

Abstract
In the absence of oxygen human life is measured in minutes. In the presence of oxygen, normal metabolism generates reactive species (ROS) that have the potential to cause cell injury contributing to human aging and disease. Between these extremes, organisms have developed means for sensing oxygen and ROS and regulating their cellular processes in response. Redox signaling contributes to the control of cell proliferation and death. Aberrant redox signaling underlies many human diseases. The attributes acquired by altered redox homeostasis in cancer cells illustrate this particularly well. This teaching review and the accompanying illustrations provide an introduction to redox biology and signaling aimed at instructors of graduate and medical students.
Keywords: Superoxide, Hydrogen peroxide, Proliferation, Apoptosis, Mitochondria, Cancer
Graphical abstract

Highlights
-
•
The ability to sense oxygen and respond to oxidative stress is ancient.
-
•
Chemical and kinetic properties of ROS are key to understanding redox signaling.
-
•
Redox signaling participates in normal control of cell proliferation and death.
-
•
Aberrant redox signaling contributes to the hallmarks of cancer.
-
•
Novel redox-based chemotherapeutics are being developed.
Oxygen and ROS sensing by the earliest life on earth
A logical assumption is that antioxidant enzymes evolved with, or after the appearance of, aerobic metabolism on earth. The thinking is that these enzymes became necessary only when cells had to protect themselves from increased levels of reactive oxygen species (ROS) that were produced as by-products of respiration. Results from the quest for the earliest form of life on earth challenge this assumption. With modern sequencing technology, rRNAs from multiple species have been compared and used to construct a phylogenetic tree based on molecular rather than morphological similarities between organisms (Fig. 1). The tree has three domains: Bacteria, Archaea and Eucarya [1,2]. At the base of the tree is the last universal common ancestor (LUCA), which is estimated to have appeared 3.5–4 billion years ago. Carl Woese postulates that LUCA was not a single organism but a community of primitive entities with a high frequency of lateral gene transfer [3]. Collectively, this community was genetically and metabolically complex, containing the molecular origins of all present life forms. Over time, the ancestors of three major domains emerged from LUCA.
Fig. 1.
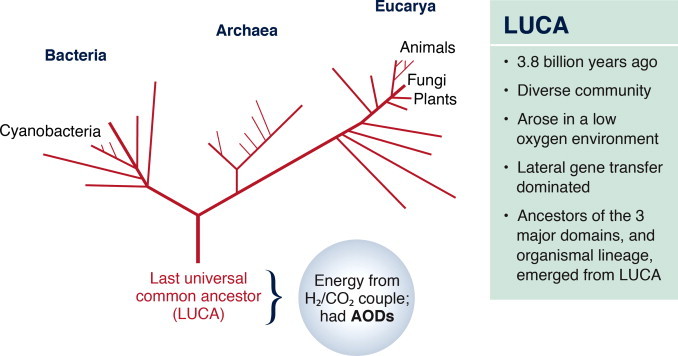
Oxidative stress preceded oxygen metabolism. A phylogenetic tree developed from sequence analysis of ribosomal RNAs [1,2]. The first semblance of life is at the base of the tree and is referred to as the last universal common ancestor, or LUCA. Oxygen levels in the atmosphere did not increase appreciably until the appearance of the cyanobacteria. Yet, LUCA had genetic material encoding for antioxidant defenses.
From Knoll A.H. (1999) Science 285: 1025–1026; http://www.sciencemag.org/content/285/5430/1025. Reprinted with permission from AAAS.
LUCA was present a billion years before the rise of oxygen levels in the atmosphere. Yet, sequence analyses suggest that it was capable of detoxifying ROS [4]. The lack of an ozone layer at the time of LUCA would have resulted in approximately thirty times more ultraviolet radiation reaching the earth’s surface. Radiation splits water into ROS. With a hemoglobin-like molecule to bind the small amount of oxygen present in the atmosphere and catalase and superoxide to detoxify ROS, LUCA was protected from oxidative damage. Importantly, LUCA teaches us that the ability to respond to oxygen, hydrogen peroxide and superoxide is ancient and was present well before the increase in atmospheric oxygen and evolution of aerobic respiration.
Interaction of modern-day organisms with oxygen-derived species
Modern day organisms have a number of regulatory systems that respond to changing levels of oxygen, hydrogen peroxide or superoxide (Fig. 2). The difference in structural complexity between unicellular and multicellular organism is accompanied by a difference in the complexity of these systems [5]. It could be argued that to a certain extent, bacteria respond to ROS primarily to protect themselves from potential damaging effects of these species. As relatively small, single-celled life forms, bacteria benefit from the ability to respond quickly to an increased level of ROS in the environment. Thus, bacteria use transcription factors that are modified by ROS to regulate genes that function to protect against oxidative stress.
Fig. 2.

Oxygen sensing and signaling in LUCA and modern-day organisms. LUCA had a hemoglobin-like protein that could have bound oxygen. Modern-day organisms have a number of regulatory systems that respond to oxygen, hydrogen peroxide or superoxide. Bacteria have redox-sensitive transcription factors that interact directly with superoxide (SoxR/SoxS) or hydrogen peroxide (OxyR, PerR) and regulate gene expression in response. In eukaryote cells, redox regulation frequently involves multiple proteins with one acting as the sensor and subsequently changing the location, activity or expression level location of a regulatory protein. The listed proteins have been shown to participate in different aspects of redox signaling.
In eukaryotic cells, redox regulation frequently involves multiple proteins with one acting as the sensor. The more complex systems seen in eukaryotes reflect evolution of redox signaling pathways, in addition to protection against oxidative stress. Oxidation (or reduction) of sensor proteins leads to changes in the location, activity or expression level location of other key regulatory molecules [5]. An example of this is regulation of protein turnover and cytoplasmic-nuclear trafficking as seen with the IκB kinase/NF-κB or KEAP/NRF2 [6] protein pairs. A multi-component system allows for greater flexibility in light of eukaryotic species having cells with multiple compartments and the higher level organization (i.e., different cell and tissue types) in multicellular eukaryotes.
Redox signaling in Bacteria
Bacteria response to increasing levels of superoxide using two proteins, SoxR and SoxS, that function in tandem to regulate gene expression [7]. SoxR acts as a sensor through a mechanism involving oxidation of an iron–sulfur group in the protein. Oxidation increases SoxR-mediated transcription of the SoxS gene by approximately 30-fold [8]. Genes regulated by SoxS function to remove superoxide and repair oxidative damage. The bacterial response to an increasing level of hydrogen peroxide involves the oxidization of critical cysteine residues in the OxyR and PerR transcription factors. Oxidation alters the proteins’ activation state. These proteins have been most well-characterized in Escherichia coli and Bacillus subtilis, respectively [9–11]. Both regulate genes encoding antioxidant defense proteins (e.g., catalase and peroxiredoxin), but with OxyR functioning as an activator of gene transcription and PerR as a repressor. Oxidation of Cys-199 in OxyR leads to the formation of an intramolecular disulfide bond with Cys-208 and, as a consequence, structural remodeling of the regulatory domain. The transcription factor is thereby activated to recruit RNA polymerase. Redox regulation of PerR involves a metal-coordinated cysteine residue. The metal group (Fe or Mn) is essential for DNA-binding. A structural change in the protein upon oxidation of the critical cysteine by hydrogen peroxide causes release of the metal group followed by release of the PerR protein from sites of gene repression.
Redox signaling in Eucarya
Distinct chemical properties of ROS
A primer on redox biology and signaling in eukaryotes might well begin with a discussion of the chemical properties of the different ROS. These have important ramifications for signal transduction [12–14]. The hydroxyl radical is the most reactive, giving it the shortest half-life in tissues (10−9 s) [15]. It oxidizes virtually any cell component that it encounters and is the primary cause of toxicity due to oxidative damage. Due to this reactivity and lack of specificity, the hydroxyl radical is not thought to have a signaling role in cells. The anionic charge on superoxide limits its diffusion through membranes. Han et al. have provided evidence, however, that superoxide can be transported from mitochondria to the cytoplasm through the voltage-dependent anion channel (VDAC) [16]. Superoxide has a high affinity for iron–sulfur clusters in proteins and reacts with these at rates limited only by diffusion. This can release free iron and promote structural changes to alter protein activity. In solutions of purified proteins, superoxide reacts with cysteine residues to generate a thiyl radical [17]. A subsequent reaction of the thiyl radical with oxygen would regenerate superoxide that then dismutates to form hydrogen peroxide. In vivo, the rapid rate of spontaneous or enzyme-catalyzed dismutation of superoxide to hydrogen peroxide makes it unlikely that oxidation of protein thiols by superoxide is used for redox signaling. Hydrogen peroxide is the most stable ROS, with an estimated half-life in cells of approximately 1 ms. As a nonpolar molecule, it can diffuse through membranes. Membrane transport of hydrogen peroxide is further facilitated by aquaporin channels [18,19]. Thus, chemical considerations point to hydrogen peroxide as the major ROS involved in redox signaling in eukaryotes.
Cellular targets of ROS
Differences between superoxide and hydrogen peroxide are further reinforced through an appreciation of their cellular targets (Fig. 3). As mentioned above for the SoxR/S system in Bacteria, one target of superoxide is iron–sulfur clusters in proteins as is seen with superoxide-mediated inactivation of the mitochondrial enzyme, aconitase [20]. Other iron–sulfur proteins are involved in DNA replication, transcription and repair [21]. Superoxide can react with nitric oxide to produce peroxynitrite, with various sequelae [22]. The primary target of redox signaling by hydrogen peroxide is cysteine residues in proteins, as discussed further below.
Fig. 3.

Targets of superoxide versus hydrogen peroxide in cells. One source of ROS in cells is NADPH oxidases (Nox). These proteins transfer electrons from a NADPH on one side of the membrane to oxygen on the other, generating the superoxide anion radical. The negative change on superoxide precludes diffusion through membranes; voltage dependent anion channels (VDAC) can facilitate transfer. Superoxide reacts readily with iron–sulfur groups in proteins. Its reaction with nitric oxide produces peroxynitrite (ONOO−). Interaction of peroxynitrite with proteins results in tyrosine nitration or S-glutathiolation. Spontaneous or enzyme-mediated dismutation of superoxide generates hydrogen peroxide. Although hydrogen peroxide can diffuse through membranes, aquaporin channels (AQP) increase the rate of transfer. Two electron oxidation of a cysteine residue in proteins by hydrogen peroxide generates a sulfenic acid group. Subsequent reactions lead to the formation of inter- or intra-molecular disulfides, or glutathionylated proteins.
Reversibility and specificity in redox signaling
One rule in signal transduction pathways is that the reactions should be reversible; that is, the signaling components have on and off states. Efforts to elucidate redox signaling pathways have focused on oxidation of the amino acid cysteine, as this can be reversed in contrast to oxidation of many other amino acids [12]. The two electron oxidation of cysteine residues by hydrogen peroxide generates sulfenic acid (Fig. 3). This intermediate can go on to form inter- or intra-molecular disulfides or glutathionylated proteins. Tyrosine phosphatases are well-recognized targets of hydrogen peroxide [23]. These proteins generally turn off kinase signaling pathways and thus, their inactivation can result in constitutive signaling.
A second rule in signal transduction pathways is specificity. Central questions in the redox field with regards to this rule are: (1) what proteins are targeted by ROS to initiate a signaling pathway and (2) how is specificity achieved? It is worth emphasizing one key difference between redox signaling and traditional signal transduction pathways [13]. The latter begin with a ligand (e.g., growth factors, cytokines or steroid hormones) binding to a surface or intracellular receptor. This is a non-covalent interaction of large molecular structures: a lock and key mechanism. In contrast, redox signaling is initiated at the atomic level through a covalent interaction; hydrogen peroxide reacts with the sulfur group of a specific cysteine residue. The size of the cysteine proteome in cells gives a large number of potential receptors, thus raising the question of how specificity can possibly be achieved.
The limitations placed by signaling criteria provide an answer to how specificity is achieved in redox signaling: key interactions are limited to those that are fast, reversible and target a specific cysteine in the target protein [24]. The likelihood of the oxidation of a unique cysteine residue in a particular protein depends on the concentration of this residue/protein in the cell, the amino acids surrounding the cysteine, the location of the protein relative to the source of hydrogen peroxide generation and the rate of reaction with hydrogen peroxide. Proteomic approaches indeed indicate that specificity is achievable, as only a small fraction of proteins become oxidized when cells are subjected to oxidative stress [25–27].
Peroxiredoxins are a family of proteins that exhibit particularly high reaction rate constants with hydrogen peroxide, on the order of 105–107 M−1 s−1 [14]. They are present in cells at a relatively high concentration for proteins of approximately 20 µM. Modeling studies indicate that the unique protein environment around the peroxiredoxin catalytic cysteine residue stabilizes the transition state of the reaction to make these proteins particularly well-designed sensors of hydrogen peroxide [28]. The high reactivity, relative abundance and the distribution of peroxiredoxin family members across different cell compartments fits the expected criteria for proteins that participate in cell signaling pathways.
The location of potential downstream targets relative to the source of hydrogen peroxide generation is a further component of specificity in redox signaling. This is illustrated with the protein tyrosine phosphatase, PTP1B, and redox signaling downstream of ligand binding to tyrosine kinase receptors. PTP1B must compete with peroxiredoxins as a target for hydrogen peroxide. The reaction rate constants of hydrogen peroxide with Prx2 and PTP1B are 2×107 M−1 s−1 [29] and 20 M−1 s−1 [23], respectively. According to the ‘floodgate model’ (Fig. 4), signaling is achieved through local inactivation of peroxiredoxins [30,31]. Ligand binding to tyrosine kinase receptors results in phosphorylation and inactivation of Prx1. Overoxidation of a critical cysteine residue in Prx2 converts the thiol group to sulfinic acid and inactivates this enzyme. As a result, sufficiently high levels of hydrogen peroxide can accumulate to target signaling pathway proteins such as PTP1B.
Fig. 4.
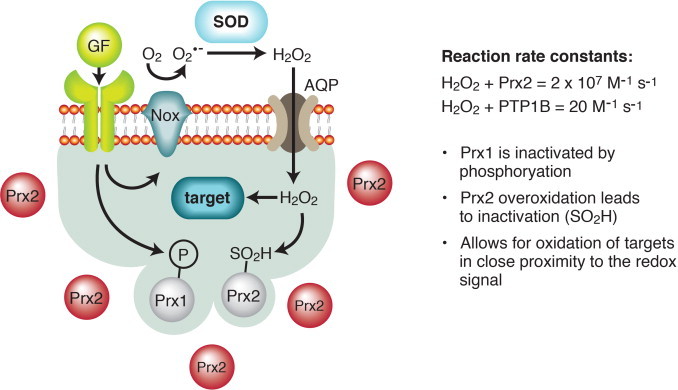
Redox signaling – specificity by location. In what has been called the floodgate model, apparent kinetic limitations of redox signaling are overcome and specificity is achieved through close proximity of a signaling target and the source of hydrogen peroxide generation [30,181]. The floodgate model is shown here with protein tyrosine phosphatase 1B (PTP1B) as the signaling target. Peroxiredoxins (Prx) are present at a relatively high concentration in cells and are very efficient at reducing hydrogen peroxide, as compared to the rate of reaction for PTP1B and hydrogen peroxide. Sufficient hydrogen peroxide can accumulate to oxidize PTP1B, however, after Prx1 is inactivated by phosphorylation downstream of receptor tyrosine kinase activation by growth factors (GF) and a critical cysteine residue in Prx2 is irreversible oxidized to sulfinic acid (SO2H).
Reprinted by permission from Macmillian Publishers Ltd, Dickinson, BC and Chang C.J. (2011) Nature Chem. Biol. 7 (8):504-511; http://www.nature.com/nchembio/journal/v7/n8/full/nchembio.607.html.
ROS function at the start of life
In animals, ROS have important functions for life from the moment of fertilization. Otto Warburg was a notable German chemist/physiologist working on the embryology of sea urchins in the early 1900s [32]. He observed a rapid rise in oxygen consumption upon fertilization of the oocytes and reasoned this was due to increased respiration needed for the ensuing embryogenesis [33,34]. Subsequent studies have determined that while some oxygen is consumed in oxidative phosphorylation, the majority is used to produce nanomolar concentrations of hydrogen peroxide at the egg surface within minutes of fertilization [35]. This is accomplished by the NADPH oxidase family member designated Udx1 [36,37]. The hydrogen peroxide cross-links tyrosine residues in proteins of the extracellular matrix [38]. The functional consequence of the oxidative burst is that the fertilization envelope is rapidly converted into a physical structure that initially prevents entry of more than one sperm cell and then protects the developing embryo [39] (Fig. 5).
Fig. 5.
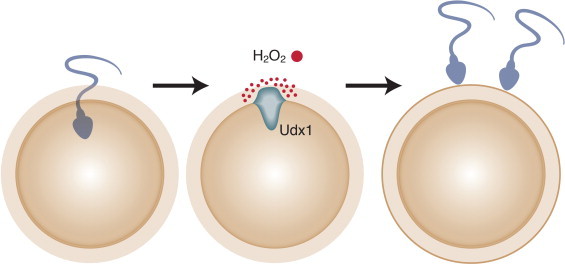
An oxidative burst at the time of fertilization prevents polyspermy. Studies of sea urchin oocytes have measured an oxidative burst within moments of fertilization [37]. Activation of the dual oxidase called Udx1 results in the generation of high local concentration of hydrogen peroxide. Proteins on the outer surface of the fertilized oocyte became cross-linked, thus preventing entry of more than one sperm [39].
Redox signaling for proliferation
ROS function in signaling pathways for proliferation that are triggered by growth factors. This was first appreciated for basic fibroblast growth factor [40], platelet-derived growth factor [41] and epidermal growth factor [42] signaling in chondrocytes, vascular smooth muscle cells and epidermoid carcinoma cells, respectively. The ROS signal is generated downstream of the growth factor receptor and upstream of mitogen-activated protein kinase (MAPK) pathways [43]. Ras is an adaptor protein bridging ligand-bound growth factor receptors and MAPKs (Fig. 6). It is activated upon GTP binding and deactivated by hydrolysis of the GTP.
Fig. 6.
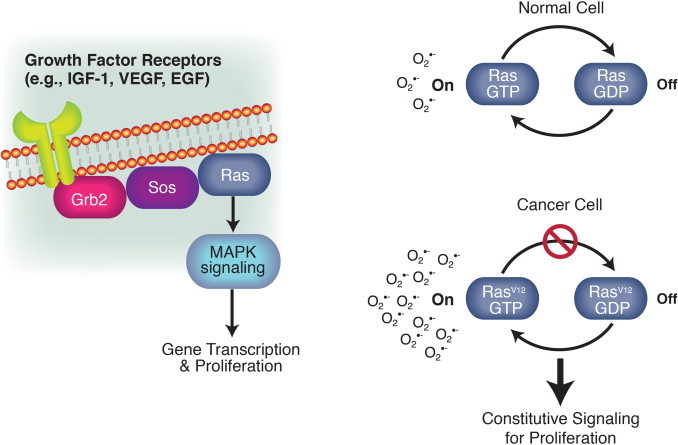
Redox signaling downstream of the Ras oncoprotein stimulates proliferation. Ras functions within signaling pathways for proliferation. It acts as a bridge between growth factor receptors and downstream kinases that ultimately regulate gene transcription. Ras is activated by binding to GTP. Hydrolysis of the GTP turns Ras off. Cells transformed with oncogenic forms of Ras (e.g., RasV12) can progress through the cell cycle in the absence of growth factors. These cells produce increased levels of superoxide as the result of increased NADPH oxidase activity [45,46]. These findings were early evidence of redox signaling for proliferation and aberrant redox signaling in cancer cells.
Oncogenic forms of Ras (e.g., H-Rasv12) cause constitutive signaling for proliferation, independent of the binding of growth factors to their ligands. The mutations inhibit GTPase activity and thus, keep Ras in its active state [44]. Transfection of NIH 3T3 cells with H-Rasv12 leads to constitutive superoxide production [45] (Fig. 6). This can be prevented by treatment of the cells with a flavoprotein inhibitor, or superoxide dismutase, but not catalase. This implicates an NADPH oxidase family member downstream of oncogenic Ras. Indeed, overexpression of the NADPH oxidase, Nox1 (also called mitogenic oxidase 1 or Mox1) confers cancer cell phenotypes [46].
NADPH oxidases are widely expressed and could contribute to redox signaling in many types of tissues (reviewed in [47]). The family consists of seven members. Nox2 (originally called gp91phox) is the catalytic subunit of the NADPH oxidase found in phagocytes. It is notable for: (1) being the first of these enzymes to be discovered and (2) setting a new paradigm that ROS can be beneficial to cells [48]. The enzymes function as multi-protein complexes. The core subunit transfers electrons from NADPH on one side of the membrane to oxygen on the other side, thus generating superoxide.
Redox signaling for death
The idea that free radicals are responsible for the gradual decline in function seen across species, with the increasing age of individuals, was first proposed by Denhan Harmon in 1956 [49]. His proposal was based on the idea of damage caused by the hydroxyl radical. While the source of these was not known at the time, he reasoned that respiratory enzymes were most likely responsible. Denhan was not aware of cellular production of superoxide and hydrogen peroxide through electron leakage from the respiratory chain. That knowledge did not come until after the discovery of superoxide dismutases in 1968 [50,51].
The idea that damage from ROS, as by-products of aerobic respiration, contributes to aging continues to be debated as a theory of aging [52,53]. Evidence in support of the theory includes the strong correlation seen between metabolic rate or superoxide production and maximum life-span of a species [54]. A long life-span is correlated with a lower metabolic rate and low superoxide production (Fig. 7). The life-span of the fruit fly, Drosophila melanogaster, is extended 30% by genetically increasing levels of both superoxide dismutase and catalase [55]. Caloric restriction has been shown to extend life-span in every species in which it has been tested, including rhesus monkeys [56,57]. Outcomes of caloric restriction include a lower steady-state level of oxidative stress and decreased oxidative damage accumulation with age.
Fig. 7.
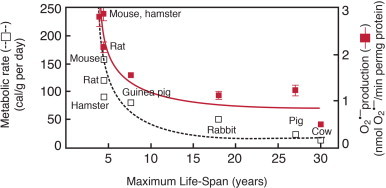
Oxygen and death – hypothesis on aging. Correlative data in support of the free radical or oxidative stress [49] and rate of living [59] theories of aging are obtained by comparing metabolic rate and superoxide production to the maximum life-span of different species [56]. A long life-span is correlated with a lower metabolic rate and low superoxide production. This fits the hypothesis that the loss of function seen with aging is due to the accumulation of molecular oxidative damage that ultimately results in cell death and aging of tissues.
From Sohal R.S. and Weindruch R. (1996) Science 273: 59–63; http://www.sciencemag.org/content/273/5271/59. Reprinted with permission from AAAS.
A prediction of the free radical, or oxidative stress, theory of aging is that manipulations that reduce oxidative damage will extend life-span. As stated above, this prediction has been borne out in studies of transgenic flies and of caloric restriction in various species. An extensive study with mouse models, however, has resulted in many cases for which a change in the accumulation of oxidative damage does not alter maximum life-span [53]. In these experiments, transgenic mice were generated to express either decreased or increased levels of CuZnSOD (encoded by Sod1), MnSOD (encoded by Sod2), Gpx1, Gpx4 or Trx2. Additional experiments examined combinations of gene knockouts: (1) CuZnSOD and MnSOD (Sod1−/−/Sod2+/−); (2) CuZnSOD and Gpx1 (Sod1−/−/Gpx1−/−); (3) CuZnSOD and Gpx4 (Sod1−/−/Gpx4+/−); (4) MnSOD and Gpx1 (Sod2+/−/Gpx1+/−); (5) MnSOD and Gpx4 (Sod2+/−/Gpx4+/−), and (6) Gpx1 and Gpx4 (Gpx1+/−/Gpx4+/−). In each of these models, the mice showed altered resistance to oxidative stress and a difference in the accumulation of oxidative damage. For example, mice overexpressing CuZnSOD were more resistant to paraquat toxicity and overexpression of MnSOD resulted in decreased levels of protein and oxidative damage with age. Despite these confirmations of altered antioxidant defenses, no difference in longevity was seen in 17 of the 18 models tested. Loss of CuZnSOD activity was the only manipulation that affected aging; Sod1−/− mice have a 30% reduction in mean and maximum life-span.
In the early 1900s, Rubner noted that different species expend a similar amount of energy over their lifetimes [58]. Tissues of small mammals such as mice use the energy allotment quickly and die sooner, while large mammals such as elephants live long lives expending the energy at a slow rate. This relationship forms the basis of the rate of living theory of aging proposed by Pearl in 1928 [59]. The oxidative stress theory of aging provides a mechanism, given that a high rate of metabolism speeds up the accumulation of damage from ROS as by-products.
Studies of birds provide insight into the relationship between ROS, the rate of aging and metabolism. Birds are an exception to the rate of living rule. The slope of the line correlating maximum life-span potential with body weight is similar for species of mammals and birds, but it is shifted upwards for birds [60]. Rats and pigeons have similar body weights, but maximum life-spans of 5 and 35 years, respectively. Montgomery and colleagues carried out an extensive comparison to gain insight into the 7-fold difference in longevity for these two species [61]. They measured multiple antioxidants defenses, ROS and markers of oxidative damage in seven different tissues or isolated mitochondria. The only consistent, significant difference is a lipid peroxidation index showing that membranes in rat tissues are more susceptible to oxidation than those in pigeons (Fig. 8). The variation in membrane lipid composition across species may explain the longevity of birds [62]. The findings from the study by Montgomery et al. suggest that manipulations of antioxidant defenses in mammals that do not alter lipid peroxidation rates will not extend life-span.
Fig. 8.

Differences in the susceptibility of pigeons versus rat cell membranes to oxidative damage. Species of birds do not conform to the rate of living theory of aging as exemplified by the 7-fold greater longevity of pigeons versus rats, despite their similar sizes. As a test of the oxidative stress theory of aging Montgomery and colleagues measured antioxidants defenses, reactive oxygen species and oxidative damage in seven different tissues or isolated mitochondria from rats and pigeons [61]. The only consistent, significant difference they observed is in a membrane peroxidation index indicating that the cell membranes in rat tissues are more susceptible to oxidation.
From Montgomery MK, Hulbert AJ, and Buttemer WA. (2011) PLoS One. 6, e24138; http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0024138.
Moving from whole organisms to cells, there is evidence of hydrogen peroxide signaling for death through the mitochondrial apoptosis pathway. One mechanism involves the Src homology 2 (SH2) domain-containing protein called p66Shc. This protein is splice variant of two cytoplasmic adaptor proteins, p52Shc and p46Shc; the latter two proteins are involved in tyrosine kinase pathway signaling for proliferation. P66 Shc has a different role in the cell. P66Shc knockout mice have increased resistance to oxidative stress, live 30% longer under laboratory conditions and accumulate lower levels of oxidative damage with age [63,64]. Cells from p66Shc null mice are resistant to apoptosis induced by different stimuli. Further studies have led to a model whereby apoptotic signals cause p66Shc to dissociate from a multi-protein complex in the intermembrane space [65] (Fig. 9). It then oxidizes cytochrome c to generate hydrogen peroxide. Increased hydrogen peroxide levels ultimately affect the mitochondrial permeability transition pore, leading to apoptosis.
Fig. 9.
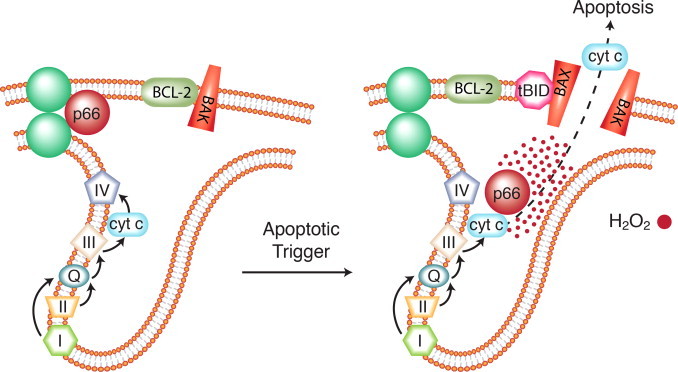
Redox signaling for cell death mediated by p66Shc in mitochondria. Knockout mice that lack the p66Shc protein have increased longevity and accumulate lower levels of markers of oxidative damage with age [63]. Cells from p66Shc knockout mice are resistant to apoptosis induced by a variety of stimuli. Studies of p66Shc have led to this model of mitochondrial-mediated apoptosis [65]. An apoptotic trigger causes p66Shc to dissociate from a multiprotein complex in the intermembrane space. It then oxidizes cytochrome c to generate hydrogen peroxide. Increased hydrogen peroxide levels ultimately affect the permeability of the outer mitochondrial membrane, leading to apoptosis.
In the intrinsic pathway to apoptosis, hydrogen peroxide can mediate cytochrome c release through a mechanism involving the mitochondrial-specific, anionic lipid, cardiolipin (Fig. 10). An early hint at this mechanism was the report from Vogelstein’s laboratory that p53-mediated apoptosis involved oxidation of mitochondrial components [66]. Cytochrome c is tethered to the outer surface of the inner mitochondrial membrane through hydrostatic and hydrophobic interactions with cardiolipin [67,68]. As levels of hydrogen peroxide increase and cardiolipin is redistributed during apoptosis, this results in partial unfolding of cytochrome c and its conversion to a peroxidase [69,70]. Cardiolipin is the target of the peroxidase activity [71]. Cytochrome c has a reduced affinity for oxidized cardiolipin and is thus released into the inter-membrane space [72,73]. Additional consequences of cardiolipin redistribution during apoptosis are the accumulation of this lipid at contact sites between the outer and inner membranes and increased negative charge of the outer membrane [74]. This facilitates the recruitment of pro-apoptotic proteins and opening of channels for the release of cytochrome c into the cytosol [75,76].
Fig. 10.
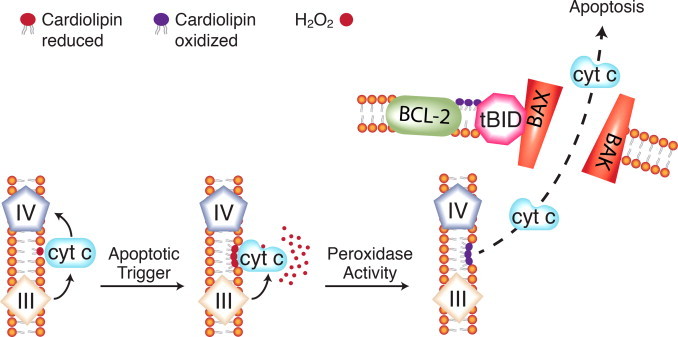
Cardiolipin oxidation and cytochrome c release in the mitochondrial pathway to apoptosis. A second, complementary model for redox signaling for apoptosis involves cytochrome c and the mitochondrial-specific phospholipid cardiolipin. Under normal conditions, cytochrome c binds cardiolipin through loose, electrostatic interactions. In the presence of hydrogen peroxide, a tighter interaction develops. This partially unfolds the cytochrome c and converts it to a peroxidase with cardiolipin as the substrate. Oxidized cardiolipin has a reduced affinity for cytochrome c, which then becomes soluble in the intermembrane space. Mitochondrial membrane lipids are reorganized during apoptosis. Oxidized cardiolipin appears in the outer mitochondrial membrane and attracts the pro-apoptotic protein tBID, thus facilitating release of cytochrome c and other apoptotic proteins from the mitochondrial intermembrane space.
Redox biology and signaling in cancer
Aberrant regulation of proliferation, migration and invasion
Cancer is a disease marked by dysregulation of many cellular processes. Early evidence that redox biology is perturbed in this disease was the finding that tumor cell lines established from different histological types of cancer show elevated constitutive production of hydrogen peroxide [77]. The study shows that although the maximal rate of production in the tumor cells is less than what is seen after activation of NADPH oxidase in phagocytic cells, the cumulative amounts released by tumor cells over 4 hours surpasses the amount produced during an oxidative burst in activated phagocytes. Altered levels of antioxidant defenses [78,79] and higher levels of 8-hydroxy-2′-deoxyguanosine [80,81], an indicator of oxidative damage to DNA, are found in cancer tissues compared to adjacent, normal tissue. Mutations resulting from oxidative damage to nuclear or mitochondrial DNA can contribute to carcinogenesis [82–85].
As could be expected from the role of hydrogen peroxide in signaling for cell proliferation and cell death, the redox changes seen in cancer cells can impact these processes. Cancer phenotypes of RasV12-transformed cells include anchorage-independent growth, an accelerated rate of proliferation and the ability to form tumors in immunocompromised mice [86]. These properties are seen in cells transfected with Nox1 [46], consistent with increased expression of Nox1 in cells carrying the RasVal12 mutation [87]. Genetic knockdown of Nox1 is sufficient to reverse the cancer cell phenotypes [46]. The conclusion is that redox signaling downstream of Nox1 is critical for transformation by oncogenic RasV12. This signaling may involve the regulation of proteins involved in cell fate decisions, including NF-κB, AP-1 and TP53 [5].
Alterations in antioxidant defenses can allow for aberrant redox signaling in cancer cells. This has been demonstrated for MnSOD, the superoxide dismutase enzyme that is found in the mitochondrial matrix. MnSOD is encoded by the SOD2 gene at chromosome 6q25 [88]. Melanomas frequently have deletions of the long arm of chromosome 6 [89,90]. When the full chromosome 6 is restored through microcell hybridization with a melanoma-derived cell line, properties of transformed cells are diminished or lost [91]. These properties include morphological features of less differentiated cells and the ability to form colonies in soft agar and tumors in athymic mice. Transfection of melanoma cells with SOD2 alone achieves the same outcome [92]. Analogous findings have been made with SV40-transformed lung fibroblasts [93]. These studies suggest that when MnSOD levels are abnormally low in cancer cells, increasing this enzyme can suppress tumor growth. In this case, an elevated level of hydrogen peroxide may push the cells into senescence [94,95].
An increase in MnSOD expression can confer on cells the deadliest of cancer properties: the ability to invade and metastasize. The majority of cancer deaths can be attributed to metastatic disease [96]. Melendez and colleagues have investigated the role of MnSOD in tumor cell migration and invasion through gene transfections with SOD2 in the absence or presence of catalase [97]. The researchers assessed the cells’ ability to migrate using a scratch assay, in which movement from a monolayer into a cell-free region is measured. The results show that the extent of migration is positively correlated to the amount of MnSOD activity in the cell population. Furthermore, cells at the leading edge of the migrating cells show the highest MnSOD expression. The SOD2 transfectants are also more invasive, as demonstrated with a transwell assay. This assay measures cell movement through a matrix in response to growth factors as a chemoattractant. Similar results are seen with fibrosarcoma cells and a bladder tumor cell line [97]. When the cells are transfected with SOD2 and catalase, however, they exhibit similar migratory ability and invasiveness as control cells transfected with an empty vector. The latter finding indicates that increased hydrogen peroxide levels in the SOD2 transfectants enhances the cells ability to migrate and invade. This can be achieved by upregulation of collagenases that are secreted from cells and break down tissue stroma [98,99].
It may seem paradoxical that decreased levels of MnSOD favor proliferation of cancer cells, while an increased level allows for cancer cell migration and invasion. Dhar and St. Clair provide a solution to this paradox; MnSOD is regulated by different factors that may each change during cancer progression [100]. Transcription factors that regulate SOD2 include NF-κB [101,102], Sp1 [103], p53 [104,105] and FOXO3 [106]. The expression or activity of these can be dysregulated during cancer as the result of mutation, chromosomal rearrangement or loss, and epigenetic alterations. NF-κB and p53 are redox-sensitive transcription factors [107]. Changes in the redox environment during cancer progression may thus impact their regulation of SOD2. MnSOD is regulated by cytokines [108,109] that may be present at various levels, depending on the inflammatory cells present in the tumor microenvironment. The mechanism of MnSOD gene repression in early stages of cancer may involve epigenetics. Hypermethylation of the gene promoter has been reported in breast [110] and pancreatic cancer [111] along with multiple myeloma [112]. In later stages of cancer, histone hyperacetylation could allow for aberrantly high expression of MnSOD.
Oxygen, ROS and cancer stem cells
A relatively new research area is focused on cancer stem cells. These are functionally defined as the subset of tumor cells that establish new tumors when transplanted into immunocompromised mice [113]. Similar to normal stem cells, they are undifferentiated but possess the relatively rare ability to self-renew or to differentiate. Hypoxia-inducible factors (HIF-1α and HIF-1β) are master regulators of stem cell properties [114]. These transcription factors are activated by aberrant growth factor-mediated signaling [115,116] or the hypoxic microenvironment [117] in cancer. Proteins whose expression is regulated by HIF-1 and that confer a stem cell phenotype include NODAL, NOTCH and MYC [118] (Fig. 11). Lower ROS levels have been reported in cancer stem cells, which may explain their resistance to oxidative stress-induced DNA damage [119].
Fig. 11.
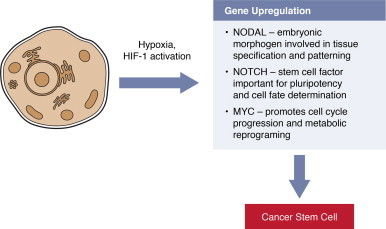
Oxygen, ROS and cancer stem cells. Hypoxia and aberrant activation of kinase signaling pathways in cancer lead to HIF-1 activation. Targets of the HIF-1 transcription factor include the NODAL, NOTCH and MYC genes that support stem cell properties. Cancer stem cells can self-renew or differentiate. These abilities allow cancer stem cells to populate tumors and are thought to be key for the metastatic spread of primary cancers.
Aberrant regulation of death
Evidence that dysregulation of redox signaling for cell death contributes to cancer comes from studies of the B cell lymphoma protein-2 (BCL-2). As the name implies, BCL-2 was discovered in B cell lymphomas [120]. A t(14;18) translocation in these lymphomas places the coding region of the gene under the control of an immumoglobulin gene promoter, thereby leading to aberrantly high levels of the protein. BCL-2 is the first oncogene to be discovered to work by inhibiting cell death, rather than promoting growth [121]. Early studies of BCL-2-overexpressing cells showed decreased lipid peroxidation after exposure to apoptotic triggers [122] and higher, basal levels of glutathione [123]. Bcl-2 knockout mice showed signs of chronic oxidative stress [124]. This led to the idea that BCL-2 functions like an antioxidant. The picture that has emerged from further studies is that mitochondrial ROS production is increased in cells that overexpress BCL-2 [125,126]. The cells adapt by increasing antioxidant defenses, making them resistant to oxidative stress [125,127].
BCL-2 is part of a family of proteins that regulate apoptosis via protein:protein interactions [128]. It localizes to nuclear, endoplasmic reticular and mitochondrial membranes [129]. The mitochondrion is thought to be the major site of action for BCL-2. There, it interacts with pro-apoptotic family members (e.g., BAX, BAK) to control outer membrane permeability and the release of cytochrome c and other mitochondrial intermembrane space proteins [130–133]. In the cytoplasm these proteins mediate cell destruction through activation of the caspase family of proteolytic enzymes. Caspases are distinguished by a critical cysteine residue in their active site, cleavage of substrates after an aspartate residue and a large set of target proteins that function to maintain the cell cytoskeleton, integrity of the DNA and play diverse roles in cell regulation [134].
A series of studies by Pervaiz and colleagues provides a model for how BCL-2 may confer resistance to oxidative stress and apoptosis [135–137] (Fig. 12). Under normal, non-stressed conditions, overexpression of BCL-2 in CEM leukemia cells, HCT116 colon carcinoma or HK-1 and C666-1 nasopharyngeal carcinoma cells increases the activity of cytochrome c oxidase (COX). COX is the terminal acceptor in the electron transport chain. It can influence the rate of ATP production by mitochondrial respiration and of ROS generation as the result of electron leakage from respiratory complexes I or III. The increased COX activity in BCL-2 overexpressing cells is associated with elevated oxygen consumption and superoxide generation [135]. The underlying mechanism involves enhanced transfer of the nuclear-encoded COX Va and Vb subunits to mitochondria [136]. This study shows that BCL-2 physically interacts with COX Va and thus may act as its chaperone. Intriguing findings were the differences seen when the cells were subjected to oxidative stress due to hypoxia, glucose deprivation or serum withdrawal [135]. BCL-2-overexpressing cells responded by decreasing COX activity, which kept ROS at a lower, sub-lethal level. In contrast, the response seen when cells with normal levels of BCL-2 were subjected to these stresses was increased COX activity and ROS levels.
Fig. 12.
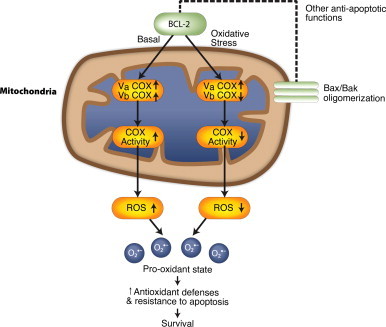
Redox-based mechanism for BCL-2’s anti-apoptotic function. BCL-2 localizes to different cellular membranes. In mitochondria, it interacts with pro-apoptotic BCL-2 family members to inhibit apoptosis. Under normal, basal conditions, BCL-2 also interacts with the COX Va subunit complex to increase electron flow, oxygen consumption and ROS levels. This results in a pro-oxidant state that promotes proliferation, while leading to adaptations that allow the cells to survive under increased oxidative stress. In contrast, under conditions of even greater oxidative stress, BCL2 inhibits the COX Vb subunit, to lower ROS production to safe levels that will not trigger apoptosis [137].
Reproduced with permission from Krishna S., Low I.C., and Pervaiz S. (2011) Biochem. J. 435, 545–551. © the Biochemical Society.
Elucidating the roles of BCL-2 family members in mitochondria may uncover secrets as to why some cancer patients are cured with current, standard-of-care chemotherapy while others succumb to the disease due to chemoresistance [138]. Pre-treatment specimens from patients with multiple myeloma, leukemia and ovarian cancer show differences in susceptibility to the mitochondrial pathway to apoptosis [139]. Sensitivity to apoptosis is measured in vitro by incubating tumor cells with peptides from pro-apoptotic BCL-2 family members and monitoring mitochondrial depolarization. A high percent of depolarization indicates mitochondria that undergo apoptosis easily. A significant, positive correlation is seen between apoptosis susceptibility and the patients’ responses to the standard chemotherapeutic regimens for these cancers. Chemotherapy is most effective in those patients whose tumor cells are close to the threshold (i.e., primed) for apoptosis. Although the basis for the different sensitivities of tumor cell mitochondria to apoptosis has not been determined, the mechanism could well have a redox component.
Aberrant regulation of metabolism
Cancer cells alter their metabolism to support growth. This metabolic reprogramming takes a number of forms, depending on the particular cancer and cancer subtype [140]. In general, though, the cellular redox environment is impacted along with proliferative capacity. Otto Warburg was the first to note that cancer cells have a high avidity for glucose and produce lactic acid even in the presence of oxygen [34]. In this initial report, Warburg postulated that cancer cells relied on glycolysis due to defective respiration. Near the end of his career he modified his position, acknowledging that the idea of damaged respiration in cancer cells had led to “fruitless controversy” [32]. Current evidence indeed indicates that mitochondria still function in cancer cells but, in addition they rely on aerobic glycolysis to support the pentose phosphate pathway and glutamine to sustain the TCA cycle [141,142]. The latter process is called glutaminolysis or anaplerosis. The oxidative arm of the pentose phosphate supplies NADPH. Overall, the metabolic rewiring of cancer cells provides precursors for growth (i.e., nucleotides, amino acids and lipids) along with NADPH to counter a more oxidized redox state [143] (Fig. 13).
Fig. 13.
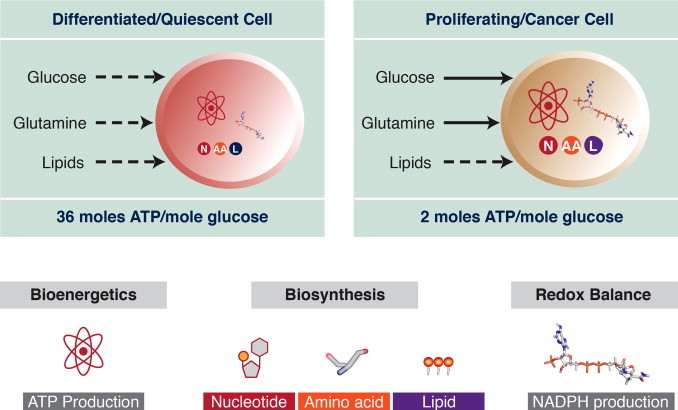
Cancer cells are metabolic opportunists. Differentiated or quiescent cells need a higher proportion of ATP than building blocks for nucleic acids, proteins and lipids. Therefore, they rely on the more energetically favorable oxidative phosphorylation. Cancer cells need to balance their need for ATP with the requirement to duplicate the cellular contents before dividing and maintain NADPH levels for reductive biosynthesis and to counter oxidative stress. Tapping into multiple metabolic pathways provides a better balance of ATP and precursors for growth. These pathways include aerobic glycolysis and glutaminolysis.
Adapted by permission from the American Association for Cancer Research: Cantor JR and Sabatini DM, cancer cell metabolism: one hallmark, many faces. Cancer Discov., 2012, 2:881-898; doi:10.1158/2159-8290.CD-12-0345.
The metabolic changes in cancer cells are brought about through oncogene activation and loss of tumor suppressors. The MYC oncoprotein increases expression of the enzyme glutaminase synthase 1 [144,145], which deaminates glutamine to produce glutamate. This reaction is the first step in glutaminolysis and it also supplies glutamate for the synthesis of glutathione. One function of the tumor suppressor p53 is to repress transcription of genes encoding glucose transporters [146]. In the cytoplasm, p53 binds to glucose-6-phosphate dehydrogenase and inactivates it [147]. This enzyme catalyzes the rate limiting step in the oxidative arm of the pentose phosphate. Thus, the loss of p53 can counter the more oxidized redox environment in cancer cells through increased glucose uptake and synthesis of NADPH.
Mitochondria and metastatic potential
Other studies have documented the contribution of mitochondria to metastases. The ability of cancer cells to metastasize in vivo is correlated with formation of colonies on soft agar media in vitro (i.e., anchorage-independent growth) [148]. Treatment of Ras-transformed cells with mitochondrially-targeted nitroxides that scavenge superoxide inhibits anchorage-independent growth [149]. Mori and colleagues used cancer cell lines to develop a gene expression signature of anchorage-independent growth; metastatic disease in melanoma, breast and lung cancer was significantly correlated with patients’ tumor specimens having this signature [150]. The signature is enriched in genes whose products are involved in the pentose phosphate pathway or are localized to mitochondria. A critical role for mitochondria in metastasis is further evidenced by studies of cybrids containing nuclear DNA of one cell type and mitochondrial DNA of a second cell type [151]. Mitochondria from highly metastatic cells are able to confer this ability onto a low metastatic potential cell type.
ROS and the hallmarks of cancer
A seminal review article by Hanahan and Weinberg was published in the first issue of the journal Cell in the new millennium. The authors persuasively present a case that “the vast catalog of cancer genotypes is a manifestation of six alterations in cell physiology that collectively dictate malignant growth” [152]. These acquired capabilities are referred to as the hallmarks of cancer: self-sufficiency in growth signals; insensitivity to anti-growth signals; evading apoptosis; limitless replicative potential; sustained angiogenesis, and tissue invasion and metastasis. While a role for ROS or redox signaling in carcinogenesis was not discussed in this review, sufficient evidence had accumulated for their role to be recognized in an updated review published by the authors [153]. An emerging hallmark added in the 2011 update is deregulated cellular energetics. As described above, the altered metabolism in cancer cells impacts redox homeostasis.
In addition to recognizing the functions that normal cells must acquire to become malignant, it is important to understand the means by which these capabilities are acquired. Hanahan and Weinberg discuss enabling characteristics for carcinogenesis: genomic instability and mutation, and tumor-promoting inflammation [152,153]. Chronic inflammatory conditions, including peptic ulcers [154], inflammatory bowel diseases [155] and hepatitis [156] are associated with increased risks of cancer developing in the affected tissue. A possible mechanism is that inflammatory cells release ROS, which are mutagenic for the resident cells [157]. Genomic instability ensues when cells lose the ability to respond appropriately to DNA damage, as is seen with loss of normal p53 function [158]. Ataxia-telangiectasia mutated (ATM) kinase orchestrates cellular responses to DNA damage and oxidative stress; the mechanism for sensing ROS involves dimerization of 2 ATM monomers via a disulfide bond [159]. Inherited mutations in the ATM gene preclude dimerization and lead to genomic instability and an increased risk of cancer [160]. Thus, loss of ATM function allows for a vicious cycle whereby the inability of the cell to sense and respond to ROS leads to genome instability and accelerated carcinogenesis.
Redox homeostasis: an old and emerging cancer target
Oxidative stress is one of the earliest mechanisms to be exploited for cancer therapy. Following the discovery of radiation around the turn of the last century, its damaging effects on tissue was recognized and put to use for the treatment of cancer. Radiation generates hydroxyl radicals and causes oxidative damage in affected tissues. In the mid-part of the last century, anthracyclines were discovered to be potent chemotherapeutic drugs (reviewed in [161]). In cells, anthracyclines participate in redox cycling reactions that generate ROS [162] and lead to oxidative DNA damage [163]. This is thought to be the major mechanism of cardiotoxicity [164], which is the dose-limiting side effect of anthracycline treatment, although it is still debated as mechanism of action in cancer cells [163,165].
As knowledge of aberrant redox homeostasis and signaling in cancer cells grows, new approaches to redox-based chemotherapeutics are being developed (reviewed in [166,167]). The approaches can be broadly categorized as: (1) inhibiting antioxidant defenses; (2) interfering with regulatory systems that are used by cells to respond to oxidative stress and, (3) targeting specific proteins that are responsible for altered redox homeostasis or signaling. Examples in the first category include novel drugs that inhibit SODs (e.g., ATN-224) [168–170] or deplete glutathione (e.g., NOV-002 and imexon) [171,172]. Agents that target Nrf2 fit into the second category [6]; aberrant Nrf2 activation appears to be a strategy whereby cancer cells are protected from elevated oxidative stress [173,174]. The third category includes drugs being developed to antagonize BCL-2’s antiapoptotic function (e.g., ABT-737) [175].
Future directions
Knowledge of redox biology provides students with insight into mechanisms by which organisms use oxygen metabolites in the control of life and death. The role of dysregulated redox biology in all major human diseases, and not just cancer, makes this knowledge an important aspect of physician training. For scientists in training, much remains to be learned in the field of redox biology and signaling. Some of the particularly promising research areas to explore might include the cross-talk between kinase and redox signaling pathways and the redox biology of stem cells. The field is well-suited for interdisciplinary collaborations as expertise in the chemistry of ROS needs to be integrated with knowledge of protein targets at the molecular, cellular and systems biology level. This is illustrated by the pursuit of novel redox-based agents for cancer treatment. A frequently encountered idea is that agents can be developed to create a redox environment in cancer cells that triggers death. That is, it pushes these oxidatively-stressed cells past their limit [176]. One challenge to this idea, however, is that thiol redox circuits in cells may not be in equilibrium [177,178]. That is, the idea of oxidative stress as a balance of oxidants versus antioxidants is a biological oversimplification. A second challenge is our limited understanding of the mechanisms by which cancer cells developed resistance to redox signaling for cell death pathways [179]. Solving these challenges will be major scientific accomplishment and can help us unlock secrets for conquering cancer (Fig. 14). In the words of Toren Finkel, “further understanding of these pathways promises to reveal to us many more secrets regarding how life begins, why it ends, and all the myriad complexities that make up the middle [180].”
Fig. 14.
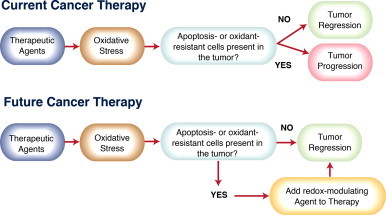
Targeting redox signaling for cancer therapy. The efficacy of current cancer therapies is limited by the presence of tumor cells with acquired resistance to apoptosis or oxidative stress. A number of strategies for targeting the altered redox biology in cancer cells are currently in development. In the current era of precision medicine, novel redox-modulating therapies will be most efficacious when matched to the specific alterations in patients’ tumors that confer resistance to apoptosis and oxidative stress.
Acknowledgements
This work was supported by NIH R01CA71768 and U54CA143924.
References
- 1.Woese C.R., Kandler O., Wheelis M.L. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(12):4576–4579. doi: 10.1073/pnas.87.12.4576. 2112744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knoll A.H. A new molecular window on early life. Science. 1999;285(5430):1025–1026. doi: 10.1126/science.285.5430.1025. 10475845 [DOI] [PubMed] [Google Scholar]
- 3.Woese C. The universal ancestor. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(12):6854–6859. doi: 10.1073/pnas.95.12.6854. 9618502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Slesak I., Slesak H., Kruk J. Oxygen and hydrogen peroxide in the early evolution of life on earth: in silico comparative analysis of biochemical pathways. Astrobiology. 2012;12(8):775–784. doi: 10.1089/ast.2011.0704. 22970865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marinho H.S., Real C., Cyrne L., Soares H., Antunes F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biology. 2014;2:535–562. doi: 10.1016/j.redox.2014.02.006. 24634836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilson A.J., Kerns J.K., Callahan J.F., Moody C.J. Keap calm, and carry on covalently. Journal of Medicinal Chemistry. 2013;56(19):7463–7476. doi: 10.1021/jm400224q. 23837912 [DOI] [PubMed] [Google Scholar]
- 7.Lushchak V.I. Adaptive response to oxidative stress: Bacteria, fungi, plants and animals. Comparative Biochemistry and Physiology C: Toxicology Pharmacology. 2011;153(2):175–190. doi: 10.1016/j.cbpc.2010.10.004. 20959147 [DOI] [PubMed] [Google Scholar]
- 8.Storz G., Tartaglia L.A., Ames B.N. Transcriptional regulator of oxidative stress-inducible genes: direct activation by oxidation. Science. 1990;248(4952):189–194. doi: 10.1126/science.2183352. 2183352 [DOI] [PubMed] [Google Scholar]
- 9.Choi H., Kim S., Mukhopadhyay P., Cho S., Woo J., Storz G. Structural basis of the redox switch in the OxyR transcription factor. Cell. 2001;105(1):103–113. doi: 10.1016/s0092-8674(01)00300-2. 11301006 [DOI] [PubMed] [Google Scholar]
- 10.Mongkolsuk S., Helmann J.D. Regulation of inducible peroxide stress responses. Molecular Microbiology. 2002;45(1):9–15. doi: 10.1046/j.1365-2958.2002.03015.x. 12100544 [DOI] [PubMed] [Google Scholar]
- 11.Dubbs J.M., Mongkolsuk S. Peroxide-sensing transcriptional regulators in bacteria. Journal of Bacteriology. 2012;194(20):5495–5503. doi: 10.1128/JB.00304-12. 22797754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corcoran A., Cotter T.G. Redox regulation of protein kinases. FEBS Journal. 2013;280(9):1944–1965. doi: 10.1111/febs.12224. 23461806 [DOI] [PubMed] [Google Scholar]
- 13.D’Autréaux B., Toledano M.B. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nature Reviews Molecular Cell Biology. 2007;8(10):813–824. doi: 10.1038/nrm2256. 17848967 [DOI] [PubMed] [Google Scholar]
- 14.Winterbourn C.C. Reconciling the chemistry and biology of reactive oxygen species. Nature Chemical Biology. 2008;4(5):278–286. doi: 10.1038/nchembio.85. 18421291 [DOI] [PubMed] [Google Scholar]
- 15.Pryor W.A. Oxy-radicals and related species: their formation, lifetimes, and reactions. Annual Review of Physiology. 1986;48:657–667. doi: 10.1146/annurev.ph.48.030186.003301. 3010829 [DOI] [PubMed] [Google Scholar]
- 16.Han D., Antunes F., Canali R., Rettori D., Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. Journal of Biological Chemistry. 2003;278(8):5557–5563. doi: 10.1074/jbc.M210269200. 12482755 [DOI] [PubMed] [Google Scholar]
- 17.Winterbourn C.C., Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radical Biology and Medicine. 1999;27(3–4):322–328. doi: 10.1016/s0891-5849(99)00051-9. 10468205 [DOI] [PubMed] [Google Scholar]
- 18.Bienert G.P., Schjoerring J.K., Jahn T.P. Membrane transport of hydrogen peroxide. Biochimica et Biophysica Acta. 2006;1758(8):994–1003. doi: 10.1016/j.bbamem.2006.02.015. 16566894 [DOI] [PubMed] [Google Scholar]
- 19.Bienert G.P., Møller A.L., Kristiansen K.A., Schulz A., Møller I.M., Schjoerring J.K. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. Journal of Biological Chemistry. 2007;282(2):1183–1192. doi: 10.1074/jbc.M603761200. 17105724 [DOI] [PubMed] [Google Scholar]
- 20.Gardner P.R., Raineri I., Epstein L.B., White C.W. Superoxide radical and iron modulate aconitase activity in mammalian cells. Journal of Biological Chemistry. 1995;270(22):13399–13405. doi: 10.1074/jbc.270.22.13399. 7768942 [DOI] [PubMed] [Google Scholar]
- 21.White M.F., Dillingham M.S. Iron–sulphur clusters in nucleic acid processing enzymes. Current Opinion in Structural Biology. 2012;22(1):94–100. doi: 10.1016/j.sbi.2011.11.004. 22169085 [DOI] [PubMed] [Google Scholar]
- 22.Pacher P., Beckman J.S., Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiological Reviews. 2007;87(1):315–424. doi: 10.1152/physrev.00029.2006. 17237348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denu J.M., Tanner K.G. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37(16):5633–5642. doi: 10.1021/bi973035t. 9548949 [DOI] [PubMed] [Google Scholar]
- 24.Winterbourn C.C., Hampton M.B. Thiol chemistry and specificity in redox signaling. Free Radical Biology and Medicine. 2008;45(5):549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. 18544350 [DOI] [PubMed] [Google Scholar]
- 25.Lind C., Gerdes R., Hamnell Y., Schuppe-Koistinen I., von Löwenhielm H.B., Holmgren A. Identification of S-glutathionylated cellular proteins during oxidative stress and constitutive metabolism by affinity purification and proteomic analysis. Archives of Biochemistry and Biophysics. 2002;406(2):229–240. doi: 10.1016/s0003-9861(02)00468-x. 12361711 [DOI] [PubMed] [Google Scholar]
- 26.Baty J.W., Hampton M.B., Winterbourn C.C. Proteomic detection of hydrogen peroxide-sensitive thiol proteins in Jurkat cells. Biochemical Journal. 2005;389(3):785–795. doi: 10.1042/BJ20050337. 15801906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McDonagh B., Ogueta S., Lasarte G., Padilla C.A., Bárcena J.A. Shotgun redox proteomics identifies specifically modified cysteines in key metabolic enzymes under oxidative stress in Saccharomyces cerevisiae. Journal of Proteomics. 2009;72(4):677–689. doi: 10.1016/j.jprot.2009.01.023. 19367685 [DOI] [PubMed] [Google Scholar]
- 28.Ferrer-Sueta G., Manta B., Botti H., Radi R., Trujillo M., Denicola A. Factors affecting protein thiol reactivity and specificity in peroxide reduction. Chemical Research in Toxicology. 2011;24(4):434–450. doi: 10.1021/tx100413v. 21391663 [DOI] [PubMed] [Google Scholar]
- 29.Peskin A.V., Low F.M., Paton L.N., Maghzal G.J., Hampton M.B., Winterbourn C.C. The high reactivity of peroxiredoxin 2 with H(2)O(2) is not reflected in its reaction with other oxidants and thiol reagents. Journal of Biological Chemistry. 2007;282(16):11885–11892. doi: 10.1074/jbc.M700339200. 17329258 [DOI] [PubMed] [Google Scholar]
- 30.Wood Z.A., Poole L.B., Karplus P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300(5619):650–653. doi: 10.1126/science.1080405. 12714747 [DOI] [PubMed] [Google Scholar]
- 31.Woo H.A., Yim S.H., Shin D.H., Kang D., Yu D.Y., Rhee S.G. Inactivation of peroxiredoxin I by phosphorylation allows localized H(2)O(2) accumulation for cell signaling. Cell. 2010;140(4):517–528. doi: 10.1016/j.cell.2010.01.009. 20178744 [DOI] [PubMed] [Google Scholar]
- 32.Koppenol W.H., Bounds P.L., Dang C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nature Reviews Cancer. 2011;11(5):325–337. doi: 10.1038/nrc3038. 21508971 [DOI] [PubMed] [Google Scholar]
- 33.Warburg O. Beobachtungen über die Oxydationsprozesse im Seeigelei. Hoppe-Seyler´s Zeitschrift für physiologische Chemie. 1908;57(1–2):1–16. [Google Scholar]
- 34.Warburg O., Notizen zur Entwicklungsphysiologie des Seeigeleies. in: Arch F & D Ges Physiol., 160(4–6) (1915) 324–332.
- 35.Foerder C.A., Klebanoff S.J., Shapiro B.M. Hydrogen peroxide production, chemiluminescence, and the respiratory burst of fertilization: interrelated events in early sea urchin development. Proceedings of the National Academy of Sciences of the United States of America. 1978;75(7):3183–3187. doi: 10.1073/pnas.75.7.3183. 277920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heinecke J.W., Shapiro B.M. Respiratory burst oxidase of fertilization. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(4):1259–1263. doi: 10.1073/pnas.86.4.1259. 2537493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wong J.L., Créton R., Wessel G.M. The oxidative burst at fertilization is dependent upon activation of the dual oxidase Udx1. Dev. Cell. 2004;7(6):801–814. doi: 10.1016/j.devcel.2004.10.014. 15572124 [DOI] [PubMed] [Google Scholar]
- 38.Hall H.G. Hardening of the sea urchin fertilization envelope by peroxidase-catalyzed phenolic coupling of tyrosines. Cell. 1978;15(2):343–355. doi: 10.1016/0092-8674(78)90003-x. 569021 [DOI] [PubMed] [Google Scholar]
- 39.Wong J.L., Wessel G.M. Free-radical crosslinking of specific proteins alters the function of the egg extracellular matrix at fertilization. Development. 2008;135(3):431–440. doi: 10.1242/dev.015503. 18094022 [DOI] [PubMed] [Google Scholar]
- 40.Lo Y.Y., Cruz T.F. Involvement of reactive oxygen species in cytokine and growth factor induction of c-fos expression in chondrocytes. Journal of Biological Chemistry. 1995;270(20):11727–11730. doi: 10.1074/jbc.270.20.11727. 7744816 [DOI] [PubMed] [Google Scholar]
- 41.Sundaresan M., Yu Z.X., Ferrans V.J., Irani K., Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270(5234):296–299. doi: 10.1126/science.270.5234.296. 7569979 [DOI] [PubMed] [Google Scholar]
- 42.Bae Y.S., Kang S.W., Seo M.S., Baines I.C., Tekle E., Chock P.B. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. Journal of Biological Chemistry. 1997;272(1):217–221. 8995250 [PubMed] [Google Scholar]
- 43.Truong T.H., Carroll K.S. Redox regulation of protein kinases. Critical Reviews in Biochemistry and Molecular Biology. 2013;48(4):332–356. doi: 10.3109/10409238.2013.790873. 23639002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scheffzek K., Ahmadian M.R., Kabsch W., Wiesmüller L., Lautwein A., Schmitz F. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277(5324):333–338. doi: 10.1126/science.277.5324.333. 9219684 [DOI] [PubMed] [Google Scholar]
- 45.Irani K., Xia Y., Zweier J.L., Sollott S.J., Der C.J., Fearon E.R. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275(5306):1649–1652. doi: 10.1126/science.275.5306.1649. 9054359 [DOI] [PubMed] [Google Scholar]
- 46.Suh Y.A., Arnold R.S., Lassegue B., Shi J., Xu X., Sorescu D. Cell transformation by the superoxide-generating oxidase Mox1. Nature. 1999;401(6748):79–82. doi: 10.1038/43459. 10485709 [DOI] [PubMed] [Google Scholar]
- 47.Bedard K., Krause K.H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological Reviews. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. 17237347 [DOI] [PubMed] [Google Scholar]
- 48.Royer-Pokora B., Kunkel L.M., Monaco A.P., Goff S.C., Newburger P.E., Baehner R.L. Cloning the gene for an inherited human disorder − chronic granulomatous disease − on the basis of its chromosomal location. Nature. 1986;322(6074):32–38. doi: 10.1038/322032a0. 2425263 [DOI] [PubMed] [Google Scholar]
- 49.Harman D. Aging: a theory based on free radical and radiation chemistry. Journal of Gerontology. 1956;11(3):298–300. doi: 10.1093/geronj/11.3.298. 13332224 [DOI] [PubMed] [Google Scholar]
- 50.McCord J.M., Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) Journal of Biological Chemistry. 1969;244(22):6049–6055. 5389100 [PubMed] [Google Scholar]
- 51.McCord J.M., Fridovich I. Superoxide dismutases: you’ve come a long way, baby. Antioxidants & Redox Signaling. 2014;20(10):1548–1549. doi: 10.1089/ars.2013.5547. 23924157 [DOI] [PubMed] [Google Scholar]
- 52.Balaban R.S., Nemoto S., Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120(4):483–495. doi: 10.1016/j.cell.2005.02.001. 15734681 [DOI] [PubMed] [Google Scholar]
- 53.Pérez V.I., Bokov A., Van Remmen H., Mele J., Ran Q., Ikeno Y. Is the oxidative stress theory of aging dead? Biochimica et Biophysica Acta. 2009;1790(10):1005–1014. doi: 10.1016/j.bbagen.2009.06.003. 19524016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ku H.H., Brunk U.T., Sohal R.S. Relationship between mitochondrial superoxide and hydrogen peroxide production and longevity of mammalian species. Free Radical Biology and Medicine. 1993;15(6):621–627. doi: 10.1016/0891-5849(93)90165-q. 8138188 [DOI] [PubMed] [Google Scholar]
- 55.Orr W.C., Sohal R.S. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science. 1994;263(5150):1128–1130. doi: 10.1126/science.8108730. 8108730 [DOI] [PubMed] [Google Scholar]
- 56.Sohal R.S., Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273(5271):59–63. doi: 10.1126/science.273.5271.59. 8658196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Colman R.J., Anderson R.M., Johnson S.C., Kastman E.K., Kosmatka K.J., Beasley T.M. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325(5937):201–204. doi: 10.1126/science.1173635. 19590001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rubner M. Das problem der lebensdauer und seine bezeihunger zum wachstum und ernarhnung. R. Oldenburg; Munchen: 1908. [Google Scholar]
- 59.Pearl R. The Rate of Living. University of London Press; UK: 1928. [Google Scholar]
- 60.Barja G. Mitochondrial free radical production and aging in mammals and birds. Annals of the New York Academy of Sciences. 1998;854:224–238. doi: 10.1111/j.1749-6632.1998.tb09905.x. 9928433 [DOI] [PubMed] [Google Scholar]
- 61.Montgomery M.K., Hulbert A.J., Buttemer W.A. The long life of birds: the rat-pigeon comparison revisited. PLOS One. 2011;6(8):e24138. doi: 10.1371/journal.pone.0024138. 21904609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hulbert A.J., Pamplona R., Buffenstein R., Buttemer W.A. Life and death: metabolic rate, membrane composition, and life span of animals. Physiological Reviews. 2007;87(4):1175–1213. doi: 10.1152/physrev.00047.2006. 17928583 [DOI] [PubMed] [Google Scholar]
- 63.Migliaccio E., Giorgio M., Mele S., Pelicci G., Reboldi P., Pandolfi P.P. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402(6759):309–313. doi: 10.1038/46311. 10580504 [DOI] [PubMed] [Google Scholar]
- 64.Trinei M., Giorgio M., Cicalese A., Barozzi S., Ventura A., Migliaccio E. A p53-p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene. 2002;21(24):3872–3878. doi: 10.1038/sj.onc.1205513. 12032825 [DOI] [PubMed] [Google Scholar]
- 65.Giorgio M., Migliaccio E., Orsini F., Paolucci D., Moroni M., Contursi C. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122(2):221–233. doi: 10.1016/j.cell.2005.05.011. 16051147 [DOI] [PubMed] [Google Scholar]
- 66.Polyak K., Xia Y., Zweier J.L., Kinzler K.W., Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389(6648):300–305. doi: 10.1038/38525. 9305847 [DOI] [PubMed] [Google Scholar]
- 67.Nicholls P. Cytochrome c binding to enzymes and membranes. Biochimica et Biophysica Acta. 1974;346(3–4):261–310. doi: 10.1016/0304-4173(74)90003-2. 4374236 [DOI] [PubMed] [Google Scholar]
- 68.Demel R.A., Jordi W., Lambrechts H., van Damme H., Hovius R., de Kruijff B. Differential interactions of apo- and holocytochrome c with acidic membrane lipids in model systems and the implications for their import into mitochondria. Journal of Biological Chemistry. 1989;264(7):3988–3997. 2537300 [PubMed] [Google Scholar]
- 69.Kagan V.E., Tyurin V.A., Jiang J., Tyurina Y.Y., Ritov V.B., Amoscato A.A. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nature Chemical Biology. 2005;1(4):223–232. doi: 10.1038/nchembio727. 16408039 [DOI] [PubMed] [Google Scholar]
- 70.Kagan V.E., Bayir H.A., Belikova N.A., Kapralov O., Tyurina Y.Y., Tyurin V.A. Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free Radical Biology and Medicine. 2009;46(11):1439–1453. doi: 10.1016/j.freeradbiomed.2009.03.004. 19285551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kagan V.E., Tyurina Y.Y., Bayir H., Chu C.T., Kapralov A.A., Vlasova I.I. The “pro-apoptotic genies” get out of mitochondria: oxidative lipidomics and redox activity of cytochrome c/cardiolipin complexes. Chem. Biol. Interact. 2006;163(1–2):15–28. doi: 10.1016/j.cbi.2006.04.019. 16797512 [DOI] [PubMed] [Google Scholar]
- 72.Shidoji Y., Hayashi K., Komura S., Ohishi N., Yagi K. Loss of molecular interaction between cytochrome c and cardiolipin due to lipid peroxidation. Biochem. Biophys. Res. Commun. 1999;264(2):343–347. doi: 10.1006/bbrc.1999.1410. 10529366 [DOI] [PubMed] [Google Scholar]
- 73.Ott M., Robertson J.D., Gogvadze V., Zhivotovsky B., Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(3):1259–1263. doi: 10.1073/pnas.241655498. 11818574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Heit B., Yeung T., Grinstein S. Changes in mitochondrial surface charge mediate recruitment of signaling molecules during apoptosis. American Journal of Physiology - Cell Physiology. 2011;300(1):C33–C41. doi: 10.1152/ajpcell.00139.2010. 20926778 [DOI] [PubMed] [Google Scholar]
- 75.Kim T.H., Zhao Y., Ding W.X., Shin J.N., He X., Seo Y.W. Bid-cardiolipin interaction at mitochondrial contact site contributes to mitochondrial cristae reorganization and cytochrome C release. Molecular Biology of the Cell. 2004;15(7):3061–3072. doi: 10.1091/mbc.E03-12-0864. 15107464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gonzalvez F., Pariselli F., Dupaigne P., Budihardjo I., Lutter M., Antonsson B. tBid interaction with cardiolipin primarily orchestrates mitochondrial dysfunctions and subsequently activates Bax and Bak. Cell Death & Differentiation. 2005;12(6):614–626. doi: 10.1038/sj.cdd.4401571. 15818416 [DOI] [PubMed] [Google Scholar]
- 77.Szatrowski T.P., Nathan C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Research. 1991;51(3):794–798. 1846317 [PubMed] [Google Scholar]
- 78.Oberley T.D., Oberley L.W. Antioxidant enzyme levels in cancer. Histology and Histopathology. 1997;12(2):525–535. 9151141 [PubMed] [Google Scholar]
- 79.Shan W., Zhong W., Zhao R., Oberley T.D. Thioredoxin 1 as a subcellular biomarker of redox imbalance in human prostate cancer progression. Free Radical Biology and Medicine. 2010;49(12):2078–2087. doi: 10.1016/j.freeradbiomed.2010.10.691. 20955789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Toyokuni S., Okamoto K., Yodoi J., Hiai H. Persistent oxidative stress in cancer. FEBS Letters. 1995;358(1):1–3. doi: 10.1016/0014-5793(94)01368-b. 7821417 [DOI] [PubMed] [Google Scholar]
- 81.Loft S., Poulsen H.E. Cancer risk and oxidative DNA damage in man. Journal of Molecular Medicine. 1996;74(6):297–312. doi: 10.1007/BF00207507. 8862511 [DOI] [PubMed] [Google Scholar]
- 82.Cheng K.C., Cahill D.S., Kasai H., Nishimura S., Loeb L.A. 8-hydroxyguanine, an abundant form of oxidative DNA damage, causes G–T and A–C substitutions. Journal of Biological Chemistry. 1992;267(1):166–172. 1730583 [PubMed] [Google Scholar]
- 83.Møller P., Wallin H. Adduct formation, mutagenesis and nucleotide excision repair of DNA damage produced by reactive oxygen species and lipid peroxidation product. Mutation Research. 1998;410(3):271–290. doi: 10.1016/s1383-5742(97)00041-0. 9630671 [DOI] [PubMed] [Google Scholar]
- 84.Jackson A.L., Loeb L.A. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutation Research. 2001;477(1–2):7–21. doi: 10.1016/s0027-5107(01)00091-4. 11376682 [DOI] [PubMed] [Google Scholar]
- 85.Brandon M., Baldi P., Wallace D.C. Mitochondrial mutations in cancer. Oncogene. 2006;25(34):4647–4662. doi: 10.1038/sj.onc.1209607. 16892079 [DOI] [PubMed] [Google Scholar]
- 86.Trachootham D., Zhou Y., Zhang H., Demizu Y., Chen Z., Pelicano H. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006;10(3):241–252. doi: 10.1016/j.ccr.2006.08.009. 16959615 [DOI] [PubMed] [Google Scholar]
- 87.Mitsushita J., Lambeth J.D., Kamata T. The superoxide-generating oxidase Nox1 is functionally required for Ras oncogene transformation. Cancer Research. 2004;64(10):3580–3585. doi: 10.1158/0008-5472.CAN-03-3909. 15150115 [DOI] [PubMed] [Google Scholar]
- 88.Church S.L., Grant J.W., Meese E.U., Trent J.M. Sublocalization of the gene encoding manganese superoxide dismutase (MnSOD/SOD2) to 6q25 by fluorescence in situ hybridization and somatic cell hybrid mapping. Genomics. 1992;14(3):823–825. doi: 10.1016/s0888-7543(05)80202-2. 1427917 [DOI] [PubMed] [Google Scholar]
- 89.Trent J.M. Cytogenetics of human malignant melanoma. Cancer and Metastasis Reviews. 1991;10(2):103–113. doi: 10.1007/BF00049408. 1873851 [DOI] [PubMed] [Google Scholar]
- 90.Bastian B.C., LeBoit P.E., Hamm H., Bröcker E.B., Pinkel D. Chromosomal gains and losses in primary cutaneous melanomas detected by comparative genomic hybridization. Cancer Research. 1998;58(10):2170–2175. 9605762 [PubMed] [Google Scholar]
- 91.Trent J.M., Stanbridge E.J., McBride H.L., Meese E.U., Casey G., Araujo D.E. Tumorigenicity in human melanoma cell lines controlled by introduction of human chromosome 6. Science. 1990;247(4942):568–571. doi: 10.1126/science.2300817. 2300817 [DOI] [PubMed] [Google Scholar]
- 92.Church S.L., Grant J.W., Ridnour L.A., Oberley L.W., Swanson P.E., Meltzer P.S. Increased manganese superoxide dismutase expression suppresses the malignant phenotype of human melanoma cells. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(7):3113–3117. doi: 10.1073/pnas.90.7.3113. 8464931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yan T., Oberley L.W., Zhong W., St Clair D.K. Manganese-containing superoxide dismutase overexpression causes phenotypic reversion in SV40-transformed human lung fibroblasts. Cancer Research. 1996;56(12):2864–2871. 8665527 [PubMed] [Google Scholar]
- 94.Sarsour E.H., Venkataraman S., Kalen A.L., Oberley L.W., Goswami P.C. Manganese superoxide dismutase activity regulates transitions between quiescent and proliferative growth. Aging Cell. 2008;7(3):405–417. doi: 10.1111/j.1474-9726.2008.00384.x. 18331617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sarsour E.H., Kalen A.L., Goswami P.C. Manganese superoxide dismutase regulates a redox cycle within the cell cycle. Antioxidants & Redox Signaling. 2014;20(10):1618–1627. doi: 10.1089/ars.2013.5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sporn M.B. The war on cancer. Lancet. 1996;347(9012):1377–1381. doi: 10.1016/s0140-6736(96)91015-6. 8637346 [DOI] [PubMed] [Google Scholar]
- 97.Connor K.M., Hempel N., Nelson K.K., Dabiri G., Gamarra A., Belarmino J. Manganese superoxide dismutase enhances the invasive and migratory activity of tumor cells. Cancer Research. 2007;67(21):10260–10267. doi: 10.1158/0008-5472.CAN-07-1204. 17974967 [DOI] [PubMed] [Google Scholar]
- 98.Brenneisen P., Briviba K., Wlaschek M., Wenk J., Scharffetter-Kochanek K. Hydrogen peroxide (H2O2) increases the steady-state mRNA levels of collagenase/MMP-1 in human dermal fibroblasts. Free Radical Biology and Medicine. 1997;22(3):515–524. doi: 10.1016/s0891-5849(96)00404-2. 8981044 [DOI] [PubMed] [Google Scholar]
- 99.Wenk J., Brenneisen P., Wlaschek M., Poswig A., Briviba K., Oberley T.D. Stable overexpression of manganese superoxide dismutase in mitochondria identifies hydrogen peroxide as a major oxidant in the AP-1-mediated induction of matrix-degrading metalloprotease-1. Journal of Biological Chemistry. 1999;274(36):25869–25876. doi: 10.1074/jbc.274.36.25869. 10464329 [DOI] [PubMed] [Google Scholar]
- 100.Dhar S.K., St Clair D.K. Manganese superoxide dismutase regulation and cancer. Free Radical Biology and Medicine. 2012;52(11–12):2209–2222. doi: 10.1016/j.freeradbiomed.2012.03.009. 22561706 [DOI] [PubMed] [Google Scholar]
- 101.Jones P.L., Ping D., Boss J.M. Tumor necrosis factor alpha and interleukin-1beta regulate the murine manganese superoxide dismutase gene through a complex intronic enhancer involving C/EBP-beta and NF-kappaB. Molecular and Cellular Biology. 1997;17(12):6970–6981. doi: 10.1128/mcb.17.12.6970. 9372929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xu Y., Kiningham K.K., Devalaraja M.N., Yeh C.C., Majima H., Kasarskis E.J. An intronic NF-kappaB element is essential for induction of the human manganese superoxide dismutase gene by tumor necrosis factor-alpha and interleukin-1beta. DNA Cell Biology. 1999;18(9):709–722. doi: 10.1089/104454999314999. 10492402 [DOI] [PubMed] [Google Scholar]
- 103.Xu Y., Porntadavity S., St Clair D.K. Transcriptional regulation of the human manganese superoxide dismutase gene: the role of specificity protein 1 (Sp1) and activating protein-2 (AP-2) Biochemical Journal. 2002;362(2):401–412. doi: 10.1042/0264-6021:3620401. 11853549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pani G., Bedogni B., Anzevino R., Colavitti R., Palazzotti B., Borrello S. Deregulated manganese superoxide dismutase expression and resistance to oxidative injury in p53-deficient cells. Cancer Research. 2000;60(16):4654–4660. 10969820 [PubMed] [Google Scholar]
- 105.Dhar S.K., Tangpong J., Chaiswing L., Oberley T.D., St Clair D.K. Manganese superoxide dismutase is a p53-regulated gene that switches cancers between early and advanced stages. Cancer Research. 2011;71(21):6684–6695. doi: 10.1158/0008-5472.CAN-11-1233. 22009531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kops G.J., Dansen T.B., Polderman P.E., Saarloos I., Wirtz K.W., Coffer P.J. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419(6904):316–321. doi: 10.1038/nature01036. 12239572 [DOI] [PubMed] [Google Scholar]
- 107.Powis G., Briehl M., Oblong J. Redox signalling and the control of cell growth and death. Pharmacology & Therapeutics. 1995;68(1):149–173. doi: 10.1016/0163-7258(95)02004-7. 8604436 [DOI] [PubMed] [Google Scholar]
- 108.Wong G.H., Elwell J.H., Oberley L.W., Goeddel D.V. Manganous superoxide dismutase is essential for cellular resistance to cytotoxicity of tumor necrosis factor. Cell. 1989;58(5):923–931. doi: 10.1016/0092-8674(89)90944-6. 2476237 [DOI] [PubMed] [Google Scholar]
- 109.Rogers R.J., Chesrown S.E., Kuo S., Monnier J.M., Nick H.S. Cytokine-inducible enhancer with promoter activity in both the rat and human manganese-superoxide dismutase genes. Biochemical Journal. 2000;347(1):233–242. 10727424 [PMC free article] [PubMed] [Google Scholar]
- 110.Huang Y., He T., Domann F.E. Decreased expression of manganese superoxide dismutase in transformed cells is associated with increased cytosine methylation of the SOD2 gene. DNA Cell Biology. 1999;18(8):643–652. doi: 10.1089/104454999315051. 10463060 [DOI] [PubMed] [Google Scholar]
- 111.Hurt E.M., Thomas S.B., Peng B., Farrar W.L. Molecular consequences of SOD2 expression in epigenetically silenced pancreatic carcinoma cell lines. British Journal of Cancer. 2007;97(8):1116–1123. doi: 10.1038/sj.bjc.6604000. 17895890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hurt E.M., Thomas S.B., Peng B., Farrar W.L. Integrated molecular profiling of SOD2 expression in multiple myeloma. Blood. 2007;109(9):3953–3962. doi: 10.1182/blood-2006-07-035162. 17192397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cho R.W., Clarke M.F. Recent advances in cancer stem cells. Current Opinion in Genetics & Development. 2008;18(1):48–53. doi: 10.1016/j.gde.2008.01.017. 18356041 [DOI] [PubMed] [Google Scholar]
- 114.Mimeault M., Batra S.K. Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer- and metastasis-initiating cells. Journal of Cellular and Molecular Medicine. 2013;17(1):30–54. doi: 10.1111/jcmm.12004. 23301832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhong H., Chiles K., Feldser D., Laughner E., Hanrahan C., Georgescu M.M. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Research. 2000;60(6):1541–1545. 10749120 [PubMed] [Google Scholar]
- 116.Stoeltzing O., Liu W., Reinmuth N., Fan F., Parikh A.A., Bucana C.D. Regulation of hypoxia-inducible factor-1alpha, vascular endothelial growth factor, and angiogenesis by an insulin-like growth factor-I receptor autocrine loop in human pancreatic cancer. American Journal of Pathology. 2003;163(3):1001–1011. doi: 10.1016/s0002-9440(10)63460-8. 12937141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wenger R.H. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB Journal. 2002;16(10):1151–1162. doi: 10.1096/fj.01-0944rev. 12153983 [DOI] [PubMed] [Google Scholar]
- 118.Quail D.F., Taylor M.J., Postovit L.M. Microenvironmental regulation of cancer stem cell phenotypes. Current Stem Cell Research & Therapy. 2012;7(3):197–216. doi: 10.2174/157488812799859838. 22329582 [DOI] [PubMed] [Google Scholar]
- 119.Diehn M., Cho R.W., Lobo N.A., Kalisky T., Dorie M.J., Kulp A.N. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458(7239):780–783. doi: 10.1038/nature07733. 19194462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tsujimoto Y., Croce C.M. Analysis of the structure, transcripts, and protein products of bcl-2, the gene involved in human follicular lymphoma. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(14):5214–5218. doi: 10.1073/pnas.83.14.5214. 3523487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Korsmeyer S.J. Bcl-2 initiates a new category of oncogenes: regulators of cell death. Blood. 1992;80(4):879–886. 1498330 [PubMed] [Google Scholar]
- 122.Hockenbery D.M., Oltvai Z.N., Yin X.M., Milliman C.L., Korsmeyer S.J. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75(2):241–251. doi: 10.1016/0092-8674(93)80066-n. 7503812 [DOI] [PubMed] [Google Scholar]
- 123.Kane D.J., Sarafian T.A., Anton R., Hahn H., Gralla E.B., Valentine J.S. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science. 1993;262(5137):1274–1277. doi: 10.1126/science.8235659. 8235659 [DOI] [PubMed] [Google Scholar]
- 124.Veis D.J., Sorenson C.M., Shutter J.R., Korsmeyer S.J. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell. 1993;75(2):229–240. doi: 10.1016/0092-8674(93)80065-m. 8402909 [DOI] [PubMed] [Google Scholar]
- 125.Esposti M.D., Hatzinisiriou I., McLennan H., Ralph S. Bcl-2 and mitochondrial oxygen radicals. New approaches with reactive oxygen species-sensitive probes. Journal of Biological Chemistry. 1999;274(42):29831–29837. doi: 10.1074/jbc.274.42.29831. 10514462 [DOI] [PubMed] [Google Scholar]
- 126.Kowaltowski A.J., Fiskum G. Redox mechanisms of cytoprotection by Bcl-2. Antioxidants & Redox Signaling. 2005;7(3–4):508–514. doi: 10.1089/ars.2005.7.508. 15706098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lee M., Hyun D.H., Marshall K.A., Ellerby L.M., Bredesen D.E., Jenner P. Effect of overexpression of BCL-2 on cellular oxidative damage, nitric oxide production, antioxidant defenses, and the proteasome. Free Radical Biology and Medicine. 2001;31(12):1550–1559. doi: 10.1016/s0891-5849(01)00633-5. 11744329 [DOI] [PubMed] [Google Scholar]
- 128.Cory S., Huang D.C., Adams J.M. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene. 2003;22(53):8590–8607. doi: 10.1038/sj.onc.1207102. 14634621 [DOI] [PubMed] [Google Scholar]
- 129.Akao Y., Otsuki Y., Kataoka S., Ito Y., Tsujimoto Y. Multiple subcellular localization of bcl-2: detection in nuclear outer membrane, endoplasmic reticulum membrane, and mitochondrial membranes. Cancer Research. 1994;54(9):2468–2471. 8162596 [PubMed] [Google Scholar]
- 130.Yang J., Liu X., Bhalla K., Kim C.N., Ibrado A.M., Cai J. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275(5303):1129–1132. doi: 10.1126/science.275.5303.1129. 9027314 [DOI] [PubMed] [Google Scholar]
- 131.Green D.R., Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305(5684):626–629. doi: 10.1126/science.1099320. 15286356 [DOI] [PubMed] [Google Scholar]
- 132.Kroemer G., Galluzzi L., Brenner C. Mitochondrial membrane permeabilization in cell death. Physiological Reviews. 2007;87(1):99–163. doi: 10.1152/physrev.00013.2006. 17237344 [DOI] [PubMed] [Google Scholar]
- 133.Llambi F., Moldoveanu T., Tait S.W., Bouchier-Hayes L., Temirov J., McCormick L.L. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Molecular Cell. 2011;44(4):517–531. doi: 10.1016/j.molcel.2011.10.001. 22036586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Li J., Yuan J. Caspases in apoptosis and beyond. Oncogene. 2008;27(48):6194–6206. doi: 10.1038/onc.2008.297. 18931687 [DOI] [PubMed] [Google Scholar]
- 135.Chen Z.X., Pervaiz S. Bcl-2 induces pro-oxidant state by engaging mitochondrial respiration in tumor cells. Cell Death & Differentiation. 2007;14(9):1617–1627. doi: 10.1038/sj.cdd.4402165. 17510660 [DOI] [PubMed] [Google Scholar]
- 136.Chen Z.X., Pervaiz S. Involvement of cytochrome c oxidase subunits Va and Vb in the regulation of cancer cell metabolism by Bcl-2. Cell Death & Differentiation. 2010;17(3):408–420. doi: 10.1038/cdd.2009.132. 19834492 [DOI] [PubMed] [Google Scholar]
- 137.Krishna S., Low I.C., Pervaiz S. Regulation of mitochondrial metabolism: yet another facet in the biology of the oncoprotein Bcl-2. Biochemical Journal. 2011;435(3):545–551. doi: 10.1042/BJ20101996. 21486225 [DOI] [PubMed] [Google Scholar]
- 138.Sarosiek K.A., Ni Chonghaile T., Letai A. Mitochondria: gatekeepers of response to chemotherapy. Trends in Cell Biology. 2013;23(12):612–619. doi: 10.1016/j.tcb.2013.08.003. 24060597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ni Chonghaile T., Sarosiek K.A., Vo T.T., Ryan J.A., Tammareddi A., Moore V.del G. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334(6059):1129–1133. doi: 10.1126/science.1206727. 22033517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cantor J.R., Sabatini D.M. Cancer cell metabolism: one hallmark, many faces. Cancer Discovery. 2012;2(10):881–898. doi: 10.1158/2159-8290.CD-12-0345. 23009760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.DeBerardinis R.J., Mancuso A., Daikhin E., Nissim I., Yudkoff M., Wehrli S. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(49):19345–19350. doi: 10.1073/pnas.0709747104. 18032601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ward P.S., Thompson C.B. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell. 2012;21(3):297–308. doi: 10.1016/j.ccr.2012.02.014. 22439925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Vander Heiden M.G., Cantley L.C., Thompson C.B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi: 10.1126/science.1160809. 19460998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wise D.R., DeBerardinis R.J., Mancuso A., Sayed N., Zhang X.Y., Pfeiffer H.K. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(48):18782–18787. doi: 10.1073/pnas.0810199105. 19033189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gao P., Tchernyshyov I., Chang T.C., Lee Y.S., Kita K., Ochi T. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458(7239):762–765. doi: 10.1038/nature07823. 19219026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Schwartzenberg-Bar-Yoseph F., Armoni M., Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Research. 2004;64(7):2627–2633. doi: 10.1158/0008-5472.can-03-0846. 15059920 [DOI] [PubMed] [Google Scholar]
- 147.Jiang P., Du W., Wang X., Mancuso A., Gao X., Wu M. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nature Cell Biology. 2011;13(3):310–316. doi: 10.1038/ncb2172. 21336310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cifone M.A., Fidler I.J. Correlation of patterns of anchorage-independent growth with in vivo behavior of cells from a murine fibrosarcoma. Proceedings of the National Academy of Sciences of the United States of America. 1980;77(2):1039–1043. doi: 10.1073/pnas.77.2.1039. 6928659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Weinberg F., Hamanaka R., Wheaton W.W., Weinberg S., Joseph J., Lopez M. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(19):8788–8793. doi: 10.1073/pnas.1003428107. 20421486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mori S., Chang J.T., Andrechek E.R., Matsumura N., Baba T., Yao G. Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene. 2009;28(31):2796–2805. doi: 10.1038/onc.2009.139. 19483725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ishikawa K., Takenaga K., Akimoto M., Koshikawa N., Yamaguchi A., Imanishi H. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320(5876):661–664. doi: 10.1126/science.1156906. 18388260 [DOI] [PubMed] [Google Scholar]
- 152.Hanahan D., Weinberg R.A. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. 10647931 [DOI] [PubMed] [Google Scholar]
- 153.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. 21376230 [DOI] [PubMed] [Google Scholar]
- 154.Uemura N., Okamoto S., Yamamoto S., Matsumura N., Yamaguchi S., Yamakido M. Helicobacter pylori infection and the development of gastric cancer. New England Journal of Medicine. 2001;345(11):784–789. doi: 10.1056/NEJMoa001999. 11556297 [DOI] [PubMed] [Google Scholar]
- 155.Askling J., Dickman P.W., Karlén P., Broström O., Lapidus A., Löfberg R. Colorectal cancer rates among first-degree relatives of patients with inflammatory bowel disease: a population-based cohort study. Lancet. 2001;357(9252):262–266. doi: 10.1016/S0140-6736(00)03612-6. 11214128 [DOI] [PubMed] [Google Scholar]
- 156.El-Serag H.B., Rudolph K.L. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–2576. doi: 10.1053/j.gastro.2007.04.061. 17570226 [DOI] [PubMed] [Google Scholar]
- 157.Grivennikov S.I., Greten F.R., Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–899. doi: 10.1016/j.cell.2010.01.025. 20303878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Smith M.L., Fornace A.J., Jr. Genomic instability and the role of p53 mutations in cancer cells. Current Opinion in Oncology. 1995;7(1):69–75. 7696366 [PubMed] [Google Scholar]
- 159.Guo Z., Kozlov S., Lavin M.F., Person M.D., Paull T.T. ATM activation by oxidative stress. Science. 2010;330(6003):517–521. doi: 10.1126/science.1192912. 20966255 [DOI] [PubMed] [Google Scholar]
- 160.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nature Reviews Cancer. 2003;3(3):155–168. doi: 10.1038/nrc1011. 12612651 [DOI] [PubMed] [Google Scholar]
- 161.Minotti G., Menna P., Salvatorelli E., Cairo G., Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacological Reviews. 2004;56(2):185–229. doi: 10.1124/pr.56.2.6. 15169927 [DOI] [PubMed] [Google Scholar]
- 162.Doroshow J.H. Anthracycline antibiotic-stimulated superoxide, hydrogen peroxide, and hydroxyl radical production by NADH dehydrogenase. Cancer Research. 1983;43(10):4543–4551. 6309369 [PubMed] [Google Scholar]
- 163.Gajewski E., Gaur S., Akman S.A., Matsumoto L., van Balgooy J.N., Doroshow J.H. Oxidative DNA base damage in MCF-10A breast epithelial cells at clinically achievable concentrations of doxorubicin. Biochem. Pharmacol. 2007;73(12):1947–1956. doi: 10.1016/j.bcp.2007.03.022. 17445777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wouters K.A., Kremer L.C., Miller T.L., Herman E.H., Lipshultz S.E. Protecting against anthracycline-induced myocardial damage: a review of the most promising strategies. British Journal of Haematology. 2005;131(5):561–578. doi: 10.1111/j.1365-2141.2005.05759.x. 16351632 [DOI] [PubMed] [Google Scholar]
- 165.Gewirtz D.A. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochemical Pharmacology. 1999;57(7):727–741. doi: 10.1016/s0006-2952(98)00307-4. 10075079 [DOI] [PubMed] [Google Scholar]
- 166.Wondrak G.T. Redox-directed cancer therapeutics: molecular mechanisms and opportunities. Antioxidants & Redox Signaling. 2009;11(12):3013–3069. doi: 10.1089/ars.2009.2541. 19496700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gorrini C., Harris I.S., Mak T.W. Modulation of oxidative stress as an anticancer strategy. Nature Reviews Drug Discovery. 2013;12(12):931–947. doi: 10.1038/nrd4002. 24287781 [DOI] [PubMed] [Google Scholar]
- 168.Huang P., Feng L., Oldham E.A., Keating M.J., Plunkett W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature. 2000;407(6802):390–395. doi: 10.1038/35030140. 11014196 [DOI] [PubMed] [Google Scholar]
- 169.Lee K., Briehl M.M., Mazar A.P., Batinic-Haberle I., Reboucas J.S., Glinsmann-Gibson B. The copper chelator ATN-224 induces peroxynitrite-dependent cell death in hematological malignancies. Free Radical Biology and Medicine. 2013;60:157–167. doi: 10.1016/j.freeradbiomed.2013.02.003. 23416365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Glasauer A., Sena L.A., Diebold L.P., Mazar A.P., Chandel N.S. Targeting SOD1 reduces experimental non–small-cell lung cancer. Journal of Clinical Investigation. 2014;124(1):117–128. doi: 10.1172/JCI71714. 24292713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Montero A.J., Jassem J. Cellular redox pathways as a therapeutic target in the treatment of cancer. Drugs. 2011;71(11):1385–1396. doi: 10.2165/11592590-000000000-00000. 21812504 [DOI] [PubMed] [Google Scholar]
- 172.Barr P.M., Miller T.P., Friedberg J.W., Peterson D.R., Baran A.M., Herr M. Phase 2 study of imexon, a prooxidant molecule, in relapsed and refractory B-cell non-Hodgkin lymphoma. Blood. 2014;124(8):1259–1265. doi: 10.1182/blood-2014-04-570044. 25016003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hayes J.D., McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends in Biochemical Sciences. 2009;34(4):176–188. doi: 10.1016/j.tibs.2008.12.008. 19321346 [DOI] [PubMed] [Google Scholar]
- 174.Jaramillo M.C., Zhang D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes & Development. 2013;27(20):2179–2191. doi: 10.1101/gad.225680.113. 24142871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Letai A. BCL-2: found bound and drugged! Trends in Molecular Medicine. 2005;11(10):442–444. doi: 10.1016/j.molmed.2005.08.007. 16150641 [DOI] [PubMed] [Google Scholar]
- 176.Schumacker P.T. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell. 2006;10(3):175–176. doi: 10.1016/j.ccr.2006.08.015. 16959608 [DOI] [PubMed] [Google Scholar]
- 177.Jones D.P. Redefining oxidative stress. Antioxidants & Redox Signaling. 2006;8(9–10):1865–1879. doi: 10.1089/ars.2006.8.1865. 16987039 [DOI] [PubMed] [Google Scholar]
- 178.Jones D.P. Radical-free biology of oxidative stress. American Journal of Physiology - Cell Physiology. 2008;295(4):C849–C868. doi: 10.1152/ajpcell.00283.2008. 18684987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Trachootham D., Alexandre J., Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nature Reviews Drug Discovery. 2009;8(7):579–591. doi: 10.1038/nrd2803. 19478820 [DOI] [PubMed] [Google Scholar]
- 180.Finkel T. Signal transduction by reactive oxygen species. Journal of Cell Biology. 2011;194(1):7–15. doi: 10.1083/jcb.201102095. 21746850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Dickinson B.C., Chang C.J. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nature Chemical Biology. 2011;7(8):504–511. doi: 10.1038/nchembio.607. 21769097 [DOI] [PMC free article] [PubMed] [Google Scholar]