

Significance
The airway pathogen Mycoplasma pneumoniae (Mp) produces a virulence factor with ADP-ribosyltransferase and vacuolating activities known as Community-Acquired Respiratory Distress Syndrome Toxin (CARDS TX). Mammalian cells exposed to recombinant CARDS TX exhibit distinct patterns of ADP-ribosylated proteins and unusual vacuolation phenotypes with disruption of monolayer integrity, leading to cell death. Recombinant CARDS TX administered to naive mice induces an allergic-type inflammatory response and airway hyperreactivity, suggesting that an analogous response in humans may play a causal role in Mp-associated asthma. Here we present the structure of CARDS TX, the newest member of the well-recognized group of ADP-ribosylating toxins. The structure enhances understanding of Mp pathogenicity and provides a platform for the development of therapies to treat Mp-associated acute and chronic airway diseases.
Keywords: mycoplasma cytotoxin, single-crystal X-ray diffraction, ADP-ribosyltransferase, vacuolation, reactive airway disease
Abstract
Mycoplasma pneumoniae (Mp) infections cause tracheobronchitis and “walking” pneumonia, and are linked to asthma and other reactive airway diseases. As part of the infectious process, the bacterium expresses a 591-aa virulence factor with both mono-ADP ribosyltransferase (mART) and vacuolating activities known as Community-Acquired Respiratory Distress Syndrome Toxin (CARDS TX). CARDS TX binds to human surfactant protein A and annexin A2 on airway epithelial cells and is internalized, leading to a range of pathogenetic events. Here we present the structure of CARDS TX, a triangular molecule in which N-terminal mART and C-terminal tandem β-trefoil domains associate to form an overall architecture distinct from other well-recognized ADP-ribosylating bacterial toxins. We demonstrate that CARDS TX binds phosphatidylcholine and sphingomyelin specifically over other membrane lipids, and that cell surface binding and internalization activities are housed within the C-terminal β-trefoil domain. The results enhance our understanding of Mp pathogenicity and suggest a novel avenue for the development of therapies to treat Mp-associated asthma and other acute and chronic airway diseases.
The Centers for Disease Control and Prevention estimate that 2 million people are infected with Mycoplasma pneumoniae (Mp) each year, with 100,000 requiring hospitalization (1). The bacterium is best known as a cause of atypical, or “walking,” pneumonia (2, 3) and is increasingly appreciated for its role in the etiology of reactive airway diseases, such as asthma and adult respiratory distress syndrome (ARDS) (4, 5). Mp infection induces cellular vacuolation and deterioration of ciliary movement, eventually leading to cell death in the airway environment, as well as a spectrum of extrapulmonary complications (6–9). Diagnosis of infection can be complicated by a latency period of up to 3 wk, difficulties in cultivating the microorganism from clinical specimens, inadequate diagnostic tools, and a broad spectrum of clinical manifestations, some of which are similar to those of other pulmonary conditions (2, 3). These characteristics, together with the ability of Mp to evade the immune system through intracellular localization and by adjusting its membrane composition to mimic that of its host (6), have hindered a comprehensive understanding of Mp infectious processes.
Mp was first detected in patients with atypical pneumonia, but was believed to be a virus because it was resistant to penicillin and sulfonamides and passed through bacteria-retaining filters (10). Mycoplasmas are the smallest self-replicating life forms identified and are postulated to have undergone genome reduction (11). Their streamlined genome size imposes nutritional requirements that dictate a parasitic lifestyle. In this context, during 50 y of study, the organism was believed to be devoid of cytotoxins, a paradigm that shifted in 2005 when a 591-aa Mp protein with mono-ADP ribosyltransferase (mART) and vacuolating activities, termed Community-Acquired Respiratory Distress Syndrome Toxin (CARDS TX), was isolated through its ability to bind with high affinity to surfactant protein A (SP-A), the most abundant protein component of pulmonary surfactant (12–14). The discovery of a previously unknown Mp virulence factor cast Mp in a different light and opened the door for a new avenue of research on what has historically been an enigmatic organism.
In the decade since the discovery of CARDS TX, advances have been made in understanding the toxin’s role in Mp pathogenicity and reactive airway disease. The gene encoding the toxin and its promoter have been analyzed, and mRNA and protein levels have been measured under various growth conditions (15). Relative to SP-4 broth-grown Mp cultures, during infection of mammalian cells, CARDS TX mRNA levels increase substantially. Analysis of infected mouse lung tissue revealed high expression of CARDS TX compared with levels when grown in SP-4 medium on a per cell basis. These data indicate CARDS TX expression is up-regulated during Mp infection, possibly owing to as-yet unidentified environmental cues (15).
When removed from the context of the bacterium, recombinant CARDS TX elicits the characteristic inflammation and vacuolated airway histopathology in intoxicated mice in a dose-dependent fashion, as observed with Mp organisms (16). Baboon tracheal organ cultures exposed to purified toxin demonstrate dose-dependent slowing and eventual cessation of cilia movement and disruption of respiratory cell integrity (12). Naive mice administered one dose respond with 30- to 80-fold increases in the expression of Th-2 cytokines and chemokines, a robust eosinophilia, accumulation of T and B cells, mucus metaplasia, and airway hyperreactivity, suggesting that an analogous allergic-type response in humans may play a causal role in Mp-associated asthma (16, 17). These studies reveal that CARDS TX recapitulates the spectrum of respiratory pathologies associated with Mp infection.
The aforementioned results are consistent with CARDS TX interaction with SP-A, which is abundant in mouse lung and baboon tracheal rings; however, recombinant CARDS TX carries out ADP-ribosylating and vacuolating activities in mammalian cell lines that lack SP-A, suggesting that it uses alternative receptors (18, 19). Using recombinant CARDS TX as bait, AnxA2 was isolated from the membrane fraction of A549 airway cells (19). HepG2 cells express low levels of AnxA2, but on transfection with a plasmid expressing AnxA2, toxin binding and cellular vacuolation are enhanced. NCI-H441 cells express both SP-A and AnxA2, but on transfection with siRNAs specific for those proteins, expression in these cells decreases relative to that in nontransfected controls, and CARDS TX binding and cellular vacuolation are proportional to AnxA2 and SP-A levels, although SP-A is preferred as a binding target (19).
The acidic vacuoles induced by CARDS TX originate from Rab9-associated compartments and are derived from the endocytic pathway (18). The V-ATPase inhibitor bafilomycin A1 and the ionophore monesin inhibit vacuole formation. The late endosomal GTPase Rab9 is enriched at the vacuole membranes, an observation confirmed by overexpression of GFP-Rab9 and the use of immunogold-labeled Rab9. The late endosomal/lysosomal-associated membrane proteins LAMP1 and LAMP2 are also localized to the vacuolar membranes, but the late endosomal protein Rab7 and early endosomal markers Rab5 and EEA1 are not. HeLa cells expressing dominant-negative Rab9 demonstrate reduced vacuole formation, whereas those expressing dominant-negative Rab7 do not. These data suggest that Rab9 is involved in CARDS TX-mediated vacuole formation (18), although how CARDS TX influences Rab9 association with vacuoles is unclear.
CARDS TX activates the NLRP3 inflammasome, an important player in regulating the immune response during infection via activation of procaspase-1, which in turn cleaves pro-IL-1β into its mature form (20). The cytokine IL-1β dictates the severity of inflammation associated with a spectrum of inflammatory diseases. CARDS TX colocalizes with and activates the inflammasome via ADP ribosylation of NLRP3 (20), but CARDS TX proteins with mutations that abrogate its mART activity and truncated molecules that cannot be internalized by macrophages do not. These results are relevant because inflammasome activation is known to contribute to lung remodeling and the development of chronic airway diseases, such as asthma and chronic obstructive pulmonary disease (21).
To better understand the molecular basis for the action of CARDS TX, we crystallized (22) and determined its structure using single-crystal X-ray diffraction. The architecture and domain organization of CARDS TX are unique relative to other ADP-ribosylating bacterial toxins. Functional data demonstrate that CARDS TX binds phosphatidylcholine (PC) and sphingomyelin (SM) specifically over other membrane lipids. Truncation of the C-terminal 20 residues abrogates cell surface binding and internalization, suggesting that receptor binding functions reside in D3. The combination of structural and functional data presented herein serves as the starting point for the design of vaccine candidates and small molecules that block CARDS TX action, providing an opportunity to reduce acute and chronic Mp-related disease manifestations.
Results and Discussion
Overall Structure of CARDS TX.
CARDS TX was crystallized as described previously (22), and its structure was determined at resolutions of 1.9 Å and 2.6 Å (Fig. 1, Fig. S1, and Table S1). The toxin consists of 17 α-helices and 43 β-strands that fold into three domains arranged in the shape of an isosceles triangle, with sides of 65 × 65 × 55 Å and a depth of 35 Å (Figs. 1 and 2). The N-terminal mART domain (D1) associates with tandem C-terminal β-trefoil subdomains (D2+D3) via a linker and an extensive interface. The contacts between D1 and D2+D3 are predominantly polar (Fig. S1 C and D), suggesting that the interface can be disrupted.
Fig. 1.

CARDS TX tertiary structure. In all figures, the D1 mART domain is shown in light blue, the steric block of the NAD+-binding pocket is violet, the linker connecting D1 to D2+D3 is dark red, D2 is dark blue, D3 is green, Cys-Sγ atoms are yellow spheres, and F591 is bright red. The aromatic patch in D3 is shown as orange sticks.
Fig. 2.
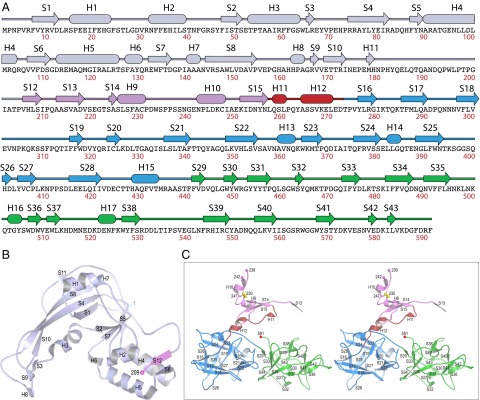
CARDS TX secondary structure. (A) Secondary structure observed crystallographically as a function of amino acid sequence. (B) The D1 mART domain lacking the steric block labeled according to A. The view is ∼90° around the horizontal relative to Fig. 1. (C) Divergent stereo pair in the same orientation as Fig. 1 showing the steric block of the NAD+-binding pocket, linker, and D2+D3 β-trefoil domain structural elements labeled as in A.
CARDS TX mART Domain.
A DALI search (23) of the Protein Data Bank (PDB) returns the mART domains of pertussis toxin (PTx; Bordetella pertussis; PDB ID code 1PRT) (24) and cholera toxin (CTx; Vibrio cholerae; PDB ID code 2A5F) (25) as those most similar to D1. The mART domains of all three toxins have the R–STS/T–E signature motif characteristic of ADP-ribosylating toxins of the cholera toxin subgroup (CTxg) (26) (Fig. 3A). When structurally aligned, the active site signature residues are conserved in 3D space despite limited overall sequence identity (Fig. 3B). The N-terminal arginine (R10) and the midregion Ser-Thr-Ser (S49-T50-S51) are predicted to bind and orient NAD+, whereas the invariant catalytic Glu (E132) is responsible for the transferase activity (12, 27). The six residues preceding and two residues following E132 form the ADP-ribosylating turn-turn (ARTT) motif (Fig. 3A and Fig. S2), which is believed to participate in target protein recognition (28). The CARDS TX ARTT motif is distinct from other bacterial mART domains (Fig. 3A), and the immediately preceding residues 94–126 form a helix-strand-helix motif (H4-S6-H5) that comprises the D1 component of the D1+D3 interface (Fig. 1 and Fig. S1 A and D).
Fig. 3.
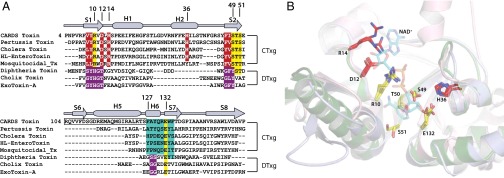
CARDS TX is a member of the CTx subgroup (CTxg) of bacterial mART domains. (A) Structure-based alignment of ADP-ribosylating toxins from the cholera (CTxg) and diphtheria toxin (DTxg) subgroups. CARDS TX secondary structure is indicated at the top. The R–STS/T–E signature motif is shared by CTxg members (yellow). ARTT residues are in cyan. The S6-H5 portion of the H4-S6-H5 motif found only in CARDS TX is boxed. (B) CARDS TX (light blue) and PTx (green) mART domains superimposed on the CTx mART domain (pink) in complex with NAD+ (cyan sticks).
Steric Blocks of CARDS TX mART Activity.
The conformation of CARDS TX shown in Fig. 1 does not appear competent to act on target proteins, because the active site and target recognition elements are inaccessible. Residues 206–256 form a physical barrier similar to that of PTx (Fig. 4 A and B and Fig. S2) that renders the NAD+-binding site inaccessible. Residues 127–134 containing the catalytic E132 residue and the target-protein recognition ARTT motif are buried by this barrier and by the extensive interface between D1 and D2+D3 (Fig. 4 C and D and Fig. S2). The C-terminal residues of the protein, R590 and F591, interact with E52 and R131 positioned adjacent to the S49-T50-S51 and catalytic E132 residues of the signature sequence, respectively (Fig. 4E and Fig. S2C).
Fig. 4.
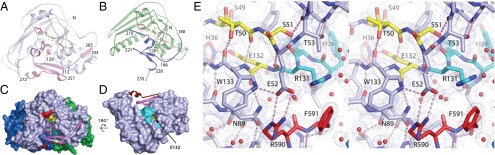
Steric blocks of CARDS TX and PTx mART active sites. The R–STS/T–E signature motif residues are colored as in Fig. 3A. (A) CARDS TX mART domain. C230 and C247 within the steric block form a disulfide bond (yellow spheres). The view is as in Fig. 2B. (B) PTx mART domain. C41 and C201 form a disulfide bond (yellow spheres) linking the steric block to the N-terminal portion of the mART domain. (C) Surface representation highlighting the steric block of the CARDS TX NAD+-binding pocket. (D) Catalytic and substrate recognition residues are buried at the D1/D2D3 interface. D2 and D3 have been removed for clarity. A tunnel into the active site runs adjacent to the ARTT motif. The yellow surface inside the tunnel is E132. (E) 1.9-Å electron density with coefficients 2mFo − DFc contoured at 1.2 σ superimposed on the refined CARDS TX structure. The C-terminal residues of the toxin, R590 and F591, project into the active site.
CARDS TX β-Trefoil Domains.
The tandem C-terminal domains D2+D3 are similar in fold and relative orientation to those of ricin B (PDB ID code 2AAI) (29). Unlike ricin B, however, the CARDS TX D2+D3 β-trefoils do not have galactose-binding sites, and the protein is negative for saccharide binding when screened against mammalian glycans in chip-based array experiments at the Consortium for Functional Glycomics. A striking feature of CARDS TX D3 is its aromatic amino acid content (35 of 152 residues), 15 of which cluster to form an aromatic patch reminiscent of that found in the cytolethal distending toxin (CDT) from Haemophilus ducreyi (PDB ID code 1SR4) (30) (Fig. 1 and Fig. S3). Mutagenesis of the aromatic patch in CDT interferes with cell surface binding and internalization (31). Similarly, and in contrast to the wild type protein, a CARDS TX truncation variant lacking residues 571–591 is not internalized by HeLa cells (Fig. S4A). Residues 571–591 are integral to the proper folding of D3 and formation of its aromatic patch (Fig. S4B). These data suggest that entry of CARDS TX into host cells is mediated by D3. Previous work indicated that CARDS TX-mediated vacuolating activity also resides in D2+D3 and is independent of D1 or mART activity, because catalytic mutants Arg10Ala, His36Ala, and Glu132Ala and N-terminal truncation variants 178CARDS591 and 264CARDS591 all induce vacuolation when incubated with HeLa cells as efficiently as the native toxin (27).
CARDS TX Binds PC and SM, But Not Other Membrane Lipids.
Previous data indicate that SP-A and AnxA2 serve as independent receptors for CARDS TX binding and vacuolation (12, 19); however, binding and vacuolation were also noted in the absence of these proteins (19), prompting a search for nonproteinaceous receptors. Although negative for glycan binding, CARDS TX binds PC and dipalmitoylphosphatidylcholine (DPPC) specifically over choline, phosphatidic acid, and other membrane lipids (Fig. 5A). Using a larger panel of membrane-associated lipids, we found that full-length CARDS TX and D2+D3 alone bind PC and SM, but D1 alone does not (Fig. 5B). PC and SM differ in their backbones, but both have phosphocholine head groups. Phosphocholine-containing membrane lipids are enriched in the outer leaflet of the plasma membrane, whereas phospholipid molecules containing a terminal primary amino group (e.g., phosphatidylethanolamine, phosphatidylserine) are present predominantly in the inner leaflet (32). Airway cells are covered by pulmonary surfactant, which is ∼40% DPPC, ∼40% PC, ∼5% surfactant proteins (i.e., SP-A, -B, -C, and -D), cholesterol, and other substances (14, 33). When adsorbing to the air–water interface of alveoli, the hydrophilic head groups are oriented in the water and the hydrophobic tails face the airspace, enabling PC and DPPC to reduce surface tension, thereby facilitating lung expansion (34). SP-A is known to associate directly with DPPC (35). Although some bacterial toxins, such as botulinum neurotoxin type A (36), use protein and membrane lipid components as coreceptors, whether PC and SM serve as coreceptors for CARDS TX remains to be determined.
Fig. 5.

CARDS TX binds PC, DPPC, and SM specifically over other membrane lipids. (A) ELISA and (B) dot blot analyses of CARDS TX binding to a panel of diverse membrane lipids and their components as described in Methods. Full-length, D1, and D2+D3 constructs were tested. The 16 lipids/lipid components on each strip are (1) triglyceride, (2) diacylglycerol, (3) phosphatidic acid, (4) phosphatidylserine, (5) phosphatidylethanolamine, (6) PC, (7) phosphatidylglycerol, (8) cardiolipin, (9) phosphatidylinositol, (10) phosphatidylinositol 4-phosphate, (11) phosphatidylinositol 4,5-bisphosphate, (12) phosphatidylinositol 3,4,5-trisphosphate, (13) cholesterol, (14) SM, (15) 3-sulfogalactosylceramide, and (16) blank.
Unique Architecture of CARDS TX.
The combination of catalytic and cell surface binding activities and the composition and spatial arrangement of CARDS TX domains is unique among the family of bacterial ADP-ribosylating toxins (reviewed in ref. 37). The mART domains of PTx (24) (Figs. 3B and 4B), CTx (38) (Fig. 3B), and heat-labile enterotoxin (39) are the most closely related to CARDS TX D1 (Fig. 2A), but in lieu of β-trefoils, they have pentameric assemblies of polypeptides that bind carbohydrates (Fig. S5 A and B). To our knowledge, mosquitocidal toxin is the only other bacterial toxin with solely mART and β-trefoil domains (40), but its organization differs, with four (nontandem) β-trefoil domains connected by flexible linkers to form “beads on a string” that encircle the catalytic mART domain (Fig. S5D). Although CARDS TX, CDT, and ricin might be considered generally similar in architecture in that each has a cytotoxic catalytic domain paired with two β-trefoil domains, CARDS TX remains unique relative to these toxins from a functional perspective, because ricin and CDT have N-glycoside hydrolase (41) and nuclease (30) catalytic activities, respectively.
Summary of CARDS TX Structure/Function Relationships.
The structural and functional data presented here provide insight into this latest addition to the group of ADP-ribosylating bacterial toxins. CARDS TX activates the NLRP3 inflammasome via ADP ribosylation of its NLRP3 component (20). The molecular basis for CARDS TX–inflammasome interactions likely involves the CARDS TX ARTT and the adjacent H4-S6-H5 motifs (Fig. 3 A and B), the latter of which mediate interactions at the D1–D3 interface (Fig. 1 and Fig. S1D). The available data do not reveal the specific CARDS TX residues that interact with cell surface lipids and protein receptors, but they do indicate that PC, DPPC, and SM binding activities are housed within D2+D3 (Fig. 5B). CARDS TX D2+D3 domains were previously reported to mediate binding, internalization, and vacuolation activities (19, 27). The crystal structure suggests that deletion of residues 571–591 in D3 or residues 274–308 in D2 will severely disrupt the integrity of the β-trefoil domains in which they reside (Fig. S4 B and C). Importantly, the former truncation abrogates binding and internalization (Fig. S4A) but the latter truncation does not (27), implicating D3 as the mediator of these activities.
The steric blocks of target recognition elements and the NAD+-binding sites suggest that CARDS TX-mediated ADP ribosylation requires a conformational change or proteolytic nicking to permit the separation of D1 from D2+D3. This notion seems plausible, given previous work demonstrating that D1 and D2+D3 in isolation retain their respective mART and binding and internalization activities (27). The solvent-exposed loop between Cys230 and Cys247 is a candidate for a proteolytic cleavage event, because residues 237–241 of this loop are disordered in all seven CARDS TX molecules in the present study (Fig. 1 and Fig. S1 A and B), and flexible loops are often targets of protease action. A cleavage event in this loop followed by reduction of the C230–C247 disulfide bond is predicted to relieve the autoinhibitory block of the NAD+-binding site in D1 and may cause dissociation of the N- and C-terminal portions; however, reduction of the Cys230–Cys247 disulfide bond itself may relieve the steric blocks of the NAD+-binding pocket, active site, and ARTT target recognition motif while leaving D2+D3 tethered to D1.
Because Mp is implicated in many airway and extrapulmonary diseases, and because CARDS TX alone replicates the cell injury associated with Mp infection, the structure serves as a starting point for the design of vaccine candidates and small molecules that block CARDS TX action, thereby affording the opportunity to eliminate/reduce a wide range of acute and chronic Mp-related disease manifestations.
Materials and Methods
Protein Expression and Purification.
“Nicked” CARDS TX was generated, purified, and crystallized as described previously (22). “Unnicked” CARDS TX was expressed and purified using the same protocol except that (i) cells were grown in Terrific Broth; (ii) the N-terminal 8× His tag was not removed; (iii) the protein was dialyzed against buffer containing 25 mM Hepes, pH 7.5, and 2 mM TCEP; and (iv) the protein was concentrated to ∼10 mg/mL for crystallization trials.
Crystallization and Data Collection.
Nicked CARDS TX crystallized in space group C2 as described previously (22). Unnicked CARDS TX crystallized in space group R3 using the sitting-drop vapor diffusion method and a Phoenix liquid-handling robot (Art Robbins Instruments). The reservoir solution (Qiagen PEG II suite) contained 30% PEG 1000 and 0.1 M Tris⋅HCl pH 8.5. The PEG 1000 present in the mother liquor was a suitable cryoprotectant, and the crystals were mounted directly onto nylon cryoloops and flash-frozen. Diffraction data from unnicked CARDS TX crystals were obtained at the Advanced Photon Source of the Argonne National Laboratory on beamline 24-ID-E using an ADSC Q315 detector (315 mm × 315 mm). Diffraction data were processed using the HKL-2000 program package (42).
Structure Determination and Refinement.
Crystals of nicked CARDS TX contain six molecules in the asymmetric unit, and crystals of unnicked CARDS TX contain one molecule in the asymmetric unit. The nicked CARDS TX structure was determined using single isomorphous replacement with anomalous scattering (SIRAS) by pressurizing a xenon chamber (Hampton Research) to 400 psi for 15 min before flash-cooling. Xenon positions were determined using SHELXD (43). Phases from 27 xenon sites were calculated and refined in SHARP (44), followed by density modification in RESOLVE (45). Sixfold noncrystallographic symmetry (NCS) averaging produced an electron density map of quality sufficient to obtain a Cα backbone trace. Phases calculated from the Cα backbone combined with the experimental phases yielded a phase set suitable for automated structure building in PHENIX (46). The six CARDS TX molecules in the asymmetric unit were refined without NCS restraints in PHENIX alternating with manual rebuilding in COOT (47).
The structure of unnicked CARDS TX was determined by molecular replacement in PHENIX using the nicked CARDS TX protomer A as the search model, followed by iterative rounds of refinement and model adjustment. Structure validation was performed by PROCHECK (48). The coordinates and structure factors are available in the PDB (ID codes 4TLV and 4TLW). All structural figures were created using PyMol version 1.7 (Schrödinger).
ELISA Lipid-Binding Analyses.
Microtiter plate (Immunoplate; Nunc) wells were coated with 50 μL of phospholipids found in cell membranes (125–500 ng/well). After drying, wells were washed twice with PBS and blocked with 200 μL of 1 mg/mL BSA in PBS for 1 h at 37 °C. Then CARDS TX (100 ng/well) was added, followed by incubation for another 1 h at 37 °C. After thorough washing with PBS, anti-CARDS TX polyclonal antibody (1:4,000 dilution in 1% skim milk/PBS) was added. The wells were again washed with PBS, and HRP-conjugated goat anti-rabbit secondary antibody (Invitrogen; 1:2,000 dilution in 1% skim milk/PBS) was added, followed by a 45-min incubation and then the addition of 3,3′,5,5′-tetramethylbenzidine substrate and measurement at 450 nm. Wells coated with BSA alone served as negative controls.
Dot Blot Lipid-Binding Analyses.
Toxin binding to membrane lipids was analyzed using membrane-associated lipids (Membrane Lipid Strips), phosphoinositides (PIP Strips), and sphingolipids (SphingoStrips) in accordance with the instructions from the manufacturer (Echelon Biosciences). Membranes were blocked for 2 h in 4% (wt/vol) skim milk or BSA dissolved in PBS. Then purified CARDS TX at 10 μg/mL was added, and membranes were incubated overnight at room temperature on a rotary shaker. Membranes were washed three times in PBS and incubated with anti-CARDS TX polyclonal antibody (1:4,000 dilution in 1% skim milk/PBS), followed by alkaline phosphatase-conjugated goat anti-rabbit secondary antibody (1:2,000 dilution in 1% skim milk/PBS).
CARDS TX Cell Surface Binding and Internalization.
The binding and internalization of the full-length and truncated 1–570 CARDS TX variant were analyzed as described previously (49).
Supplementary Material
Acknowledgments
We thank Jonathan Schuermann for technical support. J.B.B., P.J.H., and coauthors are supported by National Institutes of Health Grant U19 AI070412. J.B.B. is also supported by the Kleberg Foundation. P.J.H. is also supported by R. A. Welch Foundation Grant AQ-1399. Support for Northeastern Collaborative Access Team beamline 24-ID-E is provided by National Institutes of Health Grant P41 GM103403 and US Department of Energy Grant DE-AC02–06CH11357. The University of Texas Health Science Center at San Antonio’s X-Ray Crystallography Core Laboratory is supported in part by the Office of the Vice President for Research and by San Antonio Cancer Institute Grant P30 CA054174.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4TLV and 4TLW).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1420308112/-/DCSupplemental.
References
- 1.Centers for Disease Control and Prevention, National Center for Immunization and Respiratory Diseases Mycoplasma pneumoniae infection: Surveillance and reporting. 2014 Available at www.cdc.gov/pneumonia/atypical/mycoplasma/surv-reporting.html. Accessed March 15, 2015.
- 2.Baseman JB, Tully JG. Mycoplasmas: Sophisticated, reemerging, and burdened by their notoriety. Emerg Infect Dis. 1997;3(1):21–32. doi: 10.3201/eid0301.970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waites KB, Talkington DF. Mycoplasma pneumoniae and its role as a human pathogen. Clin Microbiol Rev. 2004;17(4):697–728. doi: 10.1128/CMR.17.4.697-728.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nisar N, Guleria R, Kumar S, Chand Chawla T, Ranjan Biswas N. Mycoplasma pneumoniae and its role in asthma. Postgrad Med J. 2007;83(976):100–104. doi: 10.1136/pgmj.2006.049023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peters J, et al. Persistence of community-acquired respiratory distress syndrome toxin-producing Mycoplasma pneumoniae in refractory asthma. Chest. 2011;140(2):401–407. doi: 10.1378/chest.11-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baseman JB, Reddy SP, Dallo SF. Interplay between mycoplasma surface proteins, airway cells, and the protean manifestations of mycoplasma-mediated human infections. Am J Respir Crit Care Med. 1996;154(4 Pt 2):S137–S144. doi: 10.1164/ajrccm/154.4_Pt_2.S137. [DOI] [PubMed] [Google Scholar]
- 7.Berg CP, et al. Mycoplasma antigens as a possible trigger for the induction of antimitochondrial antibodies in primary biliary cirrhosis. Liver Int. 2009;29(6):797–809. doi: 10.1111/j.1478-3231.2008.01942.x. [DOI] [PubMed] [Google Scholar]
- 8.Talkington DF, Waites KB, Schwartz SB, Besser RE. Emerging From Obscurity: Understanding Pulmonary and Extrapulmonary Syndromes, Pathogenesis, and Epidemiology of Human Mycoplasma pneumoniae Infections. American Society for Microbiology; Washington, DC: 2001. [Google Scholar]
- 9.Hu PC, Collier AM, Baseman JB. Interaction of virulent Mycoplasma pneumoniae with hamster tracheal organ cultures. Infect Immun. 1976;14(1):217–224. doi: 10.1128/iai.14.1.217-224.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eaton MD, Meiklejohn G, van Herick W, Corey M. Studies on the etiology of primary atypical pneumonia, I: Properties of the virus isolated and propagated in chick embryos. J Exp Med. 1945;82(5):317–328. doi: 10.1084/jem.82.5.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meseguer MA, et al. Mycoplasma pneumoniae: A reduced-genome intracellular bacterial pathogen. Infect Genet Evol. 2003;3(1):47–55. doi: 10.1016/s1567-1348(02)00151-x. [DOI] [PubMed] [Google Scholar]
- 12.Kannan TR, Baseman JB. ADP-ribosylating and vacuolating cytotoxin of Mycoplasma pneumoniae represents unique virulence determinant among bacterial pathogens. Proc Natl Acad Sci USA. 2006;103(17):6724–6729. doi: 10.1073/pnas.0510644103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kannan TR, Provenzano D, Wright JR, Baseman JB. Identification and characterization of human surfactant protein A binding protein of Mycoplasma pneumoniae. Infect Immun. 2005;73(5):2828–2834. doi: 10.1128/IAI.73.5.2828-2834.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wright JR. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol. 2005;5(1):58–68. doi: 10.1038/nri1528. [DOI] [PubMed] [Google Scholar]
- 15.Kannan TR, et al. Mycoplasma pneumoniae community-acquired respiratory distress syndrome toxin expression reveals growth phase- and infection-dependent regulation. Mol Microbiol. 2010;76(5):1127–1141. doi: 10.1111/j.1365-2958.2010.07092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hardy RD, et al. Analysis of pulmonary inflammation and function in the mouse and baboon after exposure to Mycoplasma pneumoniae CARDS toxin. PLoS ONE. 2009;4(10):e7562. doi: 10.1371/journal.pone.0007562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Medina JL, et al. Mycoplasma pneumoniae CARDS toxin induces pulmonary eosinophilic and lymphocytic inflammation. Am J Respir Cell Mol Biol. 2012;46(6):815–822. doi: 10.1165/rcmb.2011-0135OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson C, Kannan TR, Baseman JB. Cellular vacuoles induced by Mycoplasma pneumoniae CARDS toxin originate from Rab9-associated compartments. PLoS ONE. 2011;6(7):e22877. doi: 10.1371/journal.pone.0022877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Somarajan SR, et al. Annexin A2 mediates Mycoplasma pneumoniae community-acquired respiratory distress syndrome toxin binding to eukaryotic cells. MBio. 2014;5(4) doi: 10.1128/mBio.01497-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bose S, et al. ADP-ribosylation of NLRP3 by Mycoplasma pneumoniae CARDS toxin regulates inflammasome activity. MBio. 2014;5(6):e02186–14. doi: 10.1128/mBio.02186-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.dos Santos G, Kutuzov MA, Ridge KM. The inflammasome in lung diseases. Am J Physiol Lung Cell Mol Physiol. 2012;303(8):L627–L633. doi: 10.1152/ajplung.00225.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pakhomova ON, et al. Crystallization of community-acquired respiratory distress syndrome toxin from Mycoplasma pneumoniae. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66(Pt 3):294–296. doi: 10.1107/S1744309110000114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holm L, Sander C. Dali: A network tool for protein structure comparison. Trends Biochem Sci. 1995;20(11):478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- 24.Stein PE, et al. The crystal structure of pertussis toxin. Structure. 1994;2(1):45–57. doi: 10.1016/s0969-2126(00)00007-1. [DOI] [PubMed] [Google Scholar]
- 25.O’Neal CJ, Jobling MG, Holmes RK, Hol WG. Structural basis for the activation of cholera toxin by human ARF6-GTP. Science. 2005;309(5737):1093–1096. doi: 10.1126/science.1113398. [DOI] [PubMed] [Google Scholar]
- 26.Domenighini M, Rappuoli R. Three conserved consensus sequences identify the NAD-binding site of ADP-ribosylating enzymes, expressed by eukaryotes, bacteria and T-even bacteriophages. Mol Microbiol. 1996;21(4):667–674. doi: 10.1046/j.1365-2958.1996.321396.x. [DOI] [PubMed] [Google Scholar]
- 27.Kannan TR, et al. Functional mapping of community-acquired respiratory distress syndrome (CARDS) toxin of Mycoplasma pneumoniae defines regions with ADP-ribosyltransferase, vacuolating and receptor-binding activities. Mol Microbiol. 2014;93(3):568–581. doi: 10.1111/mmi.12680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han S, Tainer JA. The ARTT motif and a unified structural understanding of substrate recognition in ADP-ribosylating bacterial toxins and eukaryotic ADP-ribosyltransferases. Int J Med Microbiol. 2002;291(6-7):523–529. doi: 10.1078/1438-4221-00162. [DOI] [PubMed] [Google Scholar]
- 29.Rutenber E, Robertus JD. Structure of ricin B-chain at 2.5-A resolution. Proteins. 1991;10(3):260–269. doi: 10.1002/prot.340100310. [DOI] [PubMed] [Google Scholar]
- 30.Nesić D, Hsu Y, Stebbins CE. Assembly and function of a bacterial genotoxin. Nature. 2004;429(6990):429–433. doi: 10.1038/nature02532. [DOI] [PubMed] [Google Scholar]
- 31.Nesic D, Stebbins CE. Mechanisms of assembly and cellular interactions for the bacterial genotoxin CDT. PLoS Pathog. 2005;1(3):e28. doi: 10.1371/journal.ppat.0010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: Where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9(2):112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Veldhuizen R, Nag K, Orgeig S, Possmayer F. The role of lipids in pulmonary surfactant. Biochim Biophys Acta. 1998;1408(2-3):90–108. doi: 10.1016/s0925-4439(98)00061-1. [DOI] [PubMed] [Google Scholar]
- 34.Schurch S, Bachofenb H, Possmayer F. Pulmonary surfactant: Surface properties and function of alveolar and airway surfactant. Pure Appl Chem. 1992;64(11):1745–1750. [Google Scholar]
- 35.Shang F, et al. Crystallographic complexes of surfactant protein A and carbohydrates reveal ligand-induced conformational change. J Biol Chem. 2011;286(1):757–765. doi: 10.1074/jbc.M110.175265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stenmark P, Dupuy J, Imamura A, Kiso M, Stevens RC. Crystal structure of botulinum neurotoxin type A in complex with the cell surface co-receptor GT1b-insight into the toxin–neuron interaction. PLoS Pathog. 2008;4(8):e1000129. doi: 10.1371/journal.ppat.1000129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fieldhouse RJ, Merrill AR. Needle in the haystack: Structure-based toxin discovery. Trends Biochem Sci. 2008;33(11):546–556. doi: 10.1016/j.tibs.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 38.Zhang RG, et al. The three-dimensional crystal structure of cholera toxin. J Mol Biol. 1995;251(4):563–573. doi: 10.1006/jmbi.1995.0456. [DOI] [PubMed] [Google Scholar]
- 39.Merritt EA, Sixma TK, Kalk KH, van Zanten BA, Hol WG. Galactose-binding site in Escherichia coli heat-labile enterotoxin (LT) and cholera toxin (CT) Mol Microbiol. 1994;13(4):745–753. doi: 10.1111/j.1365-2958.1994.tb00467.x. [DOI] [PubMed] [Google Scholar]
- 40.Treiber N, Reinert DJ, Carpusca I, Aktories K, Schulz GE. Structure and mode of action of a mosquitocidal holotoxin. J Mol Biol. 2008;381(1):150–159. doi: 10.1016/j.jmb.2008.05.067. [DOI] [PubMed] [Google Scholar]
- 41.Olsnes S, Pihl A. Different biological properties of the two constituent peptide chains of ricin, a toxic protein inhibiting protein synthesis. Biochemistry. 1973;12(16):3121–3126. doi: 10.1021/bi00740a028. [DOI] [PubMed] [Google Scholar]
- 42.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 43.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64(Pt 1):112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 44.Bricogne G, Vonrhein C, Flensburg C, Schiltz M, Paciorek W. Generation, representation and flow of phase information in structure determination: Recent developments in and around SHARP 2.0. Acta Crystallogr D Biol Crystallogr. 2003;59(Pt 11):2023–2030. doi: 10.1107/s0907444903017694. [DOI] [PubMed] [Google Scholar]
- 45.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56(Pt 8):965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 48.Collaborative Computational Project, Number 4 The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(Pt 5):760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 49.Krishnan M, Kannan TR, Baseman JB. Mycoplasma pneumoniae CARDS toxin is internalized via clathrin-mediated endocytosis. PLoS ONE. 2013;8(5):e62706. doi: 10.1371/journal.pone.0062706. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.