

Significance
Idiopathic pulmonary fibrosis and emphysema are leading causes of mortality, but there are no effective therapies. Mutations in telomerase are the most common identifiable risk factor for idiopathic pulmonary fibrosis. They also predispose to severe emphysema in smokers, occurring at a frequency similar to α-1 antitrypsin deficiency. The work shown here points to alveolar stem cell senescence as a driver of these pathologies. Epithelial stem cell failure was associated with secondary inflammatory recruitment and exquisite susceptibility to injury from “second hits.” The findings suggest that efforts to reverse the stem cell failure state directly, rather than its secondary consequences, may be an effective therapy approach in telomere-mediated lung disease.
Keywords: telomerase, idiopathic pulmonary fibrosis, emphysema, senescence
Abstract
Telomere syndromes have their most common manifestation in lung disease that is recognized as idiopathic pulmonary fibrosis and emphysema. In both conditions, there is loss of alveolar integrity, but the underlying mechanisms are not known. We tested the capacity of alveolar epithelial and stromal cells from mice with short telomeres to support alveolar organoid colony formation and found that type 2 alveolar epithelial cells (AEC2s), the stem cell-containing population, were limiting. When telomere dysfunction was induced in adult AEC2s by conditional deletion of the shelterin component telomeric repeat-binding factor 2, cells survived but remained dormant and showed all the hallmarks of cellular senescence. Telomere dysfunction in AEC2s triggered an immune response, and this was associated with AEC2-derived up-regulation of cytokine signaling pathways that are known to provoke inflammation in the lung. Mice uniformly died after challenge with bleomycin, underscoring an essential role for telomere function in AEC2s for alveolar repair. Our data show that alveoloar progenitor senescence is sufficient to recapitulate the regenerative defects, inflammatory responses, and susceptibility to injury that are characteristic of telomere-mediated lung disease. They suggest alveolar stem cell failure is a driver of telomere-mediated lung disease and that efforts to reverse it may be clinically beneficial.
Mutations in telomerase and telomere genes cause abnormal telomere shortening. Clinically, this molecular abnormality manifests in a spectrum of telomere syndromes that recapitulate features of age-associated pathology (1). In highly proliferative compartments, such as the bone marrow, telomere dysfunction causes stem cell exhaustion, and hematopoietic stem cell transplantation can reverse this pathology (1). More commonly, short telomeres predispose to adult-onset disease in the lung, a tissue that has slow cell turnover (1). Idiopathic pulmonary fibrosis and emphysema are the most prevalent clinical manifestations of human telomere syndromes and account for more than 80% of presentations (1, 2). The alveolar structures are preferentially affected in these disorders, and their pathology is marked by inflammation and mesenchymal abnormalities (3, 4). Affected patients are also exquisitely sensitive to pulmonary-toxic drugs, which are fatal even when there is no detectable baseline lung disease (1, 5).
The mechanisms by which telomere defects provoke lung disease are not understood, but a number of observations have pointed to lung-intrinsic factors and epithelial dysfunction as candidate events (6–10). For example, in telomerase-null mice, DNA damage preferentially accumulates in the air-exposed epithelium after environmentally induced injury, such as with cigarette smoke (7). The additive effect of environmental injury and telomere dysfunction has been suggested to contribute to the susceptibility to emphysema seen in these mice (7). Moreover, humans that carry mutations in the surfactant protein C gene, SFTPC, which is expressed exclusively in type 2 alveolar epithelial cells (AEC2s), develop lung disease phenotypes similar to those seen in telomerase mutation carriers (10–12). Pulmonary fibrosis and emphysema patients have also been noted to have abnormally short telomeres in AEC2s (6, 7, 13). These observations, along with AEC2s’ regenerative capacity (14–16), led us to hypothesize that telomere dysfunction is sufficient to provoke AEC2 failure and that this event drives lung disease pathogenesis.
One hurdle to modeling the consequences of telomere dysfunction in a cell type-specific manner is that laboratory mice have very long telomeres (17). In the absence of telomerase, telomere dysfunction can be generated only after several generations of breeding, precluding cell type-specific studies (18). To overcome this limitation, we designed two experimental systems. First we examined the role of telomere shortening in purified AEC2s in a stem cell assay ex vivo. For an in vivo system, we generated a model in which telomere dysfunction can be induced by deleting telomeric repeat-binding factor 2 (Trf2) (19, 20) exclusively in adult AEC2s. Trf2 functions to suppress the DNA damage response, and its loss leads to telomere dysfunction by uncapping, thus allowing cell type-specific studies within a single generation (19, 20). The latter surrogate model allowed us to test the consequences of acquired DNA damage and telomere dysfunction in the adult lung. We show here, in both late-generation telomerase-null mice and in a conditional mutant model, that telomere dysfunction restricted to AEC2s impairs stem cell function by inducing senescence. This program recapitulates the inflammatory responses and susceptibility to injury that are hallmarks of telomere-mediated lung disease.
Results
Short Telomeres in AEC2s Limit Alveolosphere Formation.
We had previously shown that telomerase-null mice, mTR−/−, with short telomeres are more susceptible to cigarette smoke-induced lung injury because of lung-intrinsic defects (7). We therefore first tested the capacity of alveolar stromal cells and AEC2s derived from these mice to support alveolar regeneration in vitro. In this system, primary AEC2s are cultured with freshly isolated stromal cells marked by platelet-derived growth factor receptor α (Pdgfrα) to generate clonally derived, organoid colonies termed “alveolospheres” (Fig. 1A) (14). These structures are comprised exclusively of AEC2s that have both self-renewed and differentiated into type 1 alveolar epithelial cells (AEC1s) (14). We found that Pdgfrα+ stromal cells isolated from mTR−/− mice with short telomeres supported normal colony formation when cultured with wild-type AEC2s (Fig. 1B). In contrast, AEC2s derived from late-generation mTR−/− mice generated significantly fewer colonies when cultured with wild-type stromal cells (P = 0.038) (Fig. 1C). This defect was dependent on telomere length and did not depend on telomerase deletion, because only AEC2s from mTR−/− fourth-generation mice that had short telomeres and an up-regulated DNA damage response had this impairment, while AEC2s from first-generation mice had intact colony-forming capacity (Fig. 1 C and D and Fig. S1A). These data indicated that abnormally short telomeres preferentially limited AEC2 regenerative capacity and that this defect was cell-autonomous.
Fig. 1.

Short telomeres in AEC2s, but not in stromal cells, limit alveolosphere formation. (A) Design of experiments to test the role of telomere length in alveolosphere formation. The H&E-stained image represents a single alveolosphere that was imaged 14 d after AEC2 were plated. (B) Colony-forming efficiency for Pdgfrα+ cells that were sorted from wild-type, mTR−/− first-generation (mTR−/−G1), and mTR−/− fourth-generation (mTR−/−G4) mice that were plated with Sftpc lineage-labeled wild-type AEC2s. Alveolosphere colonies were counted in triplicate on day 14 for each mouse. (C) AEC2s were sorted from wild-type, mTR−/−G1, and mTR−/−G4 mice, mixed with lineage-labeled Pdgfrα+ cells, and counted as in B. (D) 53BP1 foci were enumerated in AEC2s marked by ATP-binding cassette subfamily A member 3 (Abca3) by immunofluorescence (n = 6 mice per group). Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01, Student's t test.
Telomere Dysfunction in Adult AEC2s Preferentially Induces Senescence.
To test the consequences of epithelial-restricted telomere dysfunction on alveolar homeostasis, we generated a model in which it can be induced in adult AEC2s. Telomere shortening is gradual in the absence of telomerase, and because laboratory mice have long telomeres, telomere dysfunction occurs only after four to six generations of breeding, thus precluding cell type-specific studies (17, 18). We generated a conditional model in which DNA damage at telomeres can be induced in a single generation by deleting Trf2. We crossed Sftpc-CreER mice with mice carrying a floxed allele of Trf2 (20–22), and compared Trf2Fl/Fl;Sftpc-CreER experimental mice with Trf2Fl+;Sftpc-CreER controls. Administration of tamoxifen efficiently deleted Trf2, as indicated by its low mRNA levels in AEC2s isolated from Trf2Fl/FlSftpc-CreER mice (4% and 7% of control levels on days 7 and 21, respectively) (Fig. S1 B–F). The deletion of Trf2 resulted in a robust induction of the DNA damage response at telomeres as evidenced by p53-binding protein 1 (53BP1) foci (Fig. 2 A and B). The telomere dysfunction was specific, because the DNA damage foci were detected only in AEC2s and not in neighboring bronchiolar epithelial cells (Fig. 2B). The p53 pathway was also activated, as evidenced by up-regulation of its target genes in freshly isolated AECs, including p21, Bax, Ccng1, and Mdm2; this signal was detected in vivo for days after Trf2 deletion (Fig. 2C). These data indicated that the induction of the DNA damage response was specific and durable in adult AEC2s.
Fig. 2.

Telomere dysfunction in AEC2s provokes senescence. (A) Telomere FISH using a labeled peptide nucleic acid probe that contains the telomere sequence (TTAGGG)4 and staining for 53BP1 shows telomere-induced DNA damage foci in sorted AEC2s 4 d after a single tamoxifen dose. (B) Representative images and quantification of 53BP1 foci. 53BP1 foci (green) were enumerated in AEC2s, identified by Abca3 staining, and in Club cells, identified by Club cell secretory protein (also known as CCSP/CC10). Immunofluorescence was performed on day 5 after tamoxifen (n = 3 mice per group). (C) mRNA levels of p53 targets in sorted AEC2s on days 7 and 21 after tamoxifen as measured by quantitative real-time PCR (qRT-PCR) and normalized to hypoxanthine phosphoribosyltransferase (Hprt) levels (n = 3 mice per group). (D) Lineage trace of AEC2s. Trf2Fl/+;mTmG;Sftpc-CreER and Trf2Fl/Fl;mTmG;Sftpc-CreER mice were given a single injection of tamoxifen (TAM), and lungs were harvested 7, 14, and 21 d later. The fraction of lineage-labeled GFP+ AEC2s, marked by Sftpc, is quantified (n = 3 mice per group). (E) The proliferating fraction of AEC2s following Trf2 deletion. Proliferation was measured after a 14-d EdU label, and proliferating AEC2s were identified by costaining for Sftpc and EdU (n = 5–7 mice per group). (F–H) Trf2Fl/+;mTmG;Sftpc-CreER and Trf2Fl/Fl;mTmG;Sftpc-CreER mice were given a single injection of tamoxifen, and lungs were harvested 7 d later. (F) Histogram of GFP+ lung cells 1 wk after lineage labeling was induced. (G) Low-power images of alveolospheres growing in the Matrigel/Transwell system. (Scale bars: 50 µm.) (H) Quantification of the colony-forming capacity of AEC2s after 7 and 14 d in culture (n = 3 or 4 mice per group). (I) Immunohistochemical staining of alveolospheres after 14 d in culture. Lineage-labeled cells were identified by GFP staining (green). AEC1s (purple), and AEC2s (red) were identified by podoplanin and prosurfactant protein C staining, respectively. (Scale bars, 50 µm.) (J) Quantification of the number of alveolospheres that contain podoplanin+ cells. Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, Student’s t test.
We examined the consequences of telomere dysfunction on AEC2 survival but found no increase in apoptosis [TUNEL assay and cleaved caspase-3 (CC3) (Fig. S1 G and H)]. Instead, the Trf2-deleted AEC2s persisted when we lineage traced their fate using a reporter line (Fig. 2D). When we measured the proliferation rate, we found that EdU (a BrdU analog) incorporation was significantly lower in AEC2s from Trf2Fl/Fl;Sftpc-CreER mice than in AEC2s from controls (14-d label, P < 0.001, Fig. 2E). Notably, although the baseline proliferation rate in tamoxifen-treated mice was higher than we had documented previously in tamoxifen-free mice (7), the proliferation in AEC2s with dysfunctional telomeres was significantly blunted (Fig. 2E). Taken together, the up-regulated DNA damage response, p53 pathway signaling, and the persistence of AEC2s suggested that AEC2s with Trf2 deletion preferentially activated a cellular senescence program in vivo.
Trf2 Deletion Limits Self-Renewal and Differentiation of Alveolar Stem Cells.
To test whether the loss of telomere function affected the regenerative potential of AEC2s, we isolated lineage-labeled cells from Trf2Fl/Fl;Sftpc-CreER lungs and examined their capacity to self-renew and differentiate in the alveolosphere assay. Cre-expressing AEC2s were labeled with a GFP reporter, allowing us to track their fate in culture. At baseline, the fraction of GFP+ AEC2s in vivo was similar in control and experimental mice (Fig. 2F). However, once the AEC2s were seeded and challenged to generate colonies, Trf2-deleted AEC2s failed to form alveolospheres (0.3 vs. 209 and 0.5 vs. 425 per 5,000 AEC2s on days 7 and 14, respectively; P < 0.001) (Fig. 2 G and H). Moreover, although control AEC2s formed clonally derived AEC2s and AEC1s, Trf2-depleted cells did not differentiate and remained arrested on day 21 (Fig. 2 I and J). These data established that telomere dysfunction induced by Trf2 deletion caused alveolar stem cell failure because of a proliferative arrest, the hallmark of cellular senescence.
Epithelial-Restricted Defects Are Sufficient to Recruit Inflammation.
We next examined whether telomere dysfunction in adult AEC2s affected lung function. Trf2Fl/Fl;Sftpc-CreER and Trf2Fl/+;Sftpc-CreER mice were treated with tamoxifen, and lung function was assessed 21 d later. Trf2Fl/Fl;Sftpc-CreER mice had no respiratory distress or weight loss, but pulmonary function studies showed they acquired an expanded total lung capacity and residual volume (Fig. 3A and Fig. S2 A and B). These abnormalities were accompanied by an increase in lung compliance (Fig. S2C). Although mean linear intercepts were statistically similar on average, in 2 of 30 (7%) of the experimental mice, there was significant air space enlargement (>2 SDs above the mean in controls) (Fig. 3B). Bronchoalveolar lavage fluid from experimental mice contained a significantly increased leukocyte fraction that was composed predominantly of macrophages but also included lymphocytes (Fig. 3 C and D). There was also parenchymal inflammation that was graded as moderate or severe in four of nine experimental mice (compared to 1 of 13 controls), and quantification of macrophages by immunohistochemistry documented an increase (P = 0.04) (Fig. 3 E–I). The inflammatory infiltrates were prominent in perivascular areas surrounding the distal bronchioles, as is consistent with systemic recruitment (Fig. 3 G and H). The macrophages were notably iron-laden (Fig. 3F); this abnormality is associated with cigarette smoke-induced lung disease and diffuse alveolar damage in humans. The lymphocytic infiltrate was CD3+, consistent with T-cell recruitment (Fig. S2 D–F), and also similar to the patterns seen in human smokers with lung disease (23). These data established that DNA damage at telomeres in adult AEC2s is sufficient to recruit a robust inflammatory response.
Fig. 3.
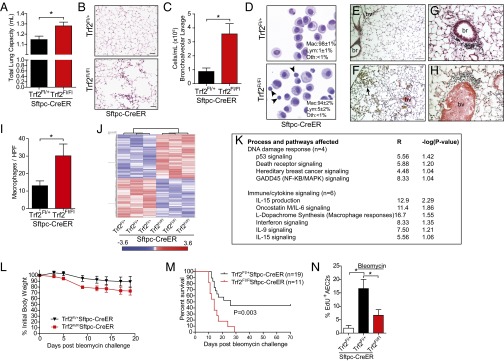
Telomere dysfunction in AEC2s recruits inflammation and impairs repair after injury. (A–I) Trf2Fl/+;Sftpc-CreER and Trf2Fl/Fl;Sftpc-CreER mice were treated with tamoxifen and examined 21 d later. (A) Total lung capacity (n = 6–8 mice per group). (B) High-power image of air space enlargement seen occasionally in Trf2Fl/Fl;Sftpc-CreER mice. (Scale bars: 50 µm.) (C) Bronchoalveolar lavage cellularity. (D) Representative Cytospin images showing macrophages and lymphocytes (marked by arrowheads). (Insets) Differential counts. For C and D, n = 5 mice per group. For differential counts, 250 cells were counted per mouse. (E–H) Representative images from control (E) and Trf2-deleted lungs showing pigmented macrophages (F, arrow) and peribronchiolar (G), and perivascular (H) inflammation. (Scale bars: 100 µm in E and F; 25 µm in G and H.) br, bronchiole; bv, blood vessel. (I) Macrophage quantification per high-powered field (HPF) by Mac-3 immunohistochemistry (n = 4 or 5 mice per group; P value is one-sided). (J) Heat map of gene-expression microarray data from purified AEC2s from Trf2Fl/+;Sftpc-CreER and Trf2Fl/Fl;Sftpc-CreER mice (n = 3 mice per group) 7 d after tamoxifen. Red indicates up-regulated genes; blue indicates down-regulated genes. The fold-change based on color is shown in the key below. (K) Pathways identified by Ingenuity analysis of the up-regulated genes. P is calculated by Fisher’s exact test (right-tailed); R is the ratio of the number of genes in the indicated pathway divided by the total number of genes that make up that pathway. (L and M) Mice treated with tamoxifen 1 wk before bleomycin challenge, were weighed every other day (L), and their survival was monitored (M). The log-rank test was used in the Kaplan–Meier survival analysis. (N) EdU incorporation of AEC2s following bleomycin challenge. Mice were challenged with bleomycin and injected with EdU for 3 d before harvest on day 14 (n = 5 mice per group). Data are expressed as mean ± SEM. *P < 0.05. Unless otherwise noted, Student’s t test was used to calculate P values.
AEC2s with Telomere Dysfunction Up-Regulate Immune-Signaling Pathways.
To define the mechanism by which epithelial-restricted telomere damage recruits inflammation, we performed a gene-expression microarray analysis on sorted AEC2s isolated from tamoxifen-treated Trf2Fl/Fl;Sftpc-CreER and Trf2Fl/+;Sftpc-CreER mice. We found an altered profile with 162 differentially up-regulated and 1,361 down-regulated genes in Trf2-deleted AEC2s (Fig. 3J). Among the differentially up-regulated genes, Il17c, encoding interleukin 17c, and Mif, encoding macrophage inhibitory factor, were notable because they are known to trigger epithelial-derived innate immune responses in the lung (24–26) (real-time PCR confirmation is shown in Fig. S2G). Moreover, the highest-fold up-regulated genes fell in immune-signaling and inflammation pathways in addition to the DNA damage response (Fig. 3K and Table S1). Specifically, one-fourth of the pathways with the highest statistical significance (6 of 23) fell in immune-cytokine signaling even though these pathways represented only a minority of the total examined (significance defined as P ≤ 0.1, Fisher’s exact test). Notably among them was Il15 signaling, which has been implicated in T-cell recruitment in the lung (Fig. 3K) (27). These observations indicated that the telomere dysfunction induced by Trf2 deletion altered the AEC2 transcriptome globally and up-regulated immune-signaling pathways.
Telomere Dysfunction in AEC2s Predisposes to Fatal Lung Disease After Injury.
To test whether telomere dysfunction in AEC2s was relevant to the response to injury, we challenged mice with bleomycin. We chose this model because patients with telomere syndrome are exquisitely susceptible to pulmonary-toxic drugs, such as bleomycin and busuflan, (1, 28). Trf2Fl/Fl;Sftpc-CreER mice that were given bleomycin developed a severe systemic illness marked by accelerated weight loss (Fig. 3L). This pathology was associated with increased mortality, and 100% of mice died by day 29 (11 of 11 in the experimental group vs. 10 of 19 in the control group; P = 0.003, log-rank test) (Fig. 3M). The early mortality in the first 2 weeks in a majority of experimental mice (64%) suggested that acute epithelial injury was a likely cause of respiratory failure. In the mice that we could study premortem, there was no difference in collagen content, apoptosis, or burden of inflammation. However, Trf2-depleted AEC2s showed proliferative defects similar to those we saw in the absence of injury (P = 0.037) (Fig. 3N).
Telomere Dysfunction in Sftpc+ Cells Signals Mesenchymal Abnormalities.
We next generated a congenital model surmising that telomere dysfunction during development in epithelial cells would cause severe lung defects and that this model would allow us to examine the downstream effectors of telomere dysfunction. We studied Trf2Fl/Fl;Sftpc-Cre mice in which Cre recombinase is expressed constitutively and Trf2 is thus deleted in epithelial progenitors during lung development. Mice were born at Mendelian ratios, but Trf2Fl/Fl;Sftpc-Cre mice died from cyanosis and a lung morphogenesis defect within hours after birth, whereas their Trf2Fl/+;Sftpc-Cre littermates survived and had no abnormalities (Fig. 4 A and B and Fig. S3A). Similar to the adult model, the DNA damage response and p53 pathway activation were restricted to epithelial cells (Fig. 4C and Fig. S3 B and C). However, in this developmental context, the response to telomere dysfunction was more severe and manifested in a profound proliferative defect as well as apoptosis (Fig. 4 F and G and Fig. S3 D and E). Importantly, there was extensive secondary mesenchymal apoptosis even though the DNA damage response was limited to epithelial cells (Fig. 4G and Fig. S3G). Trf2Fl/Fl;Sftpc-Cre mice that were bred to the Rosa-mTmG reporter line also confirmed epithelial-restricted Cre expression (Fig. S3H). These data indicated that telomere dysfunction in epithelial cells is sufficient to signal mesenchymal abnormalities.
Fig. 4.
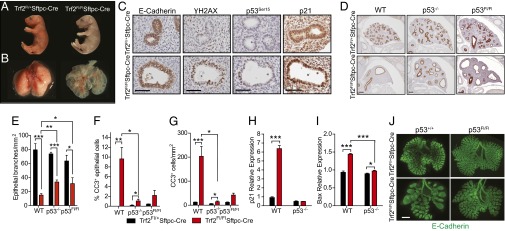
Telomere dysfunction in epithelial cells signals mesenchymal abnormalities via p53. (A) Pups showing cyanosis in mutants after birth (Right). (B) Newborn lungs showing a morphogenesis defect (Right). (C) E-cadherin staining identifies epithelial luminal cells; the remaining panels show DNA damage signaling marked by γH2AX, p53Ser15, and p21 immunohistochemistry. (Scale bars: 50 µm.) (D) E-cadherin staining identifies luminal branches during lung morphogenesis. Images were used for quantification shown in E. (Scale bars: 100 microns.) (E) Lung branching was quantified by enumerating the number of E-cadherin+ lumina per lung area (n = 3–8 mice per group). (F) Number of apoptotic epithelial and mesenchymal (G) cells (n = 2–8 mice per group). (H and I) mRNA quantification of p53 target genes (n = 4 mice per group). (J) E-cadherin whole-mount staining typical of the embryo lungs analyzed (n = 4 per group). (Scale bar: 500 μm.) Unless otherwise noted, studies shown were performed on embryonic day 14.5 lungs. Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, Student’s t test.
The Epithelial-Derived Paracrine Signal Is p53 Dependent in the Developing Lung.
Because the response to telomere dysfunction is mediated by p53 in some contexts (29–31), we generated compound mutant Trf2Fl/Fl;Sftpc-Cre mice that were also null for p53. The p53 deletion resulted in significant amelioration of the lung morphogenesis defect (Fig. 4 D and E). The rescue occurred because p53 deletion abrogated the apoptotic response in epithelial and mesenchymal cells simultaneously, while effectively abolishing p53 downstream effectors including p21 and Bax (Fig. 4 F–I). However, the p53-mediated rescue was partial and did not sustain the survival of newborn pups. To test whether the mesenchymal survival advantage was the result of an epithelial-derived p53 signal, we generated Trf2Fl/Fl;Sftpc-Cre;p53Fl/Fl mice in which p53 deletion was limited to Sftpc-expressing cells. In this experiment, we found the conditional deletion recapitulated the lung morphogenesis rescue seen in constitutively p53−/− mice and was also sufficient to rescue mesenchymal cell survival (Fig. 4 D–G). These data, taken together, indicated that telomere dysfunction in Sftpc+ cells signaled mesenchymal abnormalities in a paracrine manner that was p53 dependent.
Discussion
Telomere Dysfunction in AEC2s Recapitulates Features of Telomere-Mediated Disease.
The data we report here shed light on how telomere dysfunction disturbs alveolar homeostasis in disease and point to an important role for alveolar stem cell failure. Short telomeres limited alveolar regenerative capacity in telomerase-null mice, indicating a cell-autonomous defect. In a conditional model, telomere dysfunction and the consequent induction of DNA damage provoked alveolar stem cell senescence. The conditional model we developed allowed us to overcome an intrinsic hurdle to studies in telomerase-null mice and to dissect the driving events in telomere-mediated lung disease (Table S2). Induction of telomere dysfunction in adult AEC2s was sufficient to provoke lung function abnormalities, secondary inflammation, and catastrophic responses to pulmonary toxic drugs, all features of telomere-mediated lung disease. These observations, in light of our data in telomerase-null mice, indicate that alveolar stem cell failure alone can recapitulate key features of telomere-mediated lung pathology. Dysfunctional telomeres and biomarkers of senescence have been well documented in alveolar epithelial cells from diseased human lungs (6, 13). However, the complexity of the human pathology, in both fibrosis and emphysema, does not distinguish driver from secondary bystander events. Our data in genetically defined animal models indicate that AEC2-dependent telomere dysfunction and senescence limit alveolar repair and can signal mesenchymal abnormalities.
Senescence Is a Preferred Response to Telomere Dysfunction in Alveolar Epithelial Cells.
The data we present here highlight a contrast in the response to telomere dysfunction between high- and low-turnover tissues. In the hematopoietic system, telomere dysfunction causes stem cell failure because of replicative exhaustion, and this phenotype is clinically evident in mice and limits their survival (32–35). In contrast, when telomere dysfunction is restricted to AEC2s, a population with a comparatively slow turnover, the clinical phenotype is fairly well tolerated de novo. However, after injury with a “second hit,” the response is catastrophic, as we saw after bleomycin challenge. Human telomere-mediated lung disease similarly represents an attenuated clinical phenotype that becomes symptomatic in late adulthood, on average four decades later than bone marrow failure, reflecting a more slowly evolving process (36). However, in telomerase mutation carriers, exposure to cigarette smoke and pulmonary toxic drugs are known to provoke exacerbations and functional declines that culminate in respiratory failure (7, 8, 36, 37). The cellular responses to telomere dysfunction are also distinct. In high-turnover epithelial tissues such as the intestinal tract, telomere dysfunction preferentially induces apoptosis (32, 33, 38). On the other hand, AEC2s, in both short-telomere mice (7) and in the conditional model we generated, preferentially undergo senescence. The cellular senescence response may be particularly advantageous in the alveolar space because epithelial cells serve a critical barrier function, and their persistence, even when they have activated a senescence program, preserves the integrity of air exchange.
AEC2 Senescence Signals Secondary Paracrine Abnormalities.
A synthesis of the data we show supports a model in which AEC2 senescence simultaneously causes cell-autonomous defects and up-regulates secondary paracrine signals. In addition to the profound proliferative defects we observed in and ex vivo, we documented evidence of epithelial-derived paracrine signaling that induced inflammation and secondary mesenchymal abnormalities. Telomere-mediated senescence has been associated with altered gene expression as well as an in vitro secretory phenotype (known as “SASP”) in cultured human cells (39, 40). The data we report here show that telomere dysfunction causes senescence-associated paracrine signaling in vivo within the alveolar space that is marked by global transcriptional alterations. This program may be a primary driver of the inflammatory pathology seen in telomere-mediated lung disease. Although more work is needed to characterize the immune responses driven by epithelial senescence, it is noteworthy that the macrophage and T-cell predominance of the infiltrates recapitulate those seen in cigarette smoke-induced lung disease (23). These results raise the possibility that inhibiting inflammation, without reversing the upstream alveolar epithelial defect, may not change the natural history or improve clinical outcomes significantly for patients with telomere-mediated lung disease. Instead, therapy strategies aimed at restoring telomere function or reestablishing epithelial regenerative capacity may be more beneficial.
Methods
Mice were housed at the Johns Hopkins University School of Medicine campus, and procedures were approved by its Institutional Animal Care and Use Committee. Detailed methods outlining the mouse strains, animal studies, alveolar cell isolation along with the immunostaining and RNA analyses are supplied in SI Methods.
Supplementary Material
Acknowledgments
We thank Dr. Carol Greider for critical comments on the manuscript, Mr. Connie Talbot and Dr. Haiping Hao from the Johns Hopkins Microarray Core Facility and the Johns Hopkins Flow Cytometry Core, Mr. Andre Robinson and Mr. James Watkins for assistance with tissue processing, and Dr. Alan Meeker for the software used to analyze telomere length. This work was supported by National Institutes of Health Grants K99 HL113105 (to J.K.A.), K08 HL122521 (to C.E.B.), P01 HL10342 (to W.M.), and R01 HL119476 (to M.A.); by the Ellison Medical Research Foundation (C.E.B. and B.L.M.H.); and by the Flight Attendant’s Medical Research Institute (M.A.).
Footnotes
The authors declare no conflict of interest.
Data deposition: Microarray data compliant with the minimum information about a microarray experiment (MIAME) standard have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE56892).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1504780112/-/DCSupplemental.
References
- 1.Armanios M. Telomeres and age-related disease: How telomere biology informs clinical paradigms. J Clin Invest. 2013;123(3):996–1002. doi: 10.1172/JCI66370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stanley SE, et al. Telomerase mutations in smokers with severe emphysema. J Clin Invest. 2015;125(2):563–570. doi: 10.1172/JCI78554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Noble PW, Barkauskas CE, Jiang D. Pulmonary fibrosis: Patterns and perpetrators. J Clin Invest. 2012;122(8):2756–2762. doi: 10.1172/JCI60323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tuder RM, Petrache I. Pathogenesis of chronic obstructive pulmonary disease. J Clin Invest. 2012;122(8):2749–2755. doi: 10.1172/JCI60324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de la Fuente J, Dokal I. Dyskeratosis congenita: Advances in the understanding of the telomerase defect and the role of stem cell transplantation. Pediatr Transplant. 2007;11(6):584–594. doi: 10.1111/j.1399-3046.2007.00721.x. [DOI] [PubMed] [Google Scholar]
- 6.Alder JK, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci USA. 2008;105(35):13051–13056. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alder JK, et al. Telomere length is a determinant of emphysema susceptibility. Am J Respir Crit Care Med. 2011;184(8):904–912. doi: 10.1164/rccm.201103-0520OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Armanios MY, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356(13):1317–1326. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 9.Zoz DF, Lawson WE, Blackwell TS. Idiopathic pulmonary fibrosis: A disorder of epithelial cell dysfunction. Am J Med Sci. 2011;341(6):435–438. doi: 10.1097/MAJ.0b013e31821a9d8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas AQ, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med. 2002;165(9):1322–1328. doi: 10.1164/rccm.200112-123OC. [DOI] [PubMed] [Google Scholar]
- 11.Cottin V, et al. Combined pulmonary fibrosis and emphysema syndrome associated with familial SFTPC mutation. Thorax. 2011;66(10):918–919. doi: 10.1136/thx.2010.151407. [DOI] [PubMed] [Google Scholar]
- 12.Armanios M. Telomerase and idiopathic pulmonary fibrosis. Mutat Res. 2012;730(1-2):52–58. doi: 10.1016/j.mrfmmm.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsuji T, Aoshiba K, Nagai A. Alveolar cell senescence in patients with pulmonary emphysema. Am J Respir Crit Care Med. 2006;174(8):886–893. doi: 10.1164/rccm.200509-1374OC. [DOI] [PubMed] [Google Scholar]
- 14.Barkauskas CE, et al. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123(7):3025–3036. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hogan BL, et al. Repair and regeneration of the respiratory system: Complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell. 2014;15(2):123–138. doi: 10.1016/j.stem.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desai TJ, Brownfield DG, Krasnow MA. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature. 2014;507(7491):190–194. doi: 10.1038/nature12930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hemann MT, Greider CW. Wild-derived inbred mouse strains have short telomeres. Nucleic Acids Res. 2000;28(22):4474–4478. doi: 10.1093/nar/28.22.4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blasco MA, et al. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997;91(1):25–34. doi: 10.1016/s0092-8674(01)80006-4. [DOI] [PubMed] [Google Scholar]
- 19.de Lange T. How telomeres solve the end-protection problem. Science. 2009;326(5955):948–952. doi: 10.1126/science.1170633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Celli GB, de Lange T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat Cell Biol. 2005;7(7):712–718. doi: 10.1038/ncb1275. [DOI] [PubMed] [Google Scholar]
- 21.Rock JR, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci USA. 2011;108(52):E1475–E1483. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lazzerini Denchi E, Celli G, de Lange T. Hepatocytes with extensive telomere deprotection and fusion remain viable and regenerate liver mass through endoreduplication. Genes Dev. 2006;20(19):2648–2653. doi: 10.1101/gad.1453606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360(23):2445–2454. doi: 10.1056/NEJMra0804752. [DOI] [PubMed] [Google Scholar]
- 24.Pfeifer P, et al. IL-17C is a mediator of respiratory epithelial innate immune response. Am J Respir Cell Mol Biol. 2013;48(4):415–421. doi: 10.1165/rcmb.2012-0232OC. [DOI] [PubMed] [Google Scholar]
- 25.Fallica J, et al. Macrophage migration inhibitory factor is a novel determinant of cigarette smoke-induced lung damage. Am J Respir Cell Mol Biol. 2014;51(1):94–103. doi: 10.1165/rcmb.2013-0371OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Calandra T, Roger T. Macrophage migration inhibitory factor: A regulator of innate immunity. Nat Rev Immunol. 2003;3(10):791–800. doi: 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verbist KC, Cole CJ, Field MB, Klonowski KD. A role for IL-15 in the migration of effector CD8 T cells to the lung airways following influenza infection. J Immunol. 2011;186(1):174–182. doi: 10.4049/jimmunol.1002613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dietz AC, et al. Disease-specific hematopoietic cell transplantation: Nonmyeloablative conditioning regimen for dyskeratosis congenita. Bone Marrow Transplant. 2011;46(1):98–104. doi: 10.1038/bmt.2010.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chin L, et al. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell. 1999;97(4):527–538. doi: 10.1016/s0092-8674(00)80762-x. [DOI] [PubMed] [Google Scholar]
- 30.Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science. 1999;283(5406):1321–1325. doi: 10.1126/science.283.5406.1321. [DOI] [PubMed] [Google Scholar]
- 31.Guo N, et al. Short telomeres compromise β-cell signaling and survival. PLoS ONE. 2011;6(3):e17858. doi: 10.1371/journal.pone.0017858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee HW, et al. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392(6676):569–574. doi: 10.1038/33345. [DOI] [PubMed] [Google Scholar]
- 33.Rudolph KL, et al. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 1999;96(5):701–712. doi: 10.1016/s0092-8674(00)80580-2. [DOI] [PubMed] [Google Scholar]
- 34.Hao LY, et al. Short telomeres, even in the presence of telomerase, limit tissue renewal capacity. Cell. 2005;123(6):1121–1131. doi: 10.1016/j.cell.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 35.Armanios M, et al. Short telomeres are sufficient to cause the degenerative defects associated with aging. Am J Hum Genet. 2009;85(6):823–832. doi: 10.1016/j.ajhg.2009.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parry EM, Alder JK, Qi X, Chen JJ, Armanios M. Syndrome complex of bone marrow failure and pulmonary fibrosis predicts germline defects in telomerase. Blood. 2011;117(21):5607–5611. doi: 10.1182/blood-2010-11-322149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diaz de Leon A, et al. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS ONE. 2010;5(5):e10680. doi: 10.1371/journal.pone.0010680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jonassaint NL, Guo N, Califano JA, Montgomery EA, Armanios M. The gastrointestinal manifestations of telomere-mediated disease. Aging Cell. 2013;12(2):319–323. doi: 10.1111/acel.12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coppé JP, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang H, Pan KH, Cohen SN. Senescence-specific gene expression fingerprints reveal cell-type-dependent physical clustering of up-regulated chromosomal loci. Proc Natl Acad Sci USA. 2003;100(6):3251–3256. doi: 10.1073/pnas.2627983100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.