

Abstract
The recent advances in high throughput sequencing technology accelerate possible ways for the study of genome wide variation in several organisms and associated consequences. In the present study, mutations in TGFBR3 showing significant association with FCR trait in chicken during exome sequencing were further analyzed. Out of four SNPs, one nsSNP p.Val451Leu was found in the coding region of TGFBR3. In silico tools such as SnpSift and PANTHER predicted it as deleterious (0.04) and to be tolerated, respectively, while I-Mutant revealed that protein stability decreased. The TGFBR3 I-TASSER model has a C-score of 0.85, which was validated using PROCHECK. Based on MD simulation, mutant protein structure deviated from native with RMSD 0.08 Å due to change in the H-bonding distances of mutant residue. The docking of TGFBR3 with interacting TGFBR2 inferred that mutant required more global energy. Therefore, the present study will provide useful information about functional SNPs that have an impact on FCR traits.
Abbreviations: FCR, feed conversion ratio; MD, molecular dynamics; SIFT, sorting intolerant from tolerant; SNP, single nucleotide polymorphism; nsSNPs, non-synonymous single nucleotide polymorphisms; AASs, amino acid substitutions; TGFB, transforming growth factor beta; I-TASSER, iterative threading assembly refinement; PANTHER, protein analysis through evolutionary relationships; RMSD, root mean square deviation; RMSF, root mean square fluctuation; UTR, un-translated region
Keywords: Non-synonymous SNP, TGFBR3, Modeling, Feed conversion ratio (FCR), Chicken
Highlights
-
•
Investigated functional nsSNP p.Val451Leu (rs312979494) in feed conversion ratio (FCR) associated TGFBR3 of chicken
-
•
Computational tools (SIFT and I-Mutant 2.0) predicted that this nsSNP was deleterious.
-
•
Mutant structure of TGFBR3 showed high energies and RMS deviations compared to native using MD simulation.
-
•
Molecular docking of TGFBR3 with interacting protein TGFBR2 showed an increase in global energy of mutant compared to native.
-
•
We have predicted that functional SNP has an impact on TGFBR3 of chicken and thus can be treated as candidate SNP in screening.
Introduction
TGFβ is a superfamily protein consisting of three isoforms (TGF-β1, TGF-β2 and TGF-β3) and responsible for different biological processes like growth, development, tissue homeostasis, adhesion, apoptosis and regulation of the immune system (Kubiczkova et al., 2012, Pardali and Ten Dijke, 2012). TGF-β signaling involves three cell surface receptor proteins like TGFBR1, TGFBR2 and TGFBR3 (Cheifetz et al., 1986, Cheifetz et al., 1987). Among that, transforming growth factor beta receptor type 3 (TGFBR3) also known as betaglycan, is the largest (250–350 kDa) and most abundant binding molecule using its heparan sulfate chains (Aleman-Muench et al., 2012, Cheifetz et al., 1988, Sarraj et al., 2010). TGFBR3 binds with all three isoforms of TGF-β, but with higher affinity with TGF-β2 and it acts as a reservoir of ligand for TGF-beta receptor.
The TGFBR2 and TGFBR3 receptors were both co-expressed during embryogenesis in the developing heart, lung and eye of chick (Barnett et al., 1994). They demonstrated that the binding profile of TGFBR3 was identical to the rat receptor and contained a single transmembrane domain and highly conserved short cytoplasmic tail. The TGFBR3 consists of 841 amino acid residues with 260 amino acids of the zona pellucida (ZP) domain (Ganguly et al., 2010). ZP domain contains eight strictly conserved cysteines, which form disulfide bridges and most ZP domain proteins are synthesized as precursors with carboxy-terminal transmembrane domains (Bork and Sander, 1992, Jovine et al., 2002).
It is assumed that any alteration in well-defined structural conformation may affect the functional activity of the gene. The most frequent genomic variation is exhibited as SNPs and there is a strong correlation between variation and certain traits. The advent in high throughput sequencing technologies opens up chances for a wide range of SNP studies and increasing number of genetic variations (Alanazi et al., 2011, Mohamoud et al., 2014). Among the different types of SNPs, nsSNPs found in the coding region of gene may likely affect the resulting protein structure and function with neutral or deleterious impacts (Kumar et al., 2009, Ng and Henikoff, 2006). A high computational approach such as SIFT, PolyPhen, PANTHER, I-Mutant and SNPs3D is useful for predicting the effect of nsSNPs on protein (Capriotti et al., 2005, Johnson et al., 2005, Rajith and Doss, 2011). Among them, I-Mutant predicted most reliably (Capriotti et al., 2005). These SNPs which are associated with certain traits can also be used as genetic markers (George Priya Doss and Rajith, 2012).
The previous reports showed the impact of AASs in various genes such as BRCA1 (Rajasekaran et al., 2007), IGF1R (de Alencar and Lopes, 2010), ATM (George Priya Doss and Rajith, 2012), HBB (Alanazi et al., 2011), BRAF (Hussain et al., 2012), TYRP1 (Kamaraj and Purohit, 2013) and GalNAc-T1 (Mohamoud et al., 2014). It is very important to evaluate the effect of a single base mutation that may disrupt the structural conformation of protein. In this study, we have analyzed SNPs from exome sequencing data of FCR associated genes of chicken. We identified a critical nsSNP in the TGFBR3 gene. Therefore, we studied the stability of TGFBR3 protein providing with change in residue in terms of free energy using I-Mutant. Subsequently, computational tools were used to predict the protein 3D structure from the sequence. The main advantage of this computational study is that it could be helpful to understand phenotype and genotype associations with respect to FCR traits in chicken.
Materials and methods
Collection of the TGFBR3 SNP dataset
Pure-line chicks developed by Marshall Breeders Pvt. Ltd., Nasik, Maharastra, India were used for feeding trials to access feed conversion efficiency. Thirty male and 300 female birds were mated (each male allotted ten females to produce ten progenies by each pair) to produce 3000 progenies. For the FCR trial, male chickens were weighed and transferred in individual feeding pens at the age of 35 days. All birds had ad libitum access to a broiler grower diet. Chickens continued to have free access to food and water prior to and during the experimental period. At the age of 49 days, birds were weighed individually and their feed intake was recorded. Feed consumed (grams) per gram of weight gain (difference of weight at day 49 and weight at day 35) was taken as feed conversion ratio. Seven sibling pairs were selected such that one sibling in the pair has low and the other has a high FCR (Supplementary Table 1). We sequenced the exome of each of these 14 birds individually and identified single nucleotide variants. Based on exome sequencing results, the non-synonymous SNP rs312979494 (c.1351G > C) on the TGFBR3 gene was found to be associated with FCR (unpublished data).
Assessment of the functional impact of deleterious nsSNPs
The functional impact of this nsSNP on the TGFBR3 gene was detected using SIFT (http://snpeff.sourceforge.net/SnpSift.html). The SIFT program predicted deleterious or non-tolerated SNPs on the premise that some amino acids tend to be conserved in a protein family and any substitution at these positions would affect protein function and thus have a phenotypic effect. SIFT is a sequence based homology tool which calculates the SIFT score or tolerance index (TI) score for each alteration of amino acids. The SIFT algorithm predicts the amino acid substitutions with the cut-off value 0.05, scores ≤ 0.05 are predicted to be deleterious, while scores > 0.05 are assumed to be tolerated (Ng and Henikoff, 2003). In silico functional analysis of missense mutation identified in the TGFBR3 gene was also performed using PANTHER (www.pantherdb.org/tools/csnp). PANTHER estimates the likelihood of a particular non-synonymous (AA changing) coding SNP to cause a functional effect on the protein. It calculates the subPSEC score (substitution position-specific evolutionary conservation) based on an alignment of evolutionary associated proteins (Thomas and Kejariwal, 2004, Thomas et al., 1999). The Pdeleterious estimates the probability that a given substitution will cause a deleterious effect on protein function or not, where subPSEC score ≤ − 3 classifies AASs as deleterious and score ≥ − 3 predicted as neutral substitution. Further, I-Mutant 2.0 — a support vector machine based web server was used for the automatic prediction of protein stability changes upon single-site mutations (http://folding.biofold.org/cgi-bin/i-mutant2.0). The FASTA sequence of the protein along with the residue change was provided as the input. It predicts the protein stability based on the difference between the DDG value (kcal/mol) of the unfolding Gibbs free energy value of the mutated protein and the unfolding Gibbs free energy value of the native protein (Capriotti et al., 2005, Capriotti et al., 2008).
Modeling of TGFBR3 and molecular visualization
Evaluation of the structural stability between the native and mutant proteins was performed based on the availability of a 3D structure of a protein in the PDB database. In the case of TGFBR3, a 3D crystallographic structure was not available in PDB. Hence to obtain a 3D structure of TGFBR3, the amino acid sequence in FASTA format was submitted to the I-TASSER server (http://zhanglab.ccmb.med.umich.edu/I-TASSER/). It is based on multiple threading alignments and iterative structural assembly simulations. The prediction of the accuracy of the model depends upon a confidence score (C-score) based on the quality of the threading alignments and structural assembly refinement simulations (Roy et al., 2010). The structure model identified by I-TASSER is subjected to structure validation using PROCHECK using the SAVES server (http://nihserver.mbi.ucla.edu/SAVES/). Later the mutant 3D structure of TGFBR3 was generated using a PyMOL tool (http://www.pymol.org/).
The best 3D model was selected according to rank based on the C-score, TM score and RMSD value. The 3D structure of TGFBR3 was visualized by PyMOL (http://www.pymol.org/) and UCSF CHIMERA (https://www.cgl.ucsf.edu/chimera/). The H-bonding interaction was studied using a PyMOL visualization tool.
Molecular dynamics simulation for exploring dynamic behavior of TGFBR3
The molecular dynamics simulations were undertaken to study the mutant that was predicted to destabilize TGFBR3 conformation in our case. Both native and mutant 3D structures of TGFBR3 were used for the molecular dynamics simulation. The 3D structure of native TGFBR3 was used as a control for analyzing the impact of mutations. Afterwards, all molecular dynamics simulations for this mutant were performed by GROMACS v4.6.5 with OPLS as the force field. Both mutant and native models were solvated in a dodecahedron box with SPC216 (single point charge) water molecules at marginal radius 10 Å. The energy minimization was carried out for 5000 iterations of a tolerance of 100 kJ/mol by the steepest descent algorithm. To avoid unnecessary distortion of the protein structure, we performed (10,000 steps) a 20-ps equilibration run where all the heavy atoms were restrained to their starting position, and water to equilibrate around the protein. Emtol convergence criterion was set up to 1000 kcal/mol. The coulomb interactions truncated at 1.0 nm. The resulted trajectory files were analyzed by using GROMACS packages for differentiating RMSD and RMSF between the native and mutant structures of TGFBR3. The graphs were constructed using XMGrace on Linux platform itself.
Gene ontology and protein–protein interaction study and molecular docking
Protein–protein interaction networks are important to reveal and annotate all functional interactions among cell proteins. For this study, the online database resource STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) was used (http://string-db.org). The experimental and theoretical interaction networks of TGFBR3 were provided with unique coverage and ease of access by this tool. Different gene ontology terms of TGFBR3 were accessed using the online AmiGO 2 GO server (http://amigo.geneontology.org). The PatchDock is based on surface patch matching and more reliable docking tools with fast search along with filtering and scoring. It uses advanced data structures and a spatial search pattern. The TGFBR3 protein and other interacting proteins detected by the STRING network were used as input for docking with default RMSD 4 Å. It resulted into several structures, thus further filtered through FireDock to get a refined result. The docking score global energy and atomic contact energy (ACE) of the native and mutant complexes were analyzed.
Results
A total of four SNPs in the TGFBR3 gene were found to be associated with the FCR trait, but only one of them was nsSNP while other three were intronic. The homozygous C genotype for the SNP in TGFBR3 (c.1351G > C variant) was shown to be associated with Low FCR in the exome sequencing study.
Predictions of functional impact of SNPs
We have detected a nsSNP (p.Val451Leu) in the ZP domain region of TGFBR3 of chicken (Gallus gallus) (ENSGALG00000006038). The non-synonymous SNP rs312979494 (c.1351G > C) was found to cause an alteration at amino acid 451 (p.Val451Leu) of the TGFBR3. Firstly, the conversion levels (in terms of tolerance index — TI) of the coding nsSNP in TGFBR3 were determined using the sequence homology based tool SIFT. As a result, this nsSNP was found to be deleterious (TI score 0.04). The mutation possessed a replacement of one branched chain amino acid (Val) with another one (Leu).
Analysis of deleterious nsSNPs
PANTHER characterizes likely functional effect of amino acid variation by means of HMM-based statistical modeling and evolutionary relationship. We performed PANTHER analysis of TGFBR3 nsSNP in order to add another layer of refinement in SNP characterization. The non-synonymous SNP rs312979494 (c.1351G > C) possessed the subPSEC score less than − 3 and is therefore classified as deleterious.
Prediction of stability change on mutation of nsSNP
The main aim of this study was to identify the coding nsSNP that would be expected to disrupt the native structure of the protein and thus affecting the function of TGFBR3. We found the protein stability of nsSNP upon mutation in terms of free energy using the I-Mutant 2.0 web server. There was only one mutant Val451Leu (rs312979494), which was predicted to be unstable. The mutation from Valine to Leucine at position 451 was found to be damaging because it exhibited the lowest free energy − 1.12 kcal/mol at pH 7, temperature 27 °C.
Structure modeling of mutant protein and model quality analysis
I-TASSER modeled 3D-structure included residues ranging from 1 to 841 amino acids of TGFBR3. The ten templates were used for modeling the TGFBR3 (Table 1). The top most templates covered 98% of the protein query sequence. The top model having C-score 0.85 was selected with estimated TM-score value 0.61 ± 0.14 and RMSD value 10.5 ± 4.6 Å (Fig. 1A). This substitution showed that the RMSD value was greater than 2.0 Å and referred as highly deleterious nsSNP (Fig. 1B). The SwissPDBViewer analysis of both structures of TGFBR3 showed 0.08 Å RMS deviations in mutant structure compared to native. The PROCHECK represented the Ramachandran plot of mutant protein (Fig. 2A). The plot revealed that amino acid residues distributed over 76% in the most favored region, 17.2% in additional allowed regions, 3.9% in generously allowed regions and 2.8% in disallowed regions. The bad contacts (6) also increased in mutant compared to native (5). We have used SwissPDBViewer for analyzing hydrogen bonding and distances between atoms of native or mutant residue and atoms of vicinal residues. We observed that native residue Val451, atom CG2 formed bond with Ile445 of O atom with distance 2.7 Å, while mutated residue Leu451 of CD2 formed bond with Ile451 of O atom with distance 3.1 Å (Fig. 2B). We also studied the clashes of mutated residue using a PyMOL tool and found out that these residues would also interact with vicinal residues Val454 (Fig. 1B).
Table 1.
Top 10 templates used by I-TASSER for modeling TGFBR3 protein.
| PDB hit | Ident1 | Ident2 | Coverage | Normal Z-score |
|---|---|---|---|---|
| 4acqA | 0.11 | 0.18 | 0.98 | 1.41 |
| 3nk3A | 0.16 | 0.09 | 0.32 | 1.02 |
| 4a21A | 0.08 | 0.18 | 0.85 | 2.52 |
| 3nk3A | 0.16 | 0.09 | 0.32 | 2.30 |
| 3nk3A | 0.08 | 0.09 | 0.32 | 1.31 |
| 4acqA | 0.08 | 0.18 | 0.94 | 1.38 |
| 3nk3A | 0.17 | 0.09 | 0.33 | 3.96 |
| 3nk4A | 0.15 | 0.09 | 0.31 | 6.66 |
| 3a0fA | 0.05 | 0.18 | 0.83 | 2.63 |
| 3nk3A | 0.16 | 0.09 | 0.32 | 3.54 |
Fig. 1.

Predicted TGFBR3 model. A) Predicted TGFBR3 models by I-TASSER (helix — green, strand — red and coil — yellow). B) Mutated residue Leu at 451 position in ball and socket view created by PyMoL.
Fig. 2.

A) Ramachandran plot of TGFBR3 mutant protein model. The most favored region, additional allowed regions, generously allowed regions and disallowed regions are indicated as red, yellow, light yellow and white respectively. B) H-bonding pattern and distances of (i) native Val451 and (ii) mutated residue Leu451 (yellow line indicates H-bonding distance in Å). The native residue Val451, atom CG2 formed bond with Ile445 of O atom with distance 2.7 Å, while mutated residue Leu451 of CD2 formed bond with Ile451 of O atom with distance 3.1 Å.
Analysis of MD simulation of native and mutant TGFBR3
To understand the structural differences arising due to mutations, we performed MD simulations for the native and mutant TGFBR3. For this purpose, the root mean square deviation (RMSD) of the protein backbone as a function of time was examined. We have calculated the energy difference between native and mutant structures before and after energy minimization. The before energy minimization the total energy of native and mutant structures was 65.442 kJ/mol and 49.986 kJ/mol respectively. After a 20-ps equilibration run the RMSD of native TGFBR3 was 2.8 Å, while mutant TGFBR3 had an RMSD greater than 3.06 Å, which illustrated that this mutation, might deform the structural conformation. The RMSD for all protein-H atoms from the initial structure was examined to study the convergence of the protein system (Fig. 3A). The high RMSD value indicated that there was an immense deviation between the native and mutant structures pointing towards alteration in the structural and functional stability. Further, this mutant had a total energy of − 11,299.13 kJ/mol compared to − 11,524.46 kJ/mol for the native structure. The higher the total energy, the less stable the protein structure predicted. Thus, the total energy comparison showed that mutant had higher energy and therefore is expected to be unstable. To assess the effect of mutation on the TGFBR3 structure, we also computed the RMSF (root mean square fluctuation) values. The RMSF profile of P-loop and A-loop indicated an increased flexibility for the mutant (Fig. 3B). This atypical transformation in secondary element and instability in terms of energy suggest that any substitution at position 451 might disturb the structural conformation. We thus inferred that the nsSNP (rs312979494) could affect the structure of the native TGFBR3 protein.
Fig. 3.
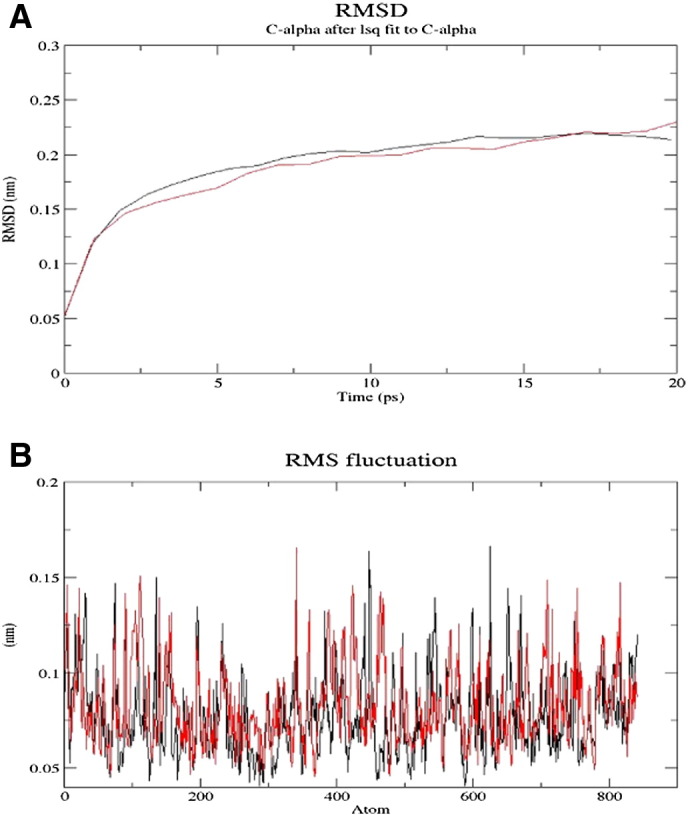
RMSD and RMSF of both models. A) RMSD value of native (black color) and mutant (red color) proteins of TGFBR3. B) RMSF of native and mutant proteins of TGFBR3 (red color indicates mutant protein and black color indicates native protein).
Analysis of gene ontology, pathway and protein–protein interaction and molecular docking of TGFBR3
Further we analyzed the gene ontology of this TGFBR3 gene and showed that TGFBR3 possessed molecular function such as TGF beta activated receptor activity, TGFBR type 2 binding, and TGF receptor binding and also contributed to protein binding and glycosaminoglycan binding (Supplementary Table 2).
The gene ontology showed that this gene involved about 35 biological processes such as blood vessel development, TGFBR signaling pathway, cell growth, and response to follicle-stimulating hormone, negative regulation of cellular component movement, pathway-restricted SMAD protein phosphorylation and positive regulation of gene expression (Supplementary Table 3). This TGFBR3 protein is involved in various pathways such as the CREB pathway, Apoptosis pathway, PAK pathway and apoptotic pathways in synovial fibroblasts.
The protein–protein interaction network of TGFBR3 (Fig. 4) was also analyzed, which revealed that TGFBR3 protein interacted with 10 proteins such as TGFB, INHA, BAMBI, CARD10, RUFY2, TGFB3, TGFBR1, GPR177, PLXND1 and TGFBR2 with scores 0.721, 0.651, 0.598, 0.581, 0.491, 0.485, 0.458, 0.457, 0.447 and 0.425 respectively (Table 2). In order to understand change in the protein interaction network, we have used PatchDock and FireDock. The docking was carried out between native TGFBR3 and TGFBR2, resulted into global energy, the binding energy of the solution and atomic contact energy (ACE) were 3.80 kJ/mol and − 5.28 kJ/mol, respectively, while mutant TGFBR3 and TGFBR2 showed global energy and ACE of 17.33 kJ/mol and 3.46 kJ/mol.
Fig. 4.
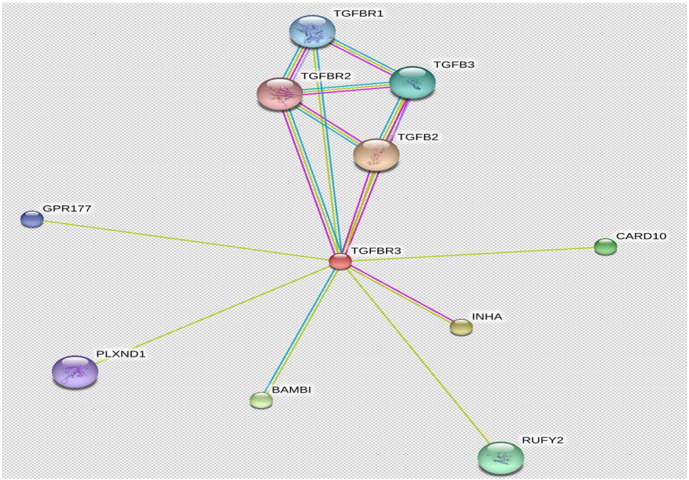
Protein–protein interaction network of TGFBR3 protein shown by STRING.
Table 2.
TGFBR3 functional partners depicted by STRING.
| Protein | Function | Score obtained |
|---|---|---|
| TGFB2 | Transforming growth factor beta-2 precursor (TGF-beta-2) | 0.721 |
| INHA | Inhibin alpha chain precursor | 0.651 |
| BAMBI | BMP and activin membrane-bound inhibitor homolog | 0.598 |
| CARD10 | caspase recruitment domain family, member 10 | 0.581 |
| RUFY2 | RUN and FYVE domain containing 2 | 0.491 |
| TGFB3 | Transforming growth factor beta-3 precursor (TGF-beta-3) | 0.485 |
| TGFBR1 | transforming growth factor, beta receptor I | 0.458 |
| GPR177 | Integral membrane protein GPR177 precursor | 0.457 |
| PLXND1 | Plexin D1 (1938 aa) | 0.447 |
| TGFBR2 | TGF-beta receptor type-2 precursor | 0.425 |
Discussion
In the present study, screening for functional genetic variant in the coding region of FCR associated TGFBR3 gene was performed using sequence and structure based algorithms such as SIFT, PANTHER, I-Mutant and I-TASSER. From that SNP analysis, SIFT predicted that nsSNP was of deleterious type, whereas PANTHER predicted that nsSNP was to be tolerated. The significant difference in outputs of SIFT and PANTHER algorithms is most likely due to the utilization of different protein sequence alignments, used to characterize the variants (de Alencar and Lopes, 2010). Every algorithm uses different sets of sequences and alignments which resulted into different outputs. The sequence based prediction analysis is more helpful over the structure based analysis, due to the fact that it reflects all types of impact on the protein. The 3D structure is very helpful for understanding the dynamic behavior of proteins. Several computational tools are available for exploring the impact of SNPs. However, variability in the prediction outputs of these tools reflects both advantages and disadvantages. Here, we have used computational tools for investigating the impact of nsSNPs on FCR associated genes of chicken. The nsSNPs which are present in the coding region of the gene may have neutral or deleterious impact (Ng and Henikoff, 2001, Ng and Henikoff, 2006).
Our results indicated that nsSNP at positions Val451Leu was found in the ZP domain of the highly conserved region of TGFBR3 and predicted to have a potential impact on its protein. The previous work showed that, mutation in the ZP domain of TGFBR3 affected its protein function (Jovine et al., 2002). The residues in the domain region are highly conserved and AASs in the ZP domain stop the inhibition and also TGF binding to this domain (Wiater et al., 2006). The SIFT calculation of Gibbs free energy provided an insight into the structural and functional impacts of AASs. The 3D models were constructed to visualize deviation among the mutant and native type TGFBR3 protein models. In Val451Leu, the required protein framework (hydrophobicity and H-bonding interactions) seems to be disturbed at this position due to side chain formations with vicinal residues. We couldn't observe a difference in the Ramachandran plot of both structures. But comparison of H-bonding pattern and distances of native and mutant residues with vicinal residues showed a considerable difference in their RMSD value. The RMSD was also observed to be about 0.008 Å in mutant compared to native. Due to a difference in total energy for native and mutant (carrying Val451Leu variant) models, the proper functioning of TGFBR3 can be disturbed. It was shown that a difference between two structures with respect to the RMSD value could affect the protein stability (George Priya Doss and Rajith, 2012, Han et al., 2006). The RMSD for all the C alpha atoms from the starting TGFBR3 structure was examined to study the convergence of both models of protein. The RMSF analysis showed that mutant protein fluctuated slightly more than native protein of TGFBR3. Finally, we confirmed that stability of TGFBR3 protein in chicken decreased due to amino acid substitutions using tools such as SIFT, I-Mutant 2.0 and I-TASSER.
Protein–protein interaction investigation is a broad way to understand the organization of the desired proteome. The functional network protein study will be helpful for drug discovery, and to understand metabolic pathways and predict or develop genotype–phenotype associations (Mohamoud et al., 2014, Wang and Moult, 2001, Wang et al., 2009). STRING maps have shown that TGFBR3 interacts with TGFB3, TGFBR2, TGFB2, TGFBR1, ACVR2A, INHA, ARR3, CARD10 and GPR177 (Fig. 4). From gene-ontology analysis, TGFBR3 has been involved in 35 different biological processes including the Apoptosis pathway, CREB pathway, and PAK pathway. Noticeably, TGFBR3 may inhibit TGFB signaling and decreased expression has been observed in various cancers. The docking results also inferred that protein–protein interaction between mutant TGFBR3 and TGFBR2 proteins has been disturbed due to mutation with an increase in global energy of mutant compared to native. Also we confirmed that RMS deviation of mutant Leu451 compared to native Val451 was 0.08 Å, and this reflected into an increase in bad contact in the mutant structure compared to native shown by the Ramachandran plot. The H-bonding distances were also disturbed in the case of the mutant residue compared to the native residue with vicinal residues between atoms. We have also observed increased expression of TGFBR3 in the ileum and cecum of High FCR birds whereas its expression was low in the liver and duodenum as compared to Low FCR birds (unpublished study, Supplementary Table 4).
In conclusion, our in silico study signifies the investigation that identified the functional SNP in TGFBR3 based on sequence and structure homology tools. Although, there was a remarkable difference in prediction of selected computational tools, our analysis validated that this nsSNP could be helpful in cellular biology of TGFBR3. We investigated the impact of the Val451Leu variant on TGFBR3 protein in the form of energy calculation and evolutionarily conserved residues. From potential SNP, the Val451Leu mutation is predicted to cause considerable change in the total energy and functional interaction behavior of TGFBR3 protein with TGFB protein. Altered TGFBR3 function due to genetic variations and mRNA expression might play a critical role in determining complex traits/diseases. The presence of nsSNP in the gene resulted in decreased protein stability. For compensating that higher expression of FCR associated TGFBR3 was detected during transcriptome sequencing. Moreover, the protein–protein interaction pathway of this TGFBR3 protein has helped us to understand the roles and associated proteins in various cellular pathways, also change in interaction was observed in mutant using molecular docking between TGFBR3 and TGFBR2. Native TGFBR3 is an accessory receptor member of the TGF-β superfamily, acting as a protector which inhibited both TGFBR1–TGFBR2 complex formation and TGF-β1 production then subsequently initiated their downstream events targeting the apoptosis related genes (Chu et al., 2011). So, it is possible that mutant TGFBR3 may not efficiently inhibit TGF-β1 production and make stable TGFBR1–TGFBR2 complex formation, which may lead to apoptosis. The analysis indicated that Val451Leu mutant of TGFBR3 could be used as candidate gene for SNP screening. Our results suggest that these novel mutants have a potential functional impact and can thus be used for poultry breeding program based on genomic selection.
Acknowledgment
This study was supported by Department of Biotechnology (BT/PR15360/AAQ/01/466/2011), Ministry of Science and Technology (DBT), Government of India, New Delhi, India. We also thank the Central Institute of Freshwater Aquaculture, Bhubaneswar and Indian Council of Agricultural Research (ICAR), New Delhi for providing deputation to KDR.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.mgene.2015.03.006.
Appendix A. Supplementary data
Supplementary material.
References
- Alanazi M., Abduljaleel Z., Khan W., Warsy A.S., Elrobh M., Khan Z., Al Amri A., Bazzi M.D. In silico analysis of single nucleotide polymorphism (SNPs) in human beta-globin gene. PLoS One. 2011;6:e25876. doi: 10.1371/journal.pone.0025876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleman-Muench G.R., Mendoza V., Stenvers K., Garcia-Zepeda E.A., Lopez-Casillas F., Raman C., Soldevila G. Betaglycan (TbetaRIII) is expressed in the thymus and regulates T cell development by protecting thymocytes from apoptosis. PLoS One. 2012;7:e44217. doi: 10.1371/journal.pone.0044217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett J.V., Moustakas A., Lin W., Wang X.F., Lin H.Y., Galper J.B., Maas R.L. Cloning and developmental expression of the chick type II and type III TGF beta receptors. Dev. Dyn. 1994;199:12–27. doi: 10.1002/aja.1001990103. [DOI] [PubMed] [Google Scholar]
- Bork P., Sander C. A large domain common to sperm receptors (Zp2 and Zp3) and TGF-beta type III receptor. FEBS Lett. 1992;300:237–240. doi: 10.1016/0014-5793(92)80853-9. [DOI] [PubMed] [Google Scholar]
- Capriotti E., Fariselli P., Casadio R. I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005;33:W306–W310. doi: 10.1093/nar/gki375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capriotti E., Fariselli P., Rossi I., Casadio R. A three-state prediction of single point mutations on protein stability changes. BMC Bioinf. 2008;9(Suppl. 2):S6. doi: 10.1186/1471-2105-9-S2-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheifetz S., Like B., Massague J. Cellular distribution of type I and type II receptors for transforming growth factor-beta. J. Biol. Chem. 1986;261:9972–9978. [PubMed] [Google Scholar]
- Cheifetz S., Weatherbee J.A., Tsang M.L., Anderson J.K., Mole J.E., Lucas R., Massague J. The transforming growth factor-beta system, a complex pattern of cross-reactive ligands and receptors. Cell. 1987;48:409–415. doi: 10.1016/0092-8674(87)90192-9. [DOI] [PubMed] [Google Scholar]
- Cheifetz S., Andres J.L., Massague J. The transforming growth factor-beta receptor type III is a membrane proteoglycan. Domain structure of the receptor. J. Biol. Chem. 1988;263:16984–16991. [PubMed] [Google Scholar]
- Chu W., Li X., Li C., Wan L., Shi H., Song X., Liu X., Chen X., Zhang C., Shan H., Lu Y., Yang B. TGFBR3, a potential negative regulator of TGF-beta signaling, protects cardiac fibroblasts from hypoxia-induced apoptosis. J. Cell. Physiol. 2011;226:2586–2594. doi: 10.1002/jcp.22604. [DOI] [PubMed] [Google Scholar]
- de Alencar S.A., Lopes J.C. A comprehensive in silico analysis of the functional and structural impact of SNPs in the IGF1R gene. J. Biomed. Biotechnol. 2010;2010:715139. doi: 10.1155/2010/715139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly A., Bansal P., Gupta T., Gupta S.K. ‘ZP domain’ of human zona pellucida glycoprotein-1 binds to human spermatozoa and induces acrosomal exocytosis. Reprod. Biol. Endocrinol. 2010;8:110. doi: 10.1186/1477-7827-8-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George Priya Doss C., Rajith B. Computational refinement of functional single nucleotide polymorphisms associated with ATM gene. PLoS One. 2012;7:e34573. doi: 10.1371/journal.pone.0034573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J.H., Kerrison N., Chothia C., Teichmann S.A. Divergence of interdomain geometry in two-domain proteins. Structure. 2006;14:935–945. doi: 10.1016/j.str.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Hussain M.R., Shaik N.A., Al-Aama J.Y., Asfour H.Z., Khan F.S., Masoodi T.A., Khan M.A., Shaik N.S. In silico analysis of Single Nucleotide Polymorphisms (SNPs) in human BRAF gene. Gene. 2012;508:188–196. doi: 10.1016/j.gene.2012.07.014. [DOI] [PubMed] [Google Scholar]
- Johnson M.M., Houck J., Chen C. Screening for deleterious nonsynonymous single-nucleotide polymorphisms in genes involved in steroid hormone metabolism and response. Cancer Epidemiol. Biomarkers Prev. 2005;14:1326–1329. doi: 10.1158/1055-9965.EPI-04-0815. [DOI] [PubMed] [Google Scholar]
- Jovine L., Qi H., Williams Z., Litscher E., Wassarman P.M. The ZP domain is a conserved module for polymerization of extracellular proteins. Nat. Cell Biol. 2002;4:457–461. doi: 10.1038/ncb802. [DOI] [PubMed] [Google Scholar]
- Kamaraj B., Purohit R. In silico screening and molecular dynamics simulation of disease-associated nsSNP in TYRP1 gene and its structural consequences in OCA3. Biomed. Res. Int. 2013;2013:697051. doi: 10.1155/2013/697051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubiczkova L., Sedlarikova L., Hajek R., Sevcikova S. TGF-beta — an excellent servant but a bad master. J. Transl. Med. 2012;10:183. doi: 10.1186/1479-5876-10-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Mohamoud H.S., Hussain M.R., El-Harouni A.A., Shaik N.A., Qasmi Z.U., Merican A.F., Baig M., Anwar Y., Asfour H., Bondagji N., Al-Aama J.Y. First comprehensive in silico analysis of the functional and structural consequences of SNPs in human GalNAc-T1 gene. Comput. Math. Methods Med. 2014;2014:904052. doi: 10.1155/2014/904052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng P.C., Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng P.C., Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng P.C., Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annu. Rev. Genomics Hum. Genet. 2006;7:61–80. doi: 10.1146/annurev.genom.7.080505.115630. [DOI] [PubMed] [Google Scholar]
- Pardali E., Ten Dijke P. TGFbeta signaling and cardiovascular diseases. Int. J. Biol. Sci. 2012;8:195–213. doi: 10.7150/ijbs.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekaran R., Sudandiradoss C., Doss C.G., Sethumadhavan R. Identification and in silico analysis of functional SNPs of the BRCA1 gene. Genomics. 2007;90:447–452. doi: 10.1016/j.ygeno.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Rajith B., Doss G.P. Path to facilitate the prediction of functional amino acid substitutions in red blood cell disorders — a computational approach. PLoS One. 2011;6:e24607. doi: 10.1371/journal.pone.0024607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A., Kucukural A., Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraj M.A., Escalona R.M., Umbers A., Chua H.K., Small C., Griswold M., Loveland K., Findlay J.K., Stenvers K.L. Fetal testis dysgenesis and compromised Leydig cell function in Tgfbr3 (beta glycan) knockout mice. Biol. Reprod. 2010;82:153–162. doi: 10.1095/biolreprod.109.078766. [DOI] [PubMed] [Google Scholar]
- Thomas P.D., Kejariwal A. Coding single-nucleotide polymorphisms associated with complex vs. Mendelian disease: evolutionary evidence for differences in molecular effects. Proc. Natl. Acad. Sci. U. S. A. 2004;101:15398–15403. doi: 10.1073/pnas.0404380101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R., McConnell R., Whittacker J., Kirkpatrick P., Bradley J., Sandford R. Identification of mutations in the repeated part of the autosomal dominant polycystic kidney disease type 1 gene, PKD1, by long-range PCR. Am. J. Hum. Genet. 1999;65:39–49. doi: 10.1086/302460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Moult J. SNPs, protein structure, and disease. Hum. Mutat. 2001;17:263–270. doi: 10.1002/humu.22. [DOI] [PubMed] [Google Scholar]
- Wang Z., Gerstein M., Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiater E., Harrison C.A., Lewis K.A., Gray P.C., Vale W.W. Identification of distinct inhibin and transforming growth factor beta-binding sites on betaglycan: functional separation of betaglycan co-receptor actions. J. Biol. Chem. 2006;281:17011–17022. doi: 10.1074/jbc.M601459200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material.