (toxin enhancing) on the formation of PA63 oligomers by nonreducing SDS-PAGE. 8A7 was preincubated with purified PA63 before the pH was reduced to 5.5 to induce oligomerization. In the presence of 8A7, the bands indicating antibody and PA63 oligomers became much fainter, while a high-molecular-mass band larger than the oligomers appeared (data not shown). These results suggested that 8A7 bound the oligomers and formed a high-molecular-mass supercomplex that could not be resolved by SDS-PAGE (Fig. 4A, lanes 13 and 14). Although binding capacity of 8A7 toward oligomers and its high affinity for PA domain 3 do not favor its individual neutralization efficacy against LeTx, these bindings contribute to the synergistic interactions of 8A7 and 2A6 (Fig. 5).
FIG 5.
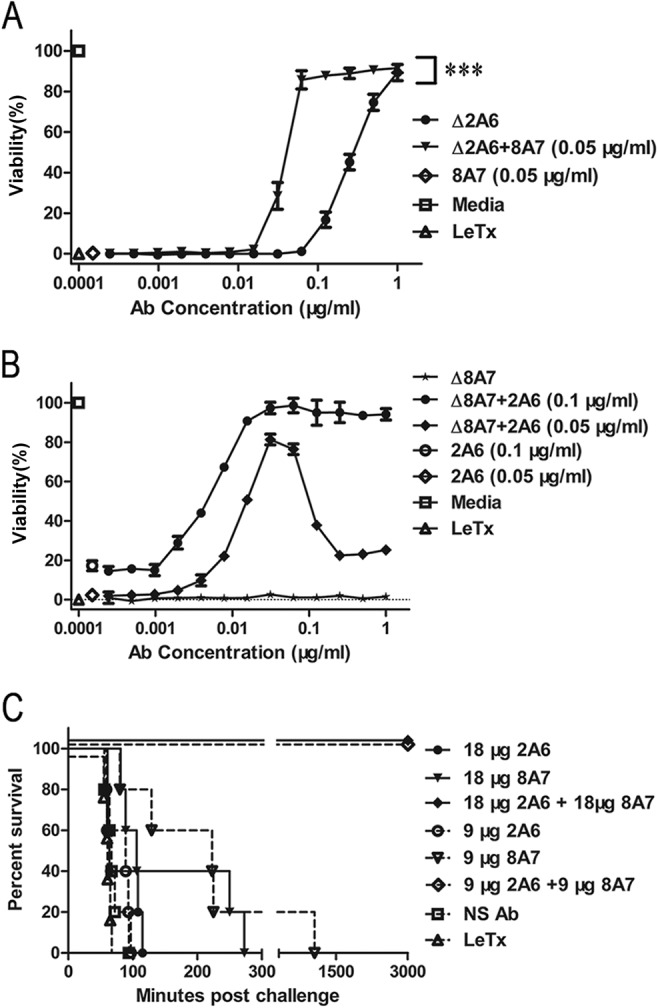
The combination of 2A6 and 8A7 enhanced protection against LeTx. (A) The viability of J774A.1 cells was measured using the MTT assay in the presence of 2A6 alone or in combination with 8A7. The concentration of 8A7 was held constant at 0.05 μg/ml, and 2A6 was titrated from 1 μg/ml. ***, P < 0.05 for absence of 8A7 versus corresponding presence of 8A7, by t test. (B) The viability of J774A.1 cells was measured using the MTT assay in the presence of 2A6 alone or in combination with 8A7. The concentration of 2A6 was held constant at 0.1 or 0.05 μg/ml, and 8A7 was titrated from 1 μg/ml. Data shown are means ± SDs from at least three independent experiments, each conducted in duplicate. (C) Fischer 344 rats (n = 5/group) were challenged with LeTx (10 μg of PA and 10 μg of LF per rat) and simultaneously injected with 2A6 alone, 8A7 alone, or the combination (the molar ratio of antibodies to PA was 1:1 or 0.5:1). Survival time is shown. NS Ab, nonspecific antibody.
We combined the five clones characterized in detail to determine whether there were synergistic interactions. Most of the antibody combinations did not show significant changes in cell neutralization levels (data not shown), with an exception. While neither 2A6 (protective in vitro) nor 8A7 (toxin-enhancing) provided in vivo protection, the combination resulted in robust toxin neutralization in vitro (Fig. 5A). To determine the optimal ratio of 2A6 and 8A7 that protected against cell death, an MTT assay was performed titrating both 2A6 and 8A7 (Fig. 5B). An excessive concentration (higher than 0.1 μg/ml) of 8A7 decreased the synergistic interactions between 2A6 and 8A7. Similarly, the augmented protection was also dependent on the concentration of 2A6. The synergistic protections given by 2A6 and 8A7 peaked at optimal molar ratio of 1:1 (Fig. 5B). The synergistic effects of 2A6 and 8A7 were then tested in vivo (Fig. 5C). The combination of 2A6 and 8A7 at the optimal molar ratio achieved 100% survival when the molar ratio of antibody to PA was 1:1 or 0.5:1.
DISCUSSION
The antibody responses induced by the AVA vaccine in humans are well understood; however, there has been very little systematic research characterizing the human antibody responses to the next-generation anthrax vaccine that is based solely on PA. To our knowledge, we are the first to characterize the domain specificities, affinities, mechanisms of neutralization, and synergistic effects of hMAbs from an individual donor immunized with the rPA vaccine. We were able to identify the specificities, function, and Ig composition of the endogenous antibody repertoire by using single-cell PCR to preserve the natural Ig VH and VL pairing from individual B cells. Despite the fact that we have characterized the response of only a single subject, the specificities and functions of the PA response in humans may be more important than the quantity of the anti-PA (10).
Of the clones we isolated from the donor, 6.4% were PA reactive, and approximately one quarter of the IgG+ ASCs was PA reactive 7 days after the donor received a boost vaccination. The total number of PA-specific clones obtained after boost vaccination was less than expected. This is likely due to low endogenous expression levels for some PA-specific clones. An alternative explanation is that the most prevalent subclasses IgG1 were used to produce linear expression cassettes, but immunoglobulin isotype has been shown to influence the V region structure and antibody affinity and/or specificity, so if there was a mismatch, we may have reduced PA-specific binding (30). However, the majority of the circulating monoclonal antibodies were not PA reactive, despite the blood sample being collected at the peak of the immune response after the boost rPA vaccination. In addition, while most of the PA-reactive hMAbs did not clearly bind to any of the recombinant PA domains that we expressed, of the antibodies that did bind, domain 4 was the most frequent target.
Based on the biological activity of PA in anthrax infection, antibody-based neutralization could occur at several different steps, including (i) receptor binding, (ii) enzymatic cleavage to PA63, (iii) PA63 oligomerization, (iv) LF or EF binding to PA63 oligomers, and (v) disruption of preformed PA oligomers through formation of a supercomplex (11). The four neutralizing antibodies obtained in this study (2A6, 4A3, 8H6, and 22F1) were characterized in detail to determine if they function through any of the mechanisms described above. Inhibition of receptor binding of both PA83 to the target cell and LF/EF to PA63 has been proposed as the primary mechanism of action for neutralizing antibodies. Vaccines based solely on PA domain 4 (cell receptor binding domain) have been reported to effectively protect mice from LeTx challenge (31, 32). However, despite the fact that domain 4 was the most frequently targeted domain among the hMAbs where domain specificity could be established, none of the neutralizing hMAbs from this donor bound domain 4 or blocked receptor binding. In fact, we could not establish clear domain specificity for any of the four neutralizing antibodies. One possibility is that these antibodies recognize conformational epitopes in PA and therefore have low affinity for individual subunits. Clone 22F1 provided the highest level of protection both in vitro and in vivo, but we were not able to establish its mechanism of action. Interestingly, 22F1 has extremely low binding affinity for PA83 but binds to PA63 with moderate affinity. Antibodies 8H6 and 4A3 were found to be strongly neutralizing both in vitro and in vivo. 8H6 inhibited furin-mediated PA cleavage with very high affinity. 4A3 acted in a previously unreported manner to interfere with PA oligomerization by promoting the degradation of PA63. Despite having low affinity for PA83, 4A3 bound to PA63 with a high affinity, indicating that 4A3 specifically interacted with PA63 after PA20 was cleaved from PA83. Finally, 2A6 required a high concentration to neutralize toxins in vitro and was not protective in vivo. No mechanism of action was identified for 2A6.
While 2A6 alone was not protective in vivo, the combination of 2A6 and 8A7, a toxin-enhancing antibody, was strongly neutralizing in vitro and in vivo. Synergism required an optimal mix of 2A6 and 8A7. This prozone-like effect has been reported previously (28, 33). 8A7 bound to PA domain 3, which is involved in oligomerization. Despite the fact 8A7 that bound to the PA63 oligomers, it did not inhibit PA-mediated cellular toxicity. Interestingly, MAbs specific to PA domain 3 derived from animals or humans generally do not provide any protection against LeTx in vivo (10, 14). Together with our findings, these results suggest that while MAbs specific for domain 3 of PA are not neutralizing when used alone, they may provide some benefit to the host in combination with other MAbs.
In summary, our study demonstrated the production of novel hMAbs following vaccination with the next-generation rPA anthrax vaccine. Although the epitope specificity could not be determined for the neutralizing hMAbs, three were able to neutralize PA-meditated toxicity in vitro and in vivo using different mechanisms. The surprising synergistic protection provided by 8A7, a toxin-enhancing antibody, paired with a mildly neutralizing antibody, 2A6, suggests that many potentially neutralizing antibodies (or combinations) may be discarded during initial screening for practical reasons. Our results provide novel insights into the humoral response to the rPA vaccine in an individual donor. Further, they illustrate the relationship between antibody binding specificity and function and suggest that a targeted monoclonal antibody cocktail might be an effective anthrax therapeutic.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (grant no. 81172980), the State Project For Essential Drug Research and Development (grant no. 2013ZX09304103), and the National High Technology Research and Development Program (863 Program) of China (grant no. 2014AA021407).
REFERENCES
- 1.Tonello F, Montecucco C. 2009. The anthrax lethal factor and its MAPK kinase-specific metalloprotease activity. Mol Aspects Med 30:431–438. doi: 10.1016/j.mam.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 2.Leppla SH. 1982. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc Natl Acad Sci U S A 79:3162–3166. doi: 10.1073/pnas.79.10.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. 2001. Identification of the cellular receptor for anthrax toxin. Nature 414:225–229. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- 4.Bradley KA, Young JA. 2003. Anthrax toxin receptor proteins. Biochem Pharmacol 65:309–314. doi: 10.1016/S0006-2952(02)01455-7. [DOI] [PubMed] [Google Scholar]
- 5.Young JA, Collier RJ. 2007. Anthrax toxin: receptor binding, internalization, pore formation, and translocation. Annu Rev Biochem 76:243–265. doi: 10.1146/annurev.biochem.75.103004.142728. [DOI] [PubMed] [Google Scholar]
- 6.Ivins BE, Pitt ML, Fellows PF, Farchaus JW, Benner GE, Waag DM, Little SF, Anderson GW Jr, Gibbs PH, Friedlander AM. 1998. Comparative efficacy of experimental anthrax vaccine candidates against inhalation anthrax in rhesus macaques. Vaccine 16:1141–1148. doi: 10.1016/S0264-410X(98)80112-6. [DOI] [PubMed] [Google Scholar]
- 7.Kaur M, Singh S, Bhatnagar R. 2013. Anthrax vaccines: present status and future prospects. Expert Rev Vaccines 12:955–970. doi: 10.1586/14760584.2013.814860. [DOI] [PubMed] [Google Scholar]
- 8.Brown BK, Cox J, Gillis A, VanCott TC, Marovich M, Milazzo M, Antonille TS, Wieczorek L, McKee KT Jr, Metcalfe K, Mallory RM, Birx D, Polonis VR, Robb ML. 2010. Phase I study of safety and immunogenicity of an Escherichia coli-derived recombinant protective antigen (rPA) vaccine to prevent anthrax in adults. PLoS One 5:e13849. doi: 10.1371/journal.pone.0013849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campbell JD, Clement KH, Wasserman SS, Donegan S, Chrisley L, Kotloff KL. 2007. Safety, reactogenicity and immunogenicity of a recombinant protective antigen anthrax vaccine given to healthy adults. Hum Vaccin 3:205–211. doi: 10.4161/hv.3.5.4459. [DOI] [PubMed] [Google Scholar]
- 10.Smith K, Crowe SR, Garman L, Guthridge CJ, Muther JJ, McKee E, Zheng NY, Farris AD, Guthridge JM, Wilson PC, James JA. 2012. Human monoclonal antibodies generated following vaccination with AVA provide neutralization by blocking furin cleavage but not by preventing oligomerization. Vaccine 30:4276–4283. doi: 10.1016/j.vaccine.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Z, Moayeri M, Purcell R. 2011. Monoclonal antibody therapies against anthrax. Toxins (Basel) 3:1004–1019. doi: 10.3390/toxins3081004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crowe SR, Ash LL, Engler RJ, Ballard JD, Harley JB, Farris AD, James JA. 2010. Select human anthrax protective antigen epitope-specific antibodies provide protection from lethal toxin challenge. J Infect Dis 202:251–260. doi: 10.1086/653495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobiler D, Gozes Y, Rosenberg H, Marcus D, Reuveny S, Altboum Z. 2002. Efficiency of protection of guinea pigs against infection with Bacillus anthracis spores by passive immunization. Infect Immun 70:544–560. doi: 10.1128/IAI.70.2.544-550.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelly-Cirino CD, Mantis NJ. 2009. Neutralizing monoclonal antibodies directed against defined linear epitopes on domain 4 of anthrax protective antigen. Infect Immun 77:4859–4867. doi: 10.1128/IAI.00117-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Little SF, Leppla SH, Cora E. 1988. Production and characterization of monoclonal antibodies to the protective antigen component of Bacillus anthracis toxin. Infect Immun 56:1807–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sawada-Hirai R, Jiang I, Wang F, Sun SM, Nedellec R, Ruther P, Alvarez A, Millis D, Morrow PR, Kang AS. 2004. Human anti-anthrax protective antigen neutralizing monoclonal antibodies derived from donors vaccinated with anthrax vaccine adsorbed. J Immune Based Ther Vaccines 2:5. doi: 10.1186/1476-8518-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Z, Moayeri M, Zhou YH, Leppla S, Emerson S, Sebrell A, Yu F, Svitel J, Schuck P, St Claire M, Purcell R. 2006. Efficient neutralization of anthrax toxin by chimpanzee monoclonal antibodies against protective antigen. J Infect Dis 193:625–633. doi: 10.1086/500148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reason D, Liberato J, Sun J, Keitel W, Zhou J. 2009. Frequency and domain specificity of toxin-neutralizing paratopes in the human antibody response to anthrax vaccine adsorbed. Infect Immun 77:2030–2035. doi: 10.1128/IAI.01254-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kummerfeldt CE. 2014. Raxibacumab: potential role in the treatment of inhalational anthrax. Infect Drug Resist 7:101–109. doi: 10.2147/IDR.S47305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu J, Cai C, Guo Q, Zhang J, Dong D, Li G, Fu L, Xu J, Chen W. 2013. Secretory expression and efficient purification of recombinant anthrax toxin lethal factor with full biological activity in E. coli. Protein Expression Purif 89:56–61. doi: 10.1016/j.pep.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 21.Xu JJ, Dong DY, Song XH, Ge M, Li GL, Fu L, Zhuang HL, Chen W. 2004. Expression, purification and characterization of the recombinant anthrax protective antigen. Sheng Wu Gong Cheng Xue Bao 20:652–655. (In Chinese.) doi: 10.3321/j.issn:1000-3061.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J, Xu J, Li G, Dong D, Song X, Guo Q, Zhao J, Fu L, Chen W. 2006. The 2beta2-2beta3 loop of anthrax protective antigen contains a dominant neutralizing epitope. Biochem Biophys Res Commun 341:1164–1171. doi: 10.1016/j.bbrc.2006.01.080. [DOI] [PubMed] [Google Scholar]
- 23.Petosa C, Collier RJ, Klimpel KR, Leppla SH, Liddington RC. 1997. Crystal structure of the anthrax toxin protective antigen. Nature 385:833–838. doi: 10.1038/385833a0. [DOI] [PubMed] [Google Scholar]
- 24.Smith K, Garman L, Wrammert J, Zheng NY, Capra JD, Ahmed R, Wilson PC. 2009. Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat Protoc 4:372–384. doi: 10.1038/nprot.2009.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crotty S, Aubert RD, Glidewell J, Ahmed R. 2004. Tracking human antigen-specific memory B cells: a sensitive and generalized ELISPOT system. J Immunol Methods 286:111–122. doi: 10.1016/j.jim.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 26.Wu S, Zhang Z, Yu R, Zhang J, Liu Y, Song X, Yi S, Liu J, Chen J, Yin Y, Xu J, Hou L, Chen W. 2014. Intramuscular delivery of adenovirus serotype 5 vector expressing humanized protective antigen induces rapid protection against anthrax that may bypass intranasally originated preexisting adenovirus immunity. Clin Vaccine Immunol 21:156–164. doi: 10.1128/CVI.00560-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liao HX, Levesque MC, Nagel A, Dixon A, Zhang R, Walter E, Parks R, Whitesides J, Marshall DJ, Hwang KK, Yang Y, Chen X, Gao F, Munshaw S, Kepler TB, Denny T, Moody MA, Haynes BF. 2009. High-throughput isolation of immunoglobulin genes from single human B cells and expression as monoclonal antibodies. J Virol Methods 158:171–179. doi: 10.1016/j.jviromet.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chow SK, Smith C, MacCarthy T, Pohl MA, Bergman A, Casadevall A. 2013. Disease-enhancing antibodies improve the efficacy of bacterial toxin-neutralizing antibodies. Cell Host Microbe 13:417–428. doi: 10.1016/j.chom.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karginov VA, Nestorovich EM, Moayeri M, Leppla SH, Bezrukov SM. 2005. Blocking anthrax lethal toxin at the protective antigen channel by using structure-inspired drug design. Proc Natl Acad Sci U S A 102:15075–15080. doi: 10.1073/pnas.0507488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Casadevall A, Janda A. 2012. Immunoglobulin isotype influences affinity and specificity. Proc Natl Acad Sci U S A 109:12272–12273. doi: 10.1073/pnas.1209750109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park YS, Lee JH, Hung CF, Wu TC, Kim TW. 2008. Enhancement of antibody responses to Bacillus anthracis protective antigen domain IV by use of calreticulin as a chimeric molecular adjuvant. Infect Immun 76:1952–1959. doi: 10.1128/IAI.01722-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chichester JA, Musiychuk K, de la Rosa P, Horsey A, Stevenson N, Ugulava N, Rabindran S, Palmer GA, Mett V, Yusibov V. 2007. Immunogenicity of a subunit vaccine against Bacillus anthracis. Vaccine 25:3111–3114. doi: 10.1016/j.vaccine.2007.01.068. [DOI] [PubMed] [Google Scholar]
- 33.Taborda CP, Rivera J, Zaragoza O, Casadevall A. 2003. More is not necessarily better: prozone-like effects in passive immunization with IgG. J Immunol 170:3621–3630. doi: 10.4049/jimmunol.170.7.3621. [DOI] [PubMed] [Google Scholar]