

Abstract
The RAS genes are critical oncogenic drivers activated by point mutation in some 20% of human malignancies. However, no pharmacological approaches to targeting RAS proteins directly have yet succeeded, leading to suggestions that these proteins may be “undruggable.” This has led to two alternative indirect approaches to targeting RAS function in cancer. One has been to target RAS signaling pathways downstream at tractable enzymes such as kinases, particularly in combination. The other, which is the focus of this review, has been to seek targets that are essential in cells bearing an activated RAS oncogene, but not those without. This synthetic lethal approach, while rooted in ideas from invertebrate genetics, has been inspired most strongly by the successful use of PARP inhibitors, such as olaparib, in the clinic to treat BRCA defective cancers. Several large-scale screens have been carried out using RNA interference-mediated expression silencing to find genes that are uniquely essential to RAS mutant but not wild type cells. These screens have been notable for the low degree of overlap between their results, with the possible exception of proteasome components, and have yet to lead to successful new clinical approaches to the treatment of RAS mutant cancers. Possible reasons for these disappointing results are discussed here, along with a re-evaluation of the approaches taken. Based on experience to date, RAS synthetic lethality has so far fallen some way short of its original promise and remains unproven as an approach to finding effective new ways of tackling RAS mutant cancers.
Introduction
Members of the RAS family of oncogenes, the closely related KRAS, NRAS and HRAS genes, are the most frequently activated drivers of human cancer (1). They were originally identified as retroviral oncogenes some fifty years ago and HRAS was the first identified human oncogene some thirty years ago (2). Extensive sequencing of human tumours has shown that KRAS is the most frequently mutated dominant oncogenic driver in human cancer, with activating mutations in one or other RAS family member being found in about 20% of human tumours (1, 2). Despite the very long history of study of RAS protein function, attempts to directly inhibit its biological activity have proved very challenging, leading to the perception that RAS proteins are “undruggable” (3). Using farnesyl transferase inhibitors to block the post-translational isoprenylation of RAS proteins, which is essential for their function, proved ineffective in clinical trials due to both alternative processing enzymes and a lack of specificity for RAS (4). Direct targeting of the guanine nucleotide binding site in RAS, which regulates its conformation and interaction with downstream effector enzymes, has proved intractable due in part to the very high affinity of RAS for GTP (5). On a more positive note, recently a number of labs have made some advances in identifying compounds that interact directly with RAS and, in some cases, perturb its ability to bind and activate downstream effectors (6-8). However, these compounds are still a very long way from being effective drugs in the clinic.
As a result of the difficulties of targeting RAS proteins directly, much attention has focused on alternative ways of selectively inhibiting RAS mutant cells by blocking the activity of enzymes in pathways controlled by RAS for which good inhibitors exist (9, 10). On the RAF pathway, one of the direct effector families of RAS, these include RAF, MEK and ERK protein kinases. On the PI 3-kinase pathway, another direct effector of RAS, these include PI 3-kinase, AKT and mTOR. Large numbers of clinical trials have been undertaken with drugs against these targets, in many cases used in combinations targeting both RAF and PI 3-kinase arms downstream of RAS (11). Although the results of most of these trials have yet to be reported, there is a concern that the toxicity of these combinations is likely to be problematic. To date, the only targeted therapy that has been proven to be effective on KRAS mutant cancer in clinical trials has been the use of a MEK inhibitor, selumetinib, in combination with a cytotoxic agent, docetaxel, in a phase II trial that demonstrated benefit in KRAS mutant non-small cell lung cancer in terms of progression-free survival only but not overall survival (12). Furthermore, single agent MEK inhibitor MEK162/binimetinib has proven effective against NRAS mutant melanoma in a phase II clinical trial (13).
While there will likely be future improvements in our ability to target RAS protein function directly and also indirectly through inhibition of downstream effector enzymes, with the exception of the direct targeting of specific mutant forms of RAS it is possible that it will be hard to achieve a good therapeutic index in many cases, as RAS signaling pathways are essential for normal development and homeostasis (14). This has led to interest in attempting to find “synthetic lethal” interactions between the expression of activated mutant RAS oncogenes and loss of expression of other genes to which the cancer cells have acquired dependence, but upon which normal cells are less dependent.
Synthetic Lethality
The concept of synthetic lethality was first described nearly a century ago by Calvin Bridges following the observation in fruit flies that two mutations could lack a phenotype individually but be lethal when combined in a single organism (15, 16) (see Fig. 1). At its simplest, two alternative pathways acting in parallel to carry out an essential function may each be quite dispensable, but loss of both pathways would have a lethal effect. In other cases, loss of one gene may have the potential to cause a damaging phenotype but the system compensates through genetic buffering to render the organism healthy, but now dependent on a second gene. Synthetic lethality was recognised as having considerable potential as an approach to targeting cancer cells, especially as loss of function of tumour suppressor genes is very common in cancer (17, 18). Still much the most impressive application of the concept of synthetic lethality to cancer therapeutics came from the observation by the labs of Ashworth (19) and of Helleday (20) that the deficiency in the BRCA1 or BRCA2 tumour suppressor genes, which are required for homologous recombination, in a subset of cancers led to them becoming dependent on Poly(ADP-ribose) polymerase (PARP1) mediated DNA repair. PARP inhibitors have made slow progress in clinical trials over the past several years, but have finally been shown to have real value in the treatment of BRCA defective cancer and are proceeding to clinical approval (21).
Figure 1.

Scheme demonstrating the basis of synthetic lethality with RAS mutation exploited in RAS synthetic lethal screens.
While the synthetic lethal interaction between BRCA defects and PARP inhibition was predicted from mechanistic knowledge of their roles in DNA repair, the availability of large-scale RNA interference libraries from about 2003 onwards allowed unbiased genome-wide screening for second genetic hits that would combine with loss of a tumour suppressor gene to cause synthetic lethality in tumour cells. Not only could this be applied to cancer cells lacking a functional tumour suppressor gene; it could equally be extended to cancer cells bearing an activating mutation in an oncogene, for example KRAS. The mechanistic underpinning for the existence of such genetic interactions, refinements of which have also been described as “induced essentiality” and “acquired dependency” (22), has remained somewhat vague, but the hope has been that the power of high throughput, large-scale functional genomics would be able to uncover novel proteins and pathways that are uniquely required by cells expressing activated mutant RAS proteins.
Possible hits that might be expected to emerge from a mutant KRAS synthetic lethal screen could fall into a number of categories. One might be genes involved in dealing with stresses resulting from the process of oncogenic transformation. It has long been recognised that oncogene signaling in cancer cells can result in elevated stresses resulting from deregulated DNA replication, redox balance, metabolic regulation, protein synthesis and other key cellular processes. The stress handling mechanisms that protect cells from these insults act orthogonally to the oncogene signaling pathways, not being controlled by them, and increased reliance on these protective systems is likely to be selected for during the micro-evolution of the tumour in vivo. These pathways are not oncogenic in themselves, but the tumour cells can become more dependent on them than their normal counterparts in a process referred to as “non-oncogene addiction” (23). As part of the process of transformation, cancer cells acquire a number of novel behaviours, the “hallmarks of cancer” (24), which also include a wider range of modifications beyond stress management that are needed for malignant growth, such as alterations in metabolic flux to support increased cell proliferation (25). Potentially any of these might emerge from mutant KRAS synthetic lethal screens.
As well as non-oncogene addiction hits, screens set up to look for unique dependencies of mutant KRAS expressing cancer cells could also yield components of signaling pathways acting downstream of RAS in cells that require continued oncogene function, as is the case for the vast majority of KRAS mutant cancer cells. Such genes would be considered to be “oncogene addiction” hits (26). As these targets lie on the same pathway as RAS, they are not usually thought of as synthetic lethal, although they clearly can show great potential for targeting RAS mutant cancers - the RAF/MEK/ERK pathway down to CDK4 has been the most studied in this regard, with a considerable body of mouse model work (27-29) and drug studies (see below) supporting the value of this approach.
As well as genetic screens, the concept of synthetic lethality could be extended to include drug library screens in which interactions between the mutant oncogene and a specific drug capable of inhibiting a protein or pathway are sought (17). Large scale screening of drugs of known mechanism of action against cell lines panels with known mutational status has been carried out by two consortia (30, 31), but has only strongly implicated inhibition of pathways downstream of RAS, particularly MEK, as showing enhanced ability to inhibit RAS mutant cells, and to some extent also the IGF-1 receptor acting via PI 3-kinase (32, 33). These targets are part of an oncogenic signaling network and would not be considered synthetic lethal by most definitions, although ultimately this is a semantic issue. It should be noted that drug induced inhibition of enzymatic activity can be functionally quite distinct from loss of expression of a protein, and drugs are likely also to be better at inhibiting multiple related isoforms, so drug screens may be expected to give significantly different insights to functional genomic screens. Due to space constraints, I will not review drug screens for KRAS mutant cell selective compounds further here, but refer to other recent reviews (5, 34, 35).
One final point about theoretical limitations of loss of function based synthetic lethal screens is that they would not be able to pick up proteins whose expression is suppressed by RAS signaling, and whose loss of function is required for the transformation process. A recent example of this is TET1, which inhibits DNA methylation resulting in expression of tumour suppressor genes and prevention of transformation, but whose expression is suppressed by mutant KRAS (36). While this would not make a potential cancer therapeutic drug target, knowledge of its identity informs about other potential targets, such as certain DNA methyltransferases.
Synthetic Lethal Screening Methodology: A Work in Progress
More than ten mutant RAS synthetic lethal genetic screens have been published over the past eight years. These have employed a number of different methodologies and reagents. Two major themes to emerge have been the use of isogenic cell lines and the use of cancer cell line panels with differing KRAS mutation status. In the isogenic approach, pairs of cell lines are used for screening that are close to identical, with the exception of the presence or absence of the mutant KRAS oncogene. This can be achieved either by deleting the mutant KRAS allele from cancer cells in which it is naturally occurring, as with the very widely used HCT-116 colon cancer cells and their KRAS deleted derivatives (37), or by adding a mutant KRAS gene to immortalised cells resulting in their transformation (38).
A number of problems are inherent in the isogenic approach that have never been adequately resolved. In the case of removal of an endogenous mutant KRAS allele from a tumour cell line, a major concern is that these cells are at least to some degree KRAS oncogene addicted (39), so cells emerging from the mutant KRAS deletion event will be heavily selected for secondary events that promote their survival. The isogenic pair of cell lines may thus be far less closely matched than initially supposed. Recently it has been shown that upregulation of YAP1 function can relieve KRAS oncogene addiction, and it is likely that KRAS deletion from HCT-116 or DLD1 colon cancer cells will have subjected them to such a selective event (40, 41).
In the case of addition of mutant KRAS to cells that are immortalised but not transformed, the resulting cells have undergone transformation in vitro without many of the selective pressures experienced by a tumour cell in vivo, and thus may have a phenotype that is significantly different to that of a cell in a naturally arising tumour. Key vulnerabilities may be lacking in these in vitro created human cancer cells, as can be demonstrated by the fact that they generally lack true addiction to the inserted driver oncogene. Most transformed cells created by addition of defined oncogenes, together with tumour suppressor gene loss of function, exhibit considerable overexpression of the oncogene. However, when oncogenic mutations in KRAS and RAS pathway genes are knocked in to, rather than over-expressed in, normal immortalised human cells, it is notable that these do not necessarily lead to acquisition of a transformed phenotype, even when occurring in combinations, pointing to further limitations of the isogenic approach (42). This result, in which activating KRAS mutations knocked into HME-1 immortalized human breast epithelial cells failed to cause transformation, is also supported by older in vivo observations that chronic low-level mutant KRAS expression results in tumour formation, but only after the spontaneous upregulation of mutant KRAS expression, along with override of senescence checkpoints (43).
An alternative KRAS synthetic lethal screening approach has been to use panels of cancer cell lines that differ in their KRAS mutation status and to seek genes whose targeting selectively kills KRAS mutant, but not wild type, cell lines. A difficulty with this approach is the huge genetic complexity of most human cancer cell lines, which means that any such analysis is done against the background of large numbers of other mutations, several of which may be of considerable significance to the screen output. As well as mutations affecting other signaling pathways that may influence response to inhibition of expression of a given gene, cells that are wild type for KRAS may also have an activated RAS phenotype due to mutations elsewhere in the pathway. While obviously problematic mutations can be avoided in the cell lines chosen (e.g. avoiding the use of EGFR mutant cells due to the partial overlap of EGFR and KRAS signaling), it is still necessary to use large numbers of cell lines to overcome noise generated by individual mutations (44). Concentrating on specific tissue types is also likely to help allow a clearer picture to emerge without the need for excessively large cell panels, but it is still likely that reliable results from such panels will require the screening of significant numbers of lines.
In addition to the choice of cell lines to be used in the screen, different methodologies for knock down of gene expression can also be used. Several large libraries of lentiviral or retroviral short hairpin RNA constructs targeting every human and mouse gene have been created and made widely available to the research community (45, 46). These can be used either in a high throughput, one gene or one hairpin at a time format, or can be used in pooled selection screens with the readout of library composition before and after selection being through microarray hybridisation or next generation sequencing analysis of hairpin or barcode sequence representation (47). shRNA driven knock down of gene expression can occur stably and allows the selection or measurement of phenotypes over time frames of weeks, which can be useful for more complex assays. By contrast, synthetic short interfering double stranded RNA molecules can also be used, giving rise to acute but transient gene expression knock down and restricted to a one gene at a time format.
Both RNA interference based screening technologies - siRNA and shRNA based - have been dogged by problems with off target effects (48-50). This has led to the need for prolonged validation of hits from KRAS synthetic lethal RNAi screens in order to eliminate false positive artefacts due to off target knock down of expression of other genes. In most cases this has been most convincing when carried out by methodologies other than RNAi itself. While awareness of the problems of off-target effects in RNA interference screens is much greater than in the past, this remains a major technical limitation of the methodology. Resolution of this will likely require the design of greatly improved RNAi libraries, using sequences with less off target activity and greater potency (51) and using greater fold redundancy so each gene is targeted by far greater numbers of sequences (52).
A consistent frustration experienced in the validation of hits from RNAi screens has been the technical difficulty of carrying out “rescue experiments”, in which the targeted gene is re-expressed from an exogenous construct with silent mutations that render the mRNA resistant to the RNAi agents. This has been thought of as the gold standard for the proof of validity of an RNA interference hit, but it is technically demanding and requires considerable attention to be paid to the level of expression of the rescued gene. In many cases massive over-expression of the gene in question results and may not adequately model the physiological function of the endogenous protein. More modest expression is preferable, which is probably best achieved through the use of bacterial artificial chromosome constructs (53). For most of the RAS synthetic lethal screens published, hits have not been validated by rescue experiments.
Another significant shortcoming of the RNAi screening technology has been the high level of false negatives due to low potency of knock down, especially by shRNA vectors. Improved library design may counter this to some extent, but the biggest improvement is likely to come from the comprehensive validation of library components, ensuring that every shRNA or siRNA was truly capable of knocking down its target (54). However, false negatives due to functional redundancy between closely related isoforms of proteins will remain a problem.
As an alternative to RNA interference, whose limitations as an analytical tool have become very well understood over the past decade, other more recently developed methods of interfering with gene expression can be used, such as CRISPR/Cas9 genome engineering libraries (55, 56). An appealing aspect of the use of CRISPR/Cas9 is that it is the DNA rather than the mRNA that is targeted, so loss of gene expression tends to be more complete, although theoretically it is possible that not all alleles of a gene in a cell are knocked out, which could be a more significant possibility in aneuploid cancer cells. By contrast, RNA interference never provides complete knockout of gene expression, but rather gives hypomorphic effects. A huge amount of the time spent validating RNA interference screen results has gone on matching up efficiency of knockdown by various si/shRNA sequences with magnitude of phenotypic effect, and in reality these never align perfectly. While CRISPR/Cas9 library screening appears very appealing at present, off target effects can clearly still occur with this methodology, although they appear to be less marked and methodological developments hold out the possibility of reducing them further (57). Other gene targeting methodologies may be applied to screens for RAS synthetic lethality: one such example is insertional mutagenesis, although the need for the use of haploid cells would make this hard to apply to typical cancer cells (58).
Finally, RAS synthetic lethal screens carried out to date have relied on cell growth in traditional two dimensional tissue culture systems, with a couple of exceptions (59, 60). It is becoming increasingly clear that such in vitro conditions are a poor mimic of the true environment of a naturally occurring tumour in vivo (61). In the future it will be important to adapt screening methodology to use three dimensional cell culture systems, such as spheroid or organoid cultures. In addition, the complex interactions between tumour cells and host tissue in vivo may be better modelled in vitro using mixed cell culture systems, possibly incorporating stromal cells, endothelial cells and immune cells as well as tumour cells. 3D multicellular tumor spheroid (MCTS) culture systems are being developed to address these issues, but as yet have not been exploited in large scale screening (62). The ultimate progression towards accurate modelling of the tumour environment comes from the use of in vivo screens; RNA interference based in vivo screens have developed rapidly in recent years (63). They have tended to be more limited in library size and have mostly been used to identify tumour suppressor genes.
RAS Synthetic Lethal Screen Results
The results of the genetic screens that have been published to date that have sought unique dependencies of RAS mutant mammalian cells are shown in Table 1, and see also Fig. 2. The most immediately obvious feature is the lack of overlap in the results from the different screens. Only the proteasome scores in multiple screens (64-66), providing evidence for a classic non-oncogene addiction phenotype of RAS mutant cancer cells on the ability to degrade damaged or excess proteins. RAS mutant cells have long been known to show elevated protein synthesis rates, with several links between RAS signaling pathways and the control of the protein synthesis machinery well established (67). However, proteasome inhibitory drugs alone show little selectivity for RAS mutant cells in vitro (30, 31). Two proteasome inhibitors are approved for clinical use in the treatment of multiple myeloma, bortezomib and carfilzomib (68). This haematological malignancy displays a high rate of RAS oncogene mutation (22% KRAS mutant and 18% NRAS mutant (69)), but it remains unclear whether RAS mutation status is in any way related to proteasome inhibitor clinical response (70). Although proteasome inhibitors have not succeeded in clinical trials in solid tumours, the synthetic lethal screen data provide some rationale for re-evaluating their possible activity in RAS mutant solid tumours. It is likely that combination with other agents would be required to provide significant response; in addition to cytotoxic agents, an interesting possible combination would be with MEK inhibitors, currently the only targeted agents to show selectivity for KRAS mutant tumours in the clinic. Indeed, the centrality of MEK signaling to RAS mediated transformation has led to interest in conducting KRAS synthetic lethal screens in combination with MEK inhibition, which has yielded Bcl-XL as a potential combination synthetic lethal hit (71).
Table 1.
Mutant RAS synthetic lethal targets from functional genomic screens
| Synthetic lethalhits | Cells | Library and screen | Reference |
|---|---|---|---|
| RAN TPX1 SDC1 |
H1299 Q61K NRAS mutant human lung cancer cells | ~3,700 druggable genes siRNA, multi well cell death |
(89) |
| Survivin | DLD1 G13D KRAS isogenic colon cancer cell lines | ~4,000 druggable genes siRNA, multi well cell death |
(90) |
| PLK1 APC/C Proteasome |
DLD1 and HCT-116 G13D KRAS isogenic colon cancer cell lines | Whole genome retroviral shRNA library, pooled proliferation screen microarray readout | (66) |
| STK33 (AKT3) (DGKZ) |
Panel of 4 KRAS mutant and 4 wild type pan-cancer cell lines, plus two normal lines | ~1,000 druggable genes, lentiviral shRNA, multi well proliferation | (76) |
| TBK1 (PSKH2) (PSMD14) (PTCH2) |
Human pan-cancer cell line panel (7 KRAS mutant, 10 wild type) | ~ 1,000 druggable genes, lentiviral shRNA, multi well proliferation | (64) |
| WT1 | Mouse lung cancer cells LKR10 and LKR13 G12D Kras | 162 KRAS relevant genes shRNA pooled screen in vivo, beadarray readout | (59) |
| SNAI2 | HCT-116 isogenic colon cancer cell lines | ~ 2,500 druggable gene retroviral shRNA pooled proliferation screen, microarray readout | (91) |
| GATA2 CDC6 Proteasome |
HCT-116 isogenic colon cancer cells and pan-cancer 26 cell line panel | ~8000 genes siRNA, multi well cell proliferation and apoptosis | (65, 79) |
| TAK1 | SW620 G12V KRAS and SW837 G12C KRAS human colon cancer cell lines | Lentiviral shRNA screen with 17 selected kinases, multi well proliferation | (92) |
| BCL-XL | KRAS mutant colon cancer cell lines HCT-116 and SW620 | ~1,200 druggable genes shRNA, pooled NGS readout, synergistic death with MEK inhibitor | (71) |
| Ctnnb1 (β-catenin) Mllt6 | Hras G12V mutant mouse keratinocytes in vivo | Whole genome lentiviral shRNA library, pooled in vivo proliferation screen NGS readout | (60) |
| COPI coatomer | KRAS plus LKB1 mutant lung cancer 17 cell line panel | Whole genome siRNA, multi well cell proliferation | (73) |
| ARHGEF2 | Human pan-cancer 72 cell line panel | Whole genome lentiviral shRNA library, pooled proliferation screen NGS readout | (93) |
Figure 2.
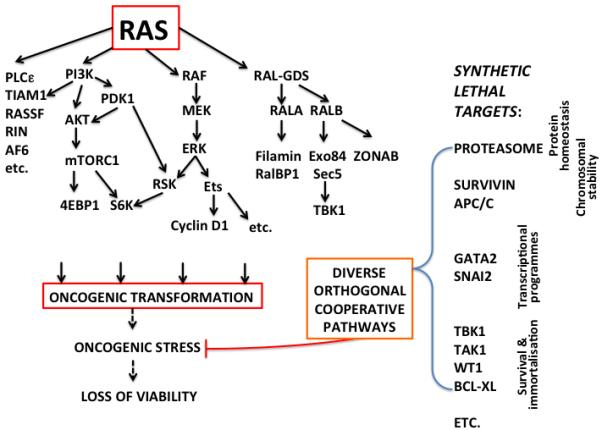
RAS signalling pathways involved in driving the transformed phenotype and their relationship to orthogonal cooperative pathways, revealed by synthetic lethal screens, that protect RAS transformed cells from oncogenic stresses. Many of these can be considered to be non-oncogene addiction effects. Some RAS synthetic lethal hits may be controlled by RAS signaling, for example, TBK1, but in most cases there does not appear to be direct control by RAS.
There are likely to be a variety of reasons for the lack of overlap between these screen results. Clearly each screen was carried out in a different way, often using fundamentally different technology and mostly on different cell lines, frequently from different tissues. Most of the screens used KRAS knockdown as a positive control, so one has to conclude at least that there are no synthetic lethal targets identified to date that can give as broadly applicable, or indeed as potent, an effect as targeting KRAS itself. When RNA interference screens have been extended to very large collections of several hundred cell lines, KRAS targeting holds up very well as an effective way of inhibiting growth of KRAS mutant cancer cells, while all of the published KRAS synthetic lethal targets fall away to statistical insignificance (W.R. Sellers, personal communication). A large part of the problem is likely to be down to the context in which the RAS mutations occur, both in terms of the presence of mutations in other genes and also tissue lineage (72-75). With the increasing level of detail available about the genetic make up of tumours upon clinical presentation, it is possible that some specific combinations of tissue type and mutational background could be identified that would render RAS mutant tumours susceptible to targeting of synthetic lethal partners. However, from the data acquired to date it is clear that there is very unlikely to exist a universal mutant RAS synthetic lethal target that comes anywhere close to the targeting of RAS proteins themselves across the broad spectrum of RAS mutant cancers.
In addition to the differences in methodology and context leading to variability between screens, it is also likely that some problems have come from experimental artefacts associated with off target effects in RNA interference technology. The synthetic lethal effect of STK33 targeting (76) has proven hard to reproduce by others (77, 78). GATA2 targeting (65, 79) has also been questioned in terms of its efficacy in KRAS mutant lung cancer cell lines in vitro (80), although in this case extensive in vivo (65, 81) and mechanistic data (82) exist to support the link.
A final concern to note with the results of the synthetic lethal screens reported so far is the magnitude of the synthetic lethal effect. The original recognition of the synthetic lethal interaction between BRCA mutation and PARP inhibition reported a differential sensitivity of around 1000 fold (19). By contrast, most of the synthetic lethal interactions reported for RAS are far weaker. While it is hard to directly compare methodologies between drug based and RNA interference mediated inhibition, it is unlikely that the magnitude of any of the RAS synthetic lethal effects found so far exceed ten fold. It is not clear that this is sufficient to form the basis of an effective therapeutic strategy.
Conclusions and Future Direction
The search for synthetic lethal interactions with RAS mutational activation has spanned more than ten years and this may be a good point at which to step back and evaluate what has been achieved. Unfortunately, it has yet to provide much basis for optimism that this will lead to a promising new therapeutic approach to tackling RAS mutant cancers in the clinic. It is certainly possible that improvements in screening technology - for example, the use of improved RNAi libraries or CRISPR/Cas9 systems, the use of expanded collections of cancer cell lines, and the use of more realistic 3D and possibly mixed cell culture systems, or further development of in vivo screening methods - could lead to major breakthroughs and the discovery of synthetic lethal targets that are relevant across reasonably broad contexts. However, at present it seems that synthetic lethal hits identified to date may have significance in only rather limited contexts. From everything we have learnt in recent years about the speed with which advanced cancers can develop resistance to therapeutic agents targeted directly at mutant oncogene products, such as BRAF and EGFR (83, 84), the concern also exists that even if initial responses to targeting of synthetic lethal partners were good, the signaling network distance between those partners and RAS itself would make it very likely that compensatory adaptation and rewiring could relatively easily occur to confer resistance. In the setting of tumours with high levels of intra-tumour heterogeneity (85, 86), which appears to be the norm for most tumours where RAS mutation levels are high, the danger is that pre-existing variation could render portions of tumours unresponsive to therapies targeting non-oncogene addiction pathways. These pathways are diverse and dependence on them is due to adaptation to selective pressures acting on the tumour and only indirectly linked to the driver oncogene. While there are certainly a number of interesting leads from the synthetic lethal approach that deserve detailed follow up, and some potential for more to come, advances elsewhere in cancer research merit a re-evaluation of priorities in the RAS field. In particular, two areas seem to be especially promising and compare very favourably relative to synthetic lethality: one is the nascent field of direct targeting of RAS proteins, especially with mutant specific inhibitors (7), and the other is immunotherapy approaches such as immune checkpoint blockade, which, although probably agnostic with regard to driver oncogene identity, is beginning to show impressive clinical activity in tumours with high rates of RAS mutation, such as lung cancer and melanoma (87, 88).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–74. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–65. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 3.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 4.Cox AD, Der CJ, Philips MR. Targeting RAS membrane association: back to the future for anti-RAS drug discovery? Clin Cancer Res. 2015;21:xxx–xxx. doi: 10.1158/1078-0432.CCR-14-3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell. 2014;25:272–81. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 6.Downward J. RAS’s cloak of invincibility slips at last? Cancer Cell. 2014;25:5–6. doi: 10.1016/j.ccr.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 7.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–51. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marcus K, Mattos C. Direct attack on RAS: intramolecular communication and mutation-specific effects. Clin Cancer Res. 2015;21:xxx–xxx. doi: 10.1158/1078-0432.CCR-14-2148. [DOI] [PubMed] [Google Scholar]
- 9.Castellano E, Sheridan C, Thin MZ, Nye E, Spencer-Dene B, Diefenbacher ME, et al. Requirement for interaction of PI3-kinase p110alpha with RAS in lung tumor maintenance. Cancer Cell. 2013;24:617–30. doi: 10.1016/j.ccr.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–6. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Britten CD. PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer Chemother Pharmacol. 2013;71:1395–409. doi: 10.1007/s00280-013-2121-1. [DOI] [PubMed] [Google Scholar]
- 12.Janne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013;14:38–47. doi: 10.1016/S1470-2045(12)70489-8. [DOI] [PubMed] [Google Scholar]
- 13.Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CM, Queirolo P, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14:249–56. doi: 10.1016/S1470-2045(13)70024-X. [DOI] [PubMed] [Google Scholar]
- 14.Drosten M, Dhawahir A, Sum EY, Urosevic J, Lechuga CG, Esteban LM, et al. Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J. 2010;29:1091–104. doi: 10.1038/emboj.2010.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nijman SM. Synthetic lethality: general principles, utility and detection using genetic screens in human cells. FEBS Lett. 2011;585:1–6. doi: 10.1016/j.febslet.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bridges CB. The origin of variation. Amer Nat. 1922;56:51–63. [Google Scholar]
- 17.Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997;278:1064–8. doi: 10.1126/science.278.5340.1064. [DOI] [PubMed] [Google Scholar]
- 18.Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689–98. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 19.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 20.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 21.Lee JM, Ledermann JA, Kohn EC. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann Oncol. 2014;25:32–40. doi: 10.1093/annonc/mdt384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tischler J, Lehner B, Fraser AG. Evolutionary plasticity of genetic interaction networks. Nat Genet. 2008;40:390–1. doi: 10.1038/ng.114. [DOI] [PubMed] [Google Scholar]
- 23.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 25.Kimmelman AC. Metabolic dependencies in RAS-driven cancers. Clin Cancer Res. 2015;21:xxx–xxx. doi: 10.1158/1078-0432.CCR-14-2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weinstein IB. Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science. 2002;297:63–4. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 27.Puyol M, Martin A, Dubus P, Mulero F, Pizcueta P, Khan G, et al. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010;18:63–73. doi: 10.1016/j.ccr.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 28.Blasco RB, Francoz S, Santamaria D, Canamero M, Dubus P, Charron J, et al. c-Raf, but not B-Raf, is essential for development of K-Ras oncogene-driven non-small cell lung carcinoma. Cancer Cell. 2011;19:652–63. doi: 10.1016/j.ccr.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karreth FA, Frese KK, DeNicola GM, Baccarini M, Tuveson DA. C-Raf is required for the initiation of lung cancer by K-Ras(G12D) Cancer Discov. 2011;1:128–36. doi: 10.1158/2159-8290.CD-10-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–5. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ebi H, Corcoran RB, Singh A, Chen Z, Song Y, Lifshits E, et al. Receptor tyrosine kinases exert dominant control over PI3K signaling in human KRAS mutant colorectal cancers. J Clin Invest. 2011;121:4311–21. doi: 10.1172/JCI57909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Molina-Arcas M, Hancock DC, Sheridan C, Kumar MS, Downward J. Coordinate direct input of both KRAS and IGF1 receptor to activation of PI3 kinase in KRAS-mutant lung cancer. Cancer Discov. 2013;3:548–63. doi: 10.1158/2159-8290.CD-12-0446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov. 2014;13:828–51. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spiegel J, Cromm PM, Zimmermann G, Grossmann TN, Waldmann H. Small-molecule modulation of Ras signaling. Nat Chem Biol. 2014;10:613–22. doi: 10.1038/nchembio.1560. [DOI] [PubMed] [Google Scholar]
- 36.Wu BK, Brenner C. Suppression of TET1-dependent DNA demethylation is essential for KRAS-mediated transformation. Cell Rep. 2014;9:1827–40. doi: 10.1016/j.celrep.2014.10.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shirasawa S, Furuse M, Yokoyama N, Sasazuki T. Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science. 1993;260:85–8. doi: 10.1126/science.8465203. [DOI] [PubMed] [Google Scholar]
- 38.Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–8. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 39.Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15:489–500. doi: 10.1016/j.ccr.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell. 2014;158:185–97. doi: 10.1016/j.cell.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shao DD, Xue W, Krall EB, Bhutkar A, Piccioni F, Wang X, et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell. 2014;158:171–84. doi: 10.1016/j.cell.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zecchin D, Arena S, Martini M, Sassi F, Pisacane A, Di Nicolantonio F, et al. Modeling tumor progression by the sequential introduction of genetic alterations into the genome of human normal cells. Hum Mutat. 2013;34:330–7. doi: 10.1002/humu.22234. [DOI] [PubMed] [Google Scholar]
- 43.Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, Chodosh LA. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol. 2007;9:493–505. doi: 10.1038/ncb1567. [DOI] [PubMed] [Google Scholar]
- 44.Boehm JS, Hahn WC. Towards systematic functional characterization of cancer genomes. Nat Rev Genet. 2011;12:487–98. doi: 10.1038/nrg3013. [DOI] [PubMed] [Google Scholar]
- 45.Root DE, Hacohen N, Hahn WC, Lander ES, Sabatini DM. Genome-scale loss-of-function screening with a lentiviral RNAi library. Nat Methods. 2006;3:715–9. doi: 10.1038/nmeth924. [DOI] [PubMed] [Google Scholar]
- 46.Paddison PJ, Silva JM, Conklin DS, Schlabach M, Li M, Aruleba S, et al. A resource for large-scale RNA-interference-based screens in mammals. Nature. 2004;428:427–31. doi: 10.1038/nature02370. [DOI] [PubMed] [Google Scholar]
- 47.Sims D, Mendes-Pereira AM, Frankum J, Burgess D, Cerone MA, Lombardelli C, et al. High-throughput RNA interference screening using pooled shRNA libraries and next generation sequencing. Genome Biol. 2011;12:R104. doi: 10.1186/gb-2011-12-10-r104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jackson AL, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov. 2010;9:57–67. doi: 10.1038/nrd3010. [DOI] [PubMed] [Google Scholar]
- 49.Franceschini A, Meier R, Casanova A, Kreibich S, Daga N, Andritschke D, et al. Specific inhibition of diverse pathogens in human cells by synthetic microRNA-like oligonucleotides inferred from RNAi screens. Proc Natl Acad Sci U S A. 2014;111:4548–53. doi: 10.1073/pnas.1402353111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sigoillot FD, Lyman S, Huckins JF, Adamson B, Chung E, Quattrochi B, et al. A bioinformatics method identifies prominent off-targeted transcripts in RNAi screens. Nat Methods. 2012;9:363–6. doi: 10.1038/nmeth.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Knott SR, Maceli AR, Erard N, Chang K, Marran K, Zhou X, et al. A Computational Algorithm to Predict shRNA Potency. Mol Cell. 2014;56:796–807. doi: 10.1016/j.molcel.2014.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kampmann M, Bassik MC, Weissman JS. Integrated platform for genome-wide screening and construction of high-density genetic interaction maps in mammalian cells. Proc Natl Acad Sci U S A. 2013;110:E2317–26. doi: 10.1073/pnas.1307002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kittler R, Pelletier L, Ma C, Poser I, Fischer S, Hyman AA, et al. RNA interference rescue by bacterial artificial chromosome transgenesis in mammalian tissue culture cells. Proc Natl Acad Sci U S A. 2005;102:2396–401. doi: 10.1073/pnas.0409861102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fellmann C, Lowe SW. Stable RNA interference rules for silencing. Nat Cell Biol. 2014;16:10–8. doi: 10.1038/ncb2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–7. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–4. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014;159:647–61. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carette JE, Guimaraes CP, Varadarajan M, Park AS, Wuethrich I, Godarova A, et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science. 2009;326:1231–5. doi: 10.1126/science.1178955. [DOI] [PubMed] [Google Scholar]
- 59.Vicent S, Chen R, Sayles LC, Lin C, Walker RG, Gillespie AK, et al. Wilms tumor 1 (WT1) regulates KRAS-driven oncogenesis and senescence in mouse and human models. J Clin Invest. 2010;120:3940–52. doi: 10.1172/JCI44165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beronja S, Janki P, Heller E, Lien WH, Keyes BE, Oshimori N, et al. RNAi screens in mice identify physiological regulators of oncogenic growth. Nature. 2013;501:185–90. doi: 10.1038/nature12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weigelt B, Ghajar CM, Bissell MJ. The need for complex 3D culture models to unravel novel pathways and identify accurate biomarkers in breast cancer. Adv Drug Deliv Rev. 2014;69-70:42–51. doi: 10.1016/j.addr.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thoma CR, Zimmermann M, Agarkova I, Kelm JM, Krek W. 3D cell culture systems modeling tumor growth determinants in cancer target discovery. Adv Drug Deliv Rev. 2014;69-70:29–41. doi: 10.1016/j.addr.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 63.Zender L, Xue W, Zuber J, Semighini CP, Krasnitz A, Ma B, et al. An oncogenomics-based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell. 2008;135:852–64. doi: 10.1016/j.cell.2008.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–12. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kumar MS, Hancock DC, Molina-Arcas M, Steckel M, East P, Diefenbacher M, et al. The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell. 2012;149:642–55. doi: 10.1016/j.cell.2012.02.059. [DOI] [PubMed] [Google Scholar]
- 66.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–48. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gao B, Roux PP. Translational control by oncogenic signaling pathways. Biochim Biophys Acta. 2014 Dec 2; doi: 10.1016/j.bbagrm.2014.11.006. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 68.Mahindra A, Laubach J, Raje N, Munshi N, Richardson PG, Anderson K. Latest advances and current challenges in the treatment of multiple myeloma. Nat Rev Clin Oncol. 2012;9:135–43. doi: 10.1038/nrclinonc.2012.15. [DOI] [PubMed] [Google Scholar]
- 69.Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25:91–101. doi: 10.1016/j.ccr.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Smith D, Armenteros E, Percy L, Kumar M, Lach A, Herledan G, et al. RAS mutation status and bortezomib therapy for relapsed multiple myeloma. Br J Haematol. 2015 Jan 12; doi: 10.1111/bjh.13258. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 71.Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell. 2013;23:121–8. doi: 10.1016/j.ccr.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Garraway LA, Sellers WR. Lineage dependency and lineage-survival oncogenes in human cancer. Nat Rev Cancer. 2006;6:593–602. doi: 10.1038/nrc1947. [DOI] [PubMed] [Google Scholar]
- 73.Kim HS, Mendiratta S, Kim J, Pecot CV, Larsen JE, Zubovych I, et al. Systematic identification of molecular subtype-selective vulnerabilities in non-small-cell lung cancer. Cell. 2013;155:552–66. doi: 10.1016/j.cell.2013.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carretero J, Shimamura T, Rikova K, Jackson AL, Wilkerson MD, Borgman CL, et al. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell. 2010;17:547–59. doi: 10.1016/j.ccr.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ou YH, Torres M, Ram R, Formstecher E, Roland C, Cheng T, et al. TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol Cell. 2011;41:458–70. doi: 10.1016/j.molcel.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Scholl C, Frohling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137:821–34. doi: 10.1016/j.cell.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 77.Babij C, Zhang Y, Kurzeja RJ, Munzli A, Shehabeldin A, Fernando M, et al. STK33 kinase activity is nonessential in KRAS-dependent cancer cells. Cancer Res. 2011;71:5818–26. doi: 10.1158/0008-5472.CAN-11-0778. [DOI] [PubMed] [Google Scholar]
- 78.Frohling S, Scholl C. STK33 kinase is not essential in KRAS-dependent cells-letter. Cancer Res. 2011;71:7716. doi: 10.1158/0008-5472.CAN-11-2495. author reply 7. [DOI] [PubMed] [Google Scholar]
- 79.Steckel M, Molina-Arcas M, Weigelt B, Marani M, Warne PH, Kuznetsov H, et al. Determination of synthetic lethal interactions in KRAS oncogene-dependent cancer cells reveals novel therapeutic targeting strategies. Cell Res. 2012;22:1227–45. doi: 10.1038/cr.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tessema M, Yingling CM, Snider AM, Do K, Juri DE, Picchi MA, et al. GATA2 is epigenetically repressed in human and mouse lung tumors and is not requisite for survival of KRAS mutant lung cancer. J Thorac Oncol. 2014;9:784–93. doi: 10.1097/JTO.0000000000000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shen S, Mao CQ, Yang XZ, Du XJ, Liu Y, Zhu YH, et al. Cationic lipid-assisted polymeric nanoparticle mediated GATA2 siRNA delivery for synthetic lethal therapy of KRAS mutant non-small-cell lung carcinoma. Mol Pharm. 2014;11:2612–22. doi: 10.1021/mp400714z. [DOI] [PubMed] [Google Scholar]
- 82.Katsumura KR, Yang C, Boyer ME, Li L, Bresnick EH. Molecular basis of crosstalk between oncogenic Ras and the master regulator of hematopoiesis GATA-2. EMBO Rep. 2014;15:938–47. doi: 10.15252/embr.201438808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer. 2014;14:455–67. doi: 10.1038/nrc3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. 2014;11:473–81. doi: 10.1038/nrclinonc.2014.104. [DOI] [PubMed] [Google Scholar]
- 85.de Bruin EC, McGranahan N, Mitter R, Salm M, Wedge DC, Yates L, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014;346:251–6. doi: 10.1126/science.1253462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gerlinger M, Santos CR, Spencer-Dene B, Martinez P, Endesfelder D, Burrell RA, et al. Genome-wide RNA interference analysis of renal carcinoma survival regulators identifies MCT4 as a Warburg effect metabolic target. J Pathol. 2012;227:146–56. doi: 10.1002/path.4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Harvey RD. Immunologic and clinical effects of targeting PD-1 in lung cancer. Clin Pharmacol Ther. 2014;96:214–23. doi: 10.1038/clpt.2014.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Girotti MR, Saturno G, Lorigan P, Marais R. No longer an untreatable disease: how targeted and immunotherapies have changed the management of melanoma patients. Mol Oncol. 2014;8:1140–58. doi: 10.1016/j.molonc.2014.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Morgan-Lappe SE, Tucker LA, Huang X, Zhang Q, Sarthy AV, Zakula D, et al. Identification of Ras-related nuclear protein, targeting protein for xenopus kinesin-like protein 2, and stearoyl-CoA desaturase 1 as promising cancer targets from an RNAi-based screen. Cancer Res. 2007;67:4390–8. doi: 10.1158/0008-5472.CAN-06-4132. [DOI] [PubMed] [Google Scholar]
- 90.Sarthy AV, Morgan-Lappe SE, Zakula D, Vernetti L, Schurdak M, Packer JC, et al. Survivin depletion preferentially reduces the survival of activated K-Ras-transformed cells. Mol Cancer Ther. 2007;6:269–76. doi: 10.1158/1535-7163.MCT-06-0560. [DOI] [PubMed] [Google Scholar]
- 91.Wang Y, Ngo VN, Marani M, Yang Y, Wright G, Staudt LM, et al. Critical role for transcriptional repressor Snail2 in transformation by oncogenic RAS in colorectal carcinoma cells. Oncogene. 2010;29:4658–70. doi: 10.1038/onc.2010.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Singh A, Sweeney MF, Yu M, Burger A, Greninger P, Benes C, et al. TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell. 2012;148:639–50. doi: 10.1016/j.cell.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cullis J, Meiri D, Sandi MJ, Radulovich N, Kent OA, Medrano M, et al. The RhoGEF GEF-H1 is required for oncogenic RAS signaling via KSR-1. Cancer Cell. 2014;25:181–95. doi: 10.1016/j.ccr.2014.01.025. [DOI] [PubMed] [Google Scholar]