

Significance
Abnormal heart development is a common birth defect. Genomic imprinting is absolutely essential for mammalian embryonic development. We found that loss of ZFP57, a master regulator of genomic imprinting, causes a number of heart morphogenetic defects. These cardiac defects are reminiscent of mutant phenotypes observed in the NOTCH signaling pathway, one of the most important pathways in development. Indeed, we demonstrate that NOTCH signaling is diminished without ZFP57. Furthermore, the maternal function of Zfp57 contributes to NOTCH signaling and embryonic heart development. Maternal and zygotic Zfp57 play redundant roles in genomic imprinting, NOTCH signaling, and heart development. Thus, our results provide mechanistic links among maternal effect, genomic imprinting, NOTCH signaling, and cardiac development.
Keywords: ZFP57, NOTCH, cardiac development, genomic imprinting, maternal-zygotic mutant
Abstract
Zfp57 is a maternal–zygotic effect gene that maintains genomic imprinting. Here we report that Zfp57 mutants exhibited a variety of cardiac defects including atrial septal defect (ASD), ventricular septal defect (VSD), thin myocardium, and reduced trabeculation. Zfp57 maternal-zygotic mutant embryos displayed more severe phenotypes with higher penetrance than the zygotic ones. Cardiac progenitor cells exhibited proliferation and differentiation defects in Zfp57 mutants. ZFP57 is a master regulator of genomic imprinting, so the DNA methylation imprint was lost in embryonic heart without ZFP57. Interestingly, the presence of imprinted DLK1, a target of ZFP57, correlated with NOTCH1 activation in cardiac cells. These results suggest that ZFP57 may modulate NOTCH signaling during cardiac development. Indeed, loss of ZFP57 caused loss of NOTCH1 activation in embryonic heart with more severe loss observed in the maternal-zygotic mutant. Maternal and zygotic functions of Zfp57 appear to play redundant roles in NOTCH1 activation and cardiomyocyte differentiation. This serves as an example of a maternal effect that can influence mammalian organ development. It also links genomic imprinting to NOTCH signaling and particular developmental functions.
Approximately 0.8% of live births carry congenital heart defects (CHDs) (1). Nearly 30% of lost pregnancies may have cardiac malformations (2–4). Cardiac septation defects are among the most common CHDs (2). Certain transcription factors including NKX2.5, GATA4, and TBX5 are required for cardiac septation (2).
LIN-12/NOTCH signaling is an important pathway in cell-fate specification (5–7). It is conserved in metazoa from Caenorhabditis elegans to humans (8). During cardiac development, it is essential for cardiomyocyte differentiation, valve development, ventricular trabeculation, and outflow tract development (9–11). It is reported that Nkx2.5 is a direct downstream target gene of NOTCH signaling in cardiac development (12).
Zfp57 is the first identified mammalian maternal-zygotic effect gene (13). Loss of zygotic Zfp57 causes partial neonatal lethality, whereas eliminating both maternal and zygotic Zfp57 results in highly penetrant embryonic lethality around midgestation (13). ZFP57 is a master regulator of many imprinted genes characterized by parental origin-dependent expression (14–16). Many imprinted genes are clustered and coregulated by a cis-acting imprinting control region (ICR) (14). ZFP57 maintains the DNA methylation imprint at ICRs (13, 17). The imprint is partially lost in the Zfp57 zygotic mutant but absent in the maternal-zygotic mutant (13). Expression of the imprinted genes is deregulated without ZFP57 (13).
Based on the literature, we hypothesized that the midgestational embryonic lethality present in the Zfp57 maternal-zygotic mutant may result from abnormalities in cardiac development (18). Indeed, Zfp57 mutants exhibited atrial septal defects (ASDs), ventricular septal defects (VSDs), thin myocardium, and reduced trabeculation, with more penetrant and worse phenotypes present in the maternal-zygotic mutant. We found NOTCH signaling was attenuated in the heart of Zfp57 mutant embryos, which may underlie these cardiac defects.
Results
Multiple Cardiac Defects Were Observed in the Zfp57 Mutant.
Zfp57 has both maternal (M) and zygotic (Z) functions (13) (SI Appendix, Fig. S1). Loss of just zygotic function of Zfp57 (M+Z−) causes partial neonatal lethality, whereas elimination of both maternal and zygotic functions of Zfp57 (M−Z−) results in highly penetrant maternal-zygotic embryonic lethality around midgestation (13) (SI Appendix, Fig. S1). Zfp57 maternal-zygotic mutant, −/−mz (M−Z−), embryos begin dying at embryonic day 11.5 (E11.5) (13). Because lethality around midgestation can be associated with cardiac defects, we examined the hearts of −/−mz (M−Z−) embryos derived from the timed matings between Zfp57 homozygous (−/−) female mice and Zfp57 heterozygous (+/−) male mice or the hearts of Zfp57 zygotic mutant, −/−z (M+Z−), embryos obtained from the matings between Zfp57 heterozygous (+/−) female mice and Zfp57 homozygous (−/−) male mice (SI Appendix, Fig. S1). As illustrated below, we found that Zfp57 mutants, −/−mz (M−Z−) and −/−z (M+Z−), exhibited ASDs, VSDs, thin myocardium, and trabeculation defects, with higher penetrance and worse defects present in the −/−mz (M−Z−) than in the −/−z (M+Z−) hearts (Fig. 1).
Fig. 1.

Loss of ZFP57 causes multiple cardiac defects in mouse embryos and neonatal pups. The Zfp57−/+ (M−Z+) and −/−mz (M−Z−) samples were generated from the crosses between homozygous (−/−) female mice and heterozygous (+/−) male mice. The +/− (M+Z+) and −/−z (M+Z−) samples were generated from the heterozygous intercrosses. ra, right atrium; rv, right ventricle; la, left atrium; lv, left ventricle; as, atrial septum (arrow); ASD, atrial septal defect (*); vs, ventricular septum; VSD, ventricular septal defect (**); vm, ventricular myocardial wall (bracket); trab, trabeculae. (A) H&E-stained transverse sections through the heart of Zfp57 −/+ and −/−mz E14.5 embryos with an image of 5× object lens (Left) and the images of 20× object lens for the enlarged area of as or ASD, vs or VSD, vm and trab, respectively. (B and C) Percentage (%) of the embryos with ASD (B) or VSD (C). The number above each bar indicates the number of embryos analyzed for each genotype. (D and E) Average ventricular wall (vm) thickness (D) or trabecular area (E). ImageJ software was used to measure multiple areas of the heart sections for each sample. Values are mean ± SEM. ANOVA was performed for statistical comparisons, followed by the post hoc independent sample t test with Bonferroni correction. *P < 0.05. n.s., statistically not significant. (F) Images (5× object lens) of live (Top) or dead (Bottom) neonatal (P0–P2) zygotic mutant, −/−z (M+Z−), pups. (G and H) Percentage of neonatal live or dead pups with ASD (G) or VSD (H). The number above each bar indicates the number of pups analyzed for each group.
ASD.
Mouse cardiac septation occurs between E11.5 and E14.5 (2). The hearts of live E14.5 embryos were subjected to serial sectioning followed by hematoxalin and eosin (H&E) staining, with the whole-heart and section heart images of −/+ (M−Z+) and −/−mz (M−Z−) embryos shown here (Fig. 1A and SI Appendix, Fig. S2 A and B). All (n = 14) live −/−mz (M−Z−) E14.5 embryos displayed secundum ASD, whereas none of the live +/− (M+Z+) (n = 7) or −/+ (M−Z+) (n = 13) E14.5 embryos had ASD (Fig. 1 A and B). About 44% (n = 9) of the live −/−z (M+Z−) E14.5 embryos also exhibited ASD (Fig. 1B).
Zfp57 zygotic mutants, −/−z (M+Z−), displayed partial neonatal lethality with 40% death by postnatal day 1 (P1) (13). Indeed, most (>80%, n = 14) dead −/−z (M+Z−) neonatal (P0–P2) pups had ASD, whereas no ASD was observed in the hearts of live −/−z (M+Z−) (n = 5) or live +/− (M+Z+) (n = 5) neonatal pups (Fig. 1 F and G and SI Appendix, Fig. S2 C–E). We also examined the hearts of E18.5 embryos. About 40% (n = 19) of live −/−z (M+Z−) E18.5 embryos displayed secundum ASDs, whereas none (n = 12) of the +/− (M+Z+) E18.5 embryos exhibited ASD (SI Appendix, Fig. S2 F–H).
VSD.
In addition to ASDs, we observed VSDs in 80% (n = 13) of live −/−mz (M−Z−) E14.5 embryos. Although none of the live −/+ (M−Z+) E14.5 embryos had ASD (Fig. 1B), 20% (n = 15) of −/+ (M−Z+) E14.5 embryos lacking maternal Zfp57 exhibited VSDs (Fig. 1C). VSDs were also observed in 20% (n = 10) of live −/−z (M+Z−) E14.5 embryos, but not in live +/− (M+Z+) E14.5 embryos (0%, n = 10) derived from heterozygous female mice (Fig. 1C). VSD was not observed in live +/− (M+Z+) (n = 5) or live −/−z (M+Z−) (n = 5) neonatal P2 pups (Fig. 1H). About 20% (n = 14) of dead −/−z (M+Z−) P2 pups had VSDs (Fig. 1H).
Thin myocardium.
The ventricular myocardium (vm) of −/−z (M+Z−) E14.5 embryos was significantly thinner than that of +/− (M+Z+) E14.5 embryos (Fig. 1D). −/−mz (M−Z−) E14.5 embryos also had a thinner vm than −/+ (M−Z+) or +/− (M+Z+) E14.5 embryos (Fig. 1 A and D). The vm of −/−mz (M−Z−) E14.5 embryos appeared to be thinner than that of −/−z (M+Z−) E14.5 embryos although the difference was not statistically significant (Fig. 1D). Interestingly, −/−z (M+Z−) and −/+ (M−Z+) E14.5 embryos had similar vm, and they both had a thinner vm than +/− (M+Z+) E14.5 embryos (Fig. 1D). Thus, loss of either maternal or zygotic Zfp57 (M− or Z−) causes reduced vm. This implies that both maternal and zygotic Zfp57 contribute to the normal development of ventricular myocardium and they have partially redundant roles.
Trabeculation defect.
Mouse cardiac trabeculation begins at E9.5 shortly after cardiac looping occurs (19). We examined and measured the trabeculae (trab) of E14.5 embryos (Fig. 1 A and E). −/−z (M+Z−) and −/−mz (M−Z−) E14.5 embryos had similar trab that was diminished compared with +/− (M+Z+) E14.5 embryos (Fig. 1E). −/+ (M−Z+) E14.5 embryos also appeared to have reduced trab compared with +/− (M+Z+) E14.5 embryos although the difference was not statistically significant (Fig. 1E). There was no significant difference in trab between −/+ (M−Z+) embryos lacking maternal Zfp57, −/−z (M+Z−) embryos lacking zygotic Zfp57, and −/−mz (M−Z−) embryos lacking both maternal and zygotic Zfp57. This finding indicates that zygotic Zfp57 is required for ventricular trabeculation and maternal Zfp57 does not appear to be redundant with zygotic Zfp57 in this process (Fig. 1E).
Expression of Zfp57 in the Heart.
We used alkaline phosphatase-conjugated digoxigenin (DIG)-labeled RNA probes to detect expression of Zfp57 in wild-type E10.5 and E13.5 embryos by RNA in situ hybridization (Fig. 2A and SI Appendix, Fig. S3). Zfp57 was highly expressed in the spine (neural tube in SI Appendix, Fig. S3A and central canal in SI Appendix, Fig. S3B) and lung (lu in SI Appendix, Fig. S3B). Zfp57 was also clearly expressed in the heart, although not as highly as in the spine or lung (Fig. 2 A and H and SI Appendix, Fig. S3). Zfp57-expressing cells were broadly but not uniformly distributed in the four heart chambers of wild-type, +/+ (M+Z+), E10.5 embryos (SI Appendix, Fig. S3C). Notable expression of Zfp57 was observed in the atrial septum (as), cushion tissue (ct), vm, and epicardium of wild-type, +/+ (M+Z+), E13.5 embryos (Fig. 2A). Some spotty expression was also detected in the ventricular septum (vs) and endocardium (SI Appendix, Fig. S3 C and D). Consistent with the presence of a weak aberrant transcript in Zfp57 mutant embryos carrying two deleted alleles of Zfp57 based on Northern blot (13), a much reduced signal was detected in −/−mz (M−Z−) E13.5 embryos including the spine, lung, and heart regions (SI Appendix, Fig. S3E). These results suggest that Zfp57 is expressed in the embryonic heart including the corresponding regions where the aforementioned cardiac defects were observed in Zfp57 mutants.
Fig. 2.

Dlk1 expression and NOTCH1 activation were both inhibited in the heart without ZFP57. The embryonic heart sections were subjected to RNA in situ hybridization (A and B) or immunostaining (C–G). Wild-type, +/+ (M+Z+), E13.5 embryos in A were derived from the cross between wild-type female mice and wild-type male mice. −/−mz (M−Z−) and −/+ (M−Z+) E13.5 embryos in B–G were derived from the cross between Zfp57 homozygous (−/−) female mice and heterozygous (+/−) male mice, whereas +/− (M+Z+) and −/−z (M+Z−) E13.5 embryos in C were generated from the timed mating between Zfp57+/− female mice and Zfp57 −/− or +/− male mice. ra, right atrium; rv, right ventricle; la, left atrium; lv, left ventricle; as, atrial septum; ct, cushion tissue; ASD, atrial septal defect (*); vs, ventricular septum; vm, ventricular myocardium. Blue signal in C, D, and F, DAPI staining. ImageJ software was used to count N1ICD-positive (N1ICD+) or DLK1-positive (DLK1+) cells. Standard Student’s t test was used to analyze datasets for −/−mz (M−Z−) (gray bar) and −/+ (M−Z+) (black bar) E13.5 embryos. Values are mean ± SEM, Student’s t test: **P < 0.01. n.s., statistically not significant. (A) Section RNA in situ hybridization (purple blue) with the antisense probe (Left) or sense probe (Right) of Zfp57 transcribed from a plasmid containing the full-length Zfp57 cDNA of 1.35 kb (13). Red arrow, atrial septum (as). Arrow, purple blue dots indicating Zfp57-positive cells. Arrowhead, epicardium. (B) Section RNA in situ hybridization (purple blue) with antisense probe of Dlk1. Red arrow, atrial septum (as). Asterisk (*), atrial septal defect (ASD). Arrow, purple blue dots indicating Dlk1-positive cells. Arrowhead, epicardium. (C) Immunostaining of DLK1 (green) on cryosections. Blue signal, DAPI staining. White arrow, cell surface staining of DLK1 surrounding blue nuclei. Yellow arrowhead, epicardium of the right ventricle (rv). (D) Immunostaining (red) of full-length NOTCH1 (Top) and cleaved NOTCH1 (N1ICD, Bottom) on paraffin-embedded sections. The Inset images represent the boxed areas in white dotted lines in each panel with a higher magnification (40×). (E) Percentage (%) of N1ICD-positive (N1ICD+) or DLK1-positive (DLK1+) cells in the heart relative to the total number of DAPI-positive cells in −/−mz (M−Z−) or −/+ (M−Z+) E13.5 embryos. (F) Coimmunostaining of DLK1 (green) and N1ICD (red) on cryosections. The boxed area of rv in white dotted lines is shown on the Right as an enlarged image for the coimmunostained heart section. White arrows, red-stained nuclei inside green-colored cell surface, i.e., N1ICD+DLK1+ cells. White arrowheads, red-stained nuclei without green-colored cell surface, i.e., N1ICD+DLK1− cells. (G) Percentage (%) of N1ICD+DLK1+ (double positive for N1ICD and DLK1) or N1ICD+DLK1− (N1ICD-positive but DLK1-negative) cells in the heart relative to the total number of DAPI-positive cells in either −/−mz (M−Z−) or −/+ (M−Z+) E13.5 embryos.
Genomic Imprinting Was Lost in the Heart of Zfp57 Mutants.
Previously, we found that ZFP57 maintains the DNA methylation imprint in mouse embryos (13). To examine if genomic imprinting was lost in the heart without ZFP57, we performed combined bisulphite restriction analysis (COBRA) of genomic DNA samples from the hearts of Zfp57 −/−mz (M−Z−) E13.5 embryos and littermate −/+ (M−Z+) embryos. Indeed, the DNA methylation imprint was lost in the embryonic heart at the intergenic germline-derived differentially methylated region (IG-DMR) of the Dlk1-Dio3 imprinted region as well as at the Peg1, Peg3, and Peg5 but not H19 imprinted regions (SI Appendix, Fig. S4A).
NOTCH Signaling Pathway Was Perturbed in the Heart of Zfp57 Mutants.
The imprinted Dlk1 gene at the Dlk1-Dio3 imprinted domain encodes a protein that shares homology with classical ligands for NOTCH receptors (20–23). However, DLK1 lacks the Delta/Serrate/LAG-2 (DSL) domain present in all canonical DSL family of ligands for NOTCH receptors (8, 24). It was reported that OSM-11, a C. elegans homolog of DLK1, facilitates LIN-12/NOTCH signaling (24, 25). However, it is controversial whether it is a positive or negative regulator of NOTCH signaling in mammals (25–27).
It was previously reported that the DNA methylation imprint at the IG-DMR of the Dlk1-Dio3 imprinted region was required for proper expression of the imprinted Dlk1 gene (22). Similarly, we found that DNA methylation imprint at the IG-DMR of the Dlk1-Dio3 imprinted region was lost and expression of the imprinted Dlk1 gene was decreased in Zfp57 mutant embryos (13). Those studies prompted us to investigate whether NOTCH signaling may be perturbed in the heart without ZFP57.
Dlk1 expression in the heart.
Based on RNA in situ hybridization, Dlk1 was highly expressed in the epicardium of −/+ (M−Z+) E13.5 embryos, with some spotty Dlk1+ cells in the myocardium (Fig. 2B). By contrast, its expression was dramatically reduced at the epicardium and myocardium of −/−mz (M−Z−) E13.5 embryos (Fig. 2B). Dlk1 was expressed in the as and vm of Zfp57 −/+ (M−Z+) embryos but not in the as and vm of −/−mz (M−Z−) embryos (Fig. 2B). The expression pattern of Dlk1 resembles that of Zfp57 in the embryonic heart (Fig. 2 A and B). We obtained similar results with DLK1 immunostaining (Fig. 2C and SI Appendix, Fig. S4B). Indeed, DLK1 was similarly expressed at the epicardium of Zfp57 +/− (M+Z+) and −/+ (M−Z+) E13.5 embryos (Fig. 2C). It was partially reduced at the epicardium in −/−z (M+Z−) but almost completely missing in −/−mz (M−Z−) E13.5 embryos (Fig. 2C). DLK1 was also reduced in the myocardium of −/−mz (M−Z−) E13.5 embryos in comparison with −/+ (M−Z+) E13.5 embryos in the repeat experiments (see the enlarged Inset images of SI Appendix, Fig. S4B). Loss of just zygotic Zfp57 led to partial loss of Dlk1 expression in the heart of −/−z (M+Z−) embryos, whereas Dlk1 expression was not impaired in the −/+ (M−Z+) embryos. By contrast, elimination of both maternal and zygotic Zfp57 resulted in almost complete loss of Dlk1 expression in the heart of −/−mz (M−Z−) embryos. These results suggest that maternal Zfp57 and zygotic Zfp57 are partially redundant in the expression of Dlk1 in the embryonic heart.
Activation of NOTCH1 was inhibited.
NOTCH1 is the predominant member of NOTCH receptors expressed in the embryonic heart (12). We used a monoclonal antibody against full-length NOTCH1 to examine its expression. We found NOTCH1 was similarly expressed in the endocardium and myocardium of Zfp57+/− (M+Z+), −/+ (M−Z+), and −/−mz (M−Z−) E13.5 embryos (Fig. 2D and SI Appendix, Fig. S4C).
A key activation event in NOTCH signaling is ligand-dependent γ-secretase–mediated cleavage of NOTCH receptors (5, 6, 11, 28–30). We performed immunostaining with a monoclonal antibody that can only detect the cleaved activated form of NOTCH1 receptor (N1ICD). Interestingly, we saw a marked decrease of N1ICD in the heart of −/−mz (M−Z−) E13.5 embryos compared with Zfp57+/− (M+Z+), −/+ (M−Z+), or −/−z (M+Z−) E13.5 embryos (SI Appendix, Fig. S4D). N1ICD was not much reduced in the heart of −/−z (M+Z−) E13.5 embryos in comparison with +/− (M+Z+) or −/+ (M−Z+) embryos (SI Appendix, Fig. S4D). N1ICD reduction in the embryonic heart without ZFP57 was confirmed in the repeat experiments comparing −/−mz (M−Z−) E13.5 embryos with −/+ (M−Z+) E13.5 embryos (Fig. 2D). Upon quantification, we found both DLK1-positive (DLK1+) cells and N1ICD-positive (N1ICD+) cells decreased dramatically in the heart of −/−mz (M−Z−) embryos compared with −/+ (M−Z+) embryos (Fig. 2E).
As expected, NOTCH1 was primarily localized on the cell surface in the heart of +/− (M+Z+), −/+ (M−Z+), or −/−mz (M−Z−) E13.5 embryos (Fig. 2D and SI Appendix, Fig. S4C). DLK1 was also present at the cell surface in the heart of +/− (M+Z+), −/+ (M−Z+), or −/−z (M+Z−) E13.5 embryos (Fig. 2C). To test how DLK1 may be involved in NOTCH1 activation, we performed coimmunostaining with monoclonal antibodies against DLK1 and full-length NOTCH1 in the heart. Interestingly, DLK1 was present at the cell surface of a subset of NOTCH1-positive cells in the heart of −/+ (M−Z+) E13.5 embryos, but not in −/−mz (M−Z−) E13.5 embryos (SI Appendix, Fig. S4E).
We also performed coimmunostaining of DLK1 and N1ICD (Fig. 2F). N1ICD was predominantly localized to the nucleus in the heart of Zfp57−/+ E13.5 embryos, whereas DLK1 was concentrated on the cell surface (Fig. 2F). Interestingly, most N1ICD+ cells also expressed DLK1, i.e., N1ICD+DLK1+ cells (Fig. 2F). Although little DLK1 was expressed, nuclear N1ICD was present in a few sparsely populated cells in the heart of −/−mz (M−Z−) E13.5 embryos (Fig. 2F). These are N1ICD+DLK1− cells that possibly resulted from NOTCH1 activation via DLK1-independent pathways. We quantified N1ICD+DLK1+ and N1ICD+DLK1− cells in the heart of −/−mz (M−Z−) and Zfp57 −/+ (M−Z+) embryos (Fig. 2G). N1ICD+DLK1+ cells accounted for about 1.5% of cells in the heart of Zfp57 −/+ (M−Z+) embryos but were absent in the heart of −/−mz (M−Z−) embryos (Fig. 2G). By contrast, the number of N1ICD+DLK1− cells remained constant, accounting for about 0.5% of cells in the heart of either −/−mz (M−Z−) or −/+ (M−Z+) embryos (Fig. 2G). These results suggest that most N1ICD generation in the heart may be dependent on DLK1.
NOTCH1 Target Genes Were Inhibited in the Heart of Zfp57 Mutants.
Hey1 and Hey2 are two direct target genes of NOTCH signaling in the cardiovascular system (31). Hey1 knockout mice do not show any obvious phenotype (31). By contrast, the Hey2 knockout mouse exhibits ASD, VSD, and thin myocardium, similar to the Zfp57 knockout mouse (31–34). We performed RNA in situ hybridization to examine the expression of Hey1 and Hey2 in the heart of E10.5 (SI Appendix, Fig. S5 A and B) and E13.5 (Fig. 3A and SI Appendix, Fig. S6 A and B) embryos. Hey1 was similarly expressed in the heart of −/−mz (M−Z−) and −/+ (M−Z+) E10.5 embryos (SI Appendix, Fig. S5A). Expression of Hey1 was also similar in the heart of Zfp57+/− (M+Z+), −/−z (M+Z−), −/+ (M−Z+), and −/−mz (M−Z−) embryos at E13.5, as is more clearly seen with the magnified right ventricle (rv) of these E13.5 embryos (Fig. 3A). This was the same in the repeat RNA in situ experiments comparing −/+ (M−Z+) with −/−mz (M−Z−) E13.5 embryos, as is exemplified by the magnified rv (SI Appendix, Fig. S6 A and B). By contrast, Hey2 expression was slightly reduced in the heart of −/−mz (M−Z−) E10.5 embryos compared with +/− (M+Z+) or −/+ (M−Z+) E10.5 embryos, whereas it was not diminished in the heart of −/−z (M+Z−) E10.5 embryos compared with +/− (M+Z+) E10.5 embryos (SI Appendix, Fig. S5 A and B). Hey2 was more profoundly down-regulated in the heart of −/−mz (M−Z−) E13.5 embryos, with the magnified image of rv as an example, although it was still similarly expressed in the heart of −/+ (M−Z+), +/− (M+Z+), and −/−z (M+Z−) E13.5 embryos (Fig. 3A). Down-regulation of Hey2 in −/−mz (M−Z−) E13.5 embryos versus −/+ (M−Z+) E13.5 embryos was verified in the repeat RNA in situ experiments, which is more obvious with the magnified image of left ventricle (lv) (SI Appendix, Fig. S6 A and B). Thus, loss of just maternal or just zygotic Zfp57 did not cause significant reduction of Hey2, whereas elimination of both maternal and zygotic Zfp57 resulted in reduced expression of Hey2 in the heart of E10.5 and E13.5 embryos. These results suggest that maternal and zygotic Zfp57 functions play a redundant role in Hey2 expression in the embryonic heart.
Fig. 3.

Some target genes of NOTCH signaling were down-regulated in the heart without ZFP57. (A) RNA in situ hybridization on the heart sections of E13.5 embryos. RNA in situ hybridization (purple blue) was performed on the heart cryosections of −/−z (M+Z−) or +/− (M+Z+) E13.5 embryos derived from heterozygous female mice and −/−mz (M−Z−) or −/+ (M−Z+) E13.5 embryos derived from homozygous female mice. Digoxigenin (DIG)-labeled antisense riboprobes used for RNA in situ were derived from the cDNA constructs of Hey1, Hey2, Nkx2.5, and Bmp10. ra, right atrium; la, left atrium; rv, right ventricle; lv, left ventricle; vs, ventricular septum. A portion of the rv is also shown as a magnified image on the Right, next to the original image for the whole heart on the Left. At least two independent embryos for each genotype were used for this RNA in situ hybridization. (B) qRT-PCR analysis of the NOTCH1 target genes in the heart of E13.5 embryos. Total RNA samples were isolated from the hearts of live Zfp57 +/− (M+Z+), −/−z (M+Z−), and −/−mz (M−Z−) E13.5 embryos. Black filled bars, +/− (M+Z+) embryos. Light gray bars, −/−z (M+Z−) embryos. Dark gray bars, −/−mz (M−Z−) embryos. The qRT-PCR data from three biological and three technical replicates were analyzed for each experimental group. Values are mean ± SEM. Student’s t test: *P < 0.05; n.s., statistically not significant.
Nkx2.5 encodes a homeodomain transcription factor essential for cardiac development (35). It is a direct target gene of NOTCH1 (12). Nuclear N1ICD can activate Nkx2.5 expression in cardiac progenitor cells (CPCs), driving CPCs to become cardiomyocytes (12). Mutations in human NKX2.5 cause ASD and VSD (35). We examined its expression in the heart by RNA in situ hybridization. Nkx2.5 was highly expressed throughout the myocardium of E10.5 and E13.5 embryos (Fig. 3A and SI Appendix, Figs. S5 and S6). Nkx2.5 was similarly expressed in the heart of +/− (M+Z+) and −/−z (M+Z−) E10.5 embryos (SI Appendix, Fig. S5B), but it was slightly down-regulated in the heart of −/−mz (M−Z−) E10.5 embryos, compared with −/+ (M−Z+), +/− (M+Z+), or −/−z (M+Z−) E10.5 embryos (SI Appendix, Fig. S5 A and B). Whereas Nkx2.5 was similarly expressed in the heart of −/+ (M−Z+), +/− (M+Z+), and −/−z (M+Z−) E13.5 embryos, its expression was significantly reduced in the heart of −/−mz (M−Z−) E13.5 embryos, as is easily seen with the magnified rv (Fig. 3A). Reduced Nkx2.5 expression in −/−mz (M−Z−) E13.5 embryos compared with −/+ (M−Z+) E13.5 embryos was verified in the repeat RNA in situ experiments, with the magnified lv as an example (SI Appendix, Fig. S6 A and B). Thus, maternal and zygotic Zfp57 are also redundant in Nkx2.5 expression in the embryonic heart.
Bmp10 is highly expressed in developing cardiac trabeculae (19). Loss of Bmp10 results in ventricular trabeculation defect due to decreased cardiomyocyte proliferation. Ablation of NOTCH signaling causes reduced Bmp10 expression and cardiomyocyte proliferation in the heart (36). Based on RNA in situ hybridization, there was no difference in Bmp10 expression in the trabeculae of the Zfp57−/+ (M−Z+) and −/−mz (M−Z−) E10.5 embryos (SI Appendix, Fig. S5A). Interestingly, Bmp10 was similarly down-regulated in the trabeculae of both −/−mz (M−Z−) and −/−z (M+Z−) E13.5 embryos, in comparison with −/+ (M−Z+) and +/− (M+Z+) E13.5 embryos, whereas its expression seemed to be similar in the trabeculae of the −/+ (M−Z+) and +/− (M+Z+) E13.5 embryos (Fig. 3A). This down-regulation is more clearly visualized by the magnified rv (Fig. 3A). Down-regulation of Bmp10 in the trabeculae of −/−mz (M−Z−) versus −/+ (M−Z+) E13.5 embryos was verified in the repeat RNA in situ experiments, with the magnified lv as an example (SI Appendix, Fig. S6 A and B). These results suggest that only zygotic Zfp57 is necessary and sufficient for proper expression of Bmp10 in the trabeculae of the embryonic heart.
We also performed quantitative RT-PCR (qRT-PCR) analysis of the total RNA samples isolated from the hearts of E10.5 and E13.5 embryos. Indeed, Nkx2.5, but not Hey1 or Bmp10, was significantly reduced in the heart of −/−mz (M−Z−) E10.5 embryos in comparison with −/+ (M−Z+) E10.5 embryos (SI Appendix, Fig. S5C). Compared with −/+ (M−Z+) E10.5 embryos, Hey2 seemed to be also inhibited in the heart of −/−mz (M−Z−) E10.5 embryos (SI Appendix, Fig. S5C). Similarly, Nkx2.5 and Hey2 but not Hey1 were down-regulated in the heart of −/−mz (M−Z−) E13.5 embryos compared with −/+ (M−Z+) or +/− (M+Z+) or −/−z (M+Z−) E13.5 embryos (Fig. 3B and SI Appendix, Fig. S6C). By contrast, no down-regulation of Nkx2.5 or Hey2 or Hey1 was observed in the heart of −/−z (M+Z−) E13.5 embryos in comparison with +/− (M+Z+) E13.5 embryos (Fig. 3B). These qRT-PCR results are entirely consistent with the results obtained with RNA in situ hybridization for Hey1, Hey2, and Nkx2.5. Therefore, we conclude that maternal and zygotic Zfp57 are redundant for expression of Hey2 and Nkx2.5 in the embryonic heart.
Based on qRT-PCR, Bmp10 appeared to be down-regulated in the heart of −/−mz (M−Z−) E13.5 embryos in comparison with −/+ (M−Z+) or +/− (M+Z+) E13.5 embryos (Fig. 3B and SI Appendix, Fig. S6C). Interestingly, Bmp10 was similarly down-regulated in the heart of −/−z (M+Z−) E13.5 embryos (Fig. 3B). These qRT-PCR results for Bmp10 are consistent with the results obtained with RNA in situ hybridization, suggesting that zygotic Zfp57 but not maternal Zfp57 is necessary and sufficient for Bmp10 expression in the embryonic heart.
Taken together, at least three NOTCH1 target genes (Hey2, Nkx2.5, and Bmp10) were down-regulated during cardiac development without ZFP57 based on RNA in situ hybridization and qRT-PCR analysis. Therefore, we conclude that the NOTCH1 signaling pathway is inhibited in the heart of Zfp57 mutant embryos.
Cardiomyocyte Proliferation Was Reduced.
Ablation of NOTCH signaling causes reduced BMP10 signaling and decreased cardiomyocyte proliferation (36). To determine if cardiomyocyte proliferation was reduced without ZFP57, we performed EdU incorporation assay in the heart of E13.5 embryos (Fig. 4). EdU labeling appeared to be reduced in the heart of −/−mz (M−Z−) E13.5 embryos in comparison with +/− (M+Z+), −/−z (M+Z−), or −/+ (M−Z+) E13.5 embryos, whereas similar EdU labeling was observed in +/− (M+Z+) and −/−z (M+Z−) E13.5 embryos, as is exemplified by the magnified heart regions (Fig. 4 A and B). When quantified, significantly fewer EdU-positive (EdU+) cells (one-third decrease) were observed in the heart of −/−mz (M−Z−) E13.5 embryos compared with −/+ (M−Z+) embryos (Fig. 4C). This finding was verified with a BrdU-incorporation assay (SI Appendix, Fig. S7A). The number of BrdU+ cells was reduced throughout the heart of −/−mz (M−Z−) E13.5 embryos in comparison with −/+ (M−Z+) E13.5 embryos. BrdU+ cells were reduced by one-third in −/−mz (M−Z−) embryos (SI Appendix, Fig. S7C), similar to what was observed with EdU labeling (Fig. 4C). However, the percentage of BrdU+ cells in the heart of −/+ (M−Z+) or −/−mz (M−Z−) E13.5 embryos was about half of the percentage of EdU+ cells in the heart of −/+ (M−Z+) or −/−mz (M−Z−) E13.5 embryos, respectively. Because our BrdU labeling protocol required hydrochloride (HCl) treatment, this led to partial destruction of the nuclei that made it difficult for us to accurately quantify cell numbers and percentages of BrdU+ cells. By contrast, our EdU labeling protocol was much simpler and the data obtained with EdU labeling could accurately reflect percentages of proliferating cells in the heart. Thus, most of our cell proliferation assays were done with the EdU labeling method (Figs. 4 A and B and 5 D and E and SI Appendix, Figs. S10 and S11). Nevertheless, the results obtained from both EdU and BrdU labeling experiments arrive at similar conclusions that the portion of the cells undergoing proliferation is reduced in the embryonic heart without ZFP57.
Fig. 4.

Loss of ZFP57 caused reduced proliferation in the heart of E13.5 embryos. Immunostaining (red) was performed on the heart cryosections of E13.5 embryos derived from EdU-injected pregnant heterozygous or homozygous female mice. Zfp57+/− (M+Z+) and −/−z (M+Z−) E13.5 embryos were generated from the timed mating between Zfp57 heterozygous female mice and homozygous or heterozygous male mice, whereas −/−mz (M−Z−) and −/+ (M−Z+) E13.5 embryos were derived from the cross between Zfp57 homozygous female mice and heterozygous male mice. Red signal, nuclei of EdU-labeled cells. Blue signal, DAPI-stained nuclei. ra, right atrium; rv, right ventricle; la, left atrium; lv, left ventricle; vs, ventricular septum; ec, endocardial cushion. (A) Anti-EdU immunostaining on the heart cryosections of +/− (M+Z+), −/−z (M+Z−), and −/−mz (M−Z−) E13.5 embryos. A portion of the magnified vs or rv is shown on the Right, next to the heart image of lower magnification. Boxes in white dotted lines show the areas of vs that are shown as the magnified images on the Right. (B) Anti-EdU immunostaining on the heart cryosections of −/+ (M−Z+) and −/−mz (M−Z−) E13.5 embryos. A portion of the magnified ra, vs, rv, or lv is shown on the Right, next to the heart image of lower magnification. Boxes in white dotted lines show the areas of vs (ii) and rv (i) that are shown as the magnified images on the Right. (C) Percentage of EdU-positive (% EdU+) cells in the heart relative to DAPI-positive cells. ImageJ software was used to quantify the numbers of immunostained cells or DAPI-stained cells. Black filled bar, Zfp57 −/+ (M−Z+) embryos. Gray filled bar, −/−mz (M−Z−) embryos. At least three independent E13.5 embryos were analyzed for each group. Values are mean ± SEM. Student’s t test: **P < 0.01.
Fig. 5.

Cardiac differentiation was inhibited in the heart of Zfp57 mutant embryos. Immunostaining was performed on the heart cryosections of E13.5 embryos. Zfp57+/− (M+Z+) and −/−z (M+Z−) E13.5 embryos were generated from the timed mating between Zfp57 heterozygous female mice and Zfp57 homozygous or heterozygous male mice, whereas −/−mz (M−Z−) and −/+ (M−Z+) E13.5 embryos were derived from homozygous female mice mated with heterozygous male mice. ra, right atrium; rv, right ventricle; la, left atrium; lv, left ventricle; vs, ventricular septum. Blue signal, DAPI staining. The small box in white dotted lines was magnified as an Inset image (the large box in white dotted lines) inside the original image of lower magnification (B and C) or as a separate image on the Right next to the original image of lower magnification (A, D, and E) in the same panel. (A) c-KIT immunostaining (red) of E13.5 embryos. (B) NKX2.5 immunostaining (red) of E13.5 embryos. (C) GATA4 immunostaining (red) of E13.5 embryos. (D) NKX2.5 (red) and EdU (green) coimmunostaining of E13.5 embryos. Arrowheads, NKX2.5 and EdU double positive (NKX2.5+EdU+) cells with yellow nuclei. (E) c-KIT (red) and EdU (green) coimmunostaining of E13.5 embryos. Arrowheads, c-KIT and EdU double positive (c-KIT+EdU+) cells with green nuclei inside red-colored cell surface. (F and G) ImageJ software was used to quantify the number of immunostained cells on the heart sections. Gray filled bars, −/−mz (M−Z−) E13.5 embryos. Black filled bars, −/+ (M−Z+) E13.5 embryos. Values are mean ± SEM. Student’s t test: *P < 0.05. (F) Percentage (%) of NKX2.5+ cells within the EdU+ cell population in the heart, i.e., no. of NKX2.5+EdU+ cells/no. of EdU+ cells. (G) Percentage (%) of c-KIT+ cells within the EdU+ cell population in the heart, i.e., no. of c-KIT+EdU+ cells/no. of EdU+ cells.
To examine whether apoptosis would play a role in the cardiac defects of Zfp57 mutant embryos, we performed immunostaining with antibodies against cleaved Caspase-3. Similar Caspase-3 immunostaining was observed in the heart of −/−mz (M−Z−) and −/+ (M−Z+) E13.5 embryos, as is exemplified by the magnified images of rv and vs (SI Appendix, Fig. S7B). When quantified, there was no significant difference in the number of Caspase-3-positive cells in the heart comparing −/−mz (M−Z−) with −/+ (M−Z+) E13.5 embryos (SI Appendix, Fig. S7D). Thus, apoptosis is not involved in the cardiac defects observed in Zfp57 mutant embryos.
N-Myc is involved in cardiomyocyte proliferation (37, 38). Interestingly, its expression was down-regulated in the heart of −/−z (M+Z−) E13.5 embryos in comparison with +/− (M+Z+) E13.5 embryos but not as much as in −/−mz (M−Z−) E13.5 embryos based on qRT-PCR analysis (SI Appendix, Fig. S8A). Indeed, N-Myc expression was significantly reduced in the heart of −/−mz (M−Z−) E13.5 embryos in comparison with −/−z (M+Z−) or −/+ (M−Z+) E13.5 embryos (SI Appendix, Fig. S8 A and B). These results indicate that both maternal and zygotic Zfp57 are required for full expression of N-Myc in the embryonic heart and their roles are not redundant in N-Myc expression. Inhibition of N-Myc may also contribute to cardiomyocyte proliferation defect in Zfp57 mutant embryos.
Differentiation of CPCs Was Inhibited.
Because NOTCH signaling is crucial for cell-fate specification, we wondered if cardiomyocyte differentiation may be affected without ZFP57. C-KIT is a common marker for hematopoietic stem cells and progenitor cells (39, 40). It is also reported to be expressed in CPCs (12). Indeed, c-Kit was increased by about threefold in the heart of −/−mz (M−Z−) E10.5 embryos versus −/+ (M−Z+) E10.5 embryos based on qRT-PCR analysis (SI Appendix, Fig. S8C). It was also increased in the heart of −/−mz (M−Z−) E13.5 embryos compared with +/− (M+Z+), −/+ (M−Z+), or −/−z (M+Z−) E13.5 embryos based on qRT-PCR analysis, whereas there was no difference in c-Kit expression comparing −/−z (M+Z−) with +/− (M+Z+) E13.5 embryos (SI Appendix, Fig. S8 D and E). These findings were confirmed by c-KIT immunostaining. Although it was similarly expressed in the heart of +/− (M+Z+), −/+ (M−Z+), and −/−z (M+Z−) E13.5 embryos, c-KIT was significantly increased in the heart of −/−mz (M−Z−) E13.5 embryos based on immunostaining (Fig. 5A). Increased expression of c-KIT in the heart of −/−mz (M−Z−) E13.5 embryos compared with −/+ (M−Z+) E13.5 embryos was confirmed in the repeat experiment and c-KIT was indeed found to be mainly localized on the cell membrane, as is more clearly visualized in the magnified inset images as well as the magnified ra, rv, and vs (SI Appendix, Fig. S9 B and E). When quantified, we observed twofold increase of c-KIT-positive (c-KIT+) cells in the heart of −/−mz (M−Z−) E13.5 embryos compared with −/+ (M−Z+) E13.5 embryos (SI Appendix, Fig. S9F). Therefore, loss of both maternal and zygotic Zfp57 caused significant increase of c-Kit expression in the embryonic heart of −/−mz (M−Z−) embryos, whereas loss of just maternal or just zygotic Zfp57 did not result in any increased c-Kit expression in either −/+ (M−Z+) or −/−z (M+Z−) embryos. These results demonstrate that maternal and zygotic Zfp57 are redundant in c-Kit expression in the embryonic heart. Accumulation of c-KIT+ cells indicates that CPC differentiation is probably inhibited without ZFP57.
NKX2.5 and GATA4 are two key transcription factors required for cardiomyocyte differentiation (12, 35, 41). Based on qRT-PCR, both Nkx2.5 and Gata4 transcripts were down-regulated in −/−mz (M−Z−) E13.5 embryos compared with −/+ (M−Z+) E10.5 embryos, although Nkx2.5 transcript but not Gata4 transcript was also reduced in −/−mz (M−Z−) E10.5 embryos in comparison with −/+ (M−Z+) E10.5 embryos (SI Appendix, Fig. S8 C and D). To test if NKX2.5 protein may be also reduced without ZFP57, we performed immunostaining with antibodies against NKX2.5 in E10.5 (SI Appendix, Fig. S9A) and E13.5 (Fig. 5B) embryos. In agreement with our RNA in situ results for Nkx2.5 in E10.5 embryos (SI Appendix, Fig. S5B), NKX2.5 was reduced in immunostaining in the heart of −/−mz (M−Z−) but not −/−z (M+Z−) E10.5 embryos compared with +/− (M+Z+) E10.5 embryos (SI Appendix, Fig. S9A). NKX2.5 was also decreased in the heart of −/−mz (M−Z−) E13.5 embryos in comparison with +/− (M+Z+), −/+ (M−Z+), or −/−z (M+Z−) E13.5 embryos, whereas it was similar in +/− (M+Z+), −/+ (M−Z+), or −/−z (M+Z−) E13.5 embryos (Fig. 5B). These results are in agreement with those obtained with qRT-PCR and RNA in situ for Nkx2.5 in E13.5 embryos (Fig. 3). Reduced NKX2.5 in the heart of −/−mz (M−Z−) versus −/+ (M−Z+) E13.5 embryos was confirmed by the repeat immunostaining experiments (SI Appendix, Fig. S9C). Both the number and the mean intensity of NKX2.5+ cells decreased in the heart of −/−mz (M−Z−) E13.5 embryos in comparison with −/+ (M−Z+) embryos (SI Appendix, Fig. S9 F and G).
Similarly, GATA4 protein was also down-regulated in the heart of −/−mz (M−Z−) E13.5 embryos but not in the heart of −/−z (M+Z−) E13.5 in comparison with +/− (M+Z+) or −/+ (M−Z+) E13.5 embryos (Fig. 5C). Down-regulation of GATA4 in the heart of −/−mz (M−Z−) E13.5 embryos versus −/+ (M−Z+) E13.5 embryos was verified by the repeat immunostaining experiments (SI Appendix, Fig. S9D). Upon quantification, the number of GATA4+ cells also decreased in the heart of −/−mz (M−Z−) E13.5 embryos in comparison with −/+ (M−Z+) E13.5 embryos (SI Appendix, Fig. S9F).
These results suggest that differentiation of CPCs into NKX2.5+ or GATA4+ cells is attenuated in the heart without ZFP57. Maternal and zygotic Zfp57 are redundant in the expression of NKX2.5 and GATA4 in the embryonic heart. In our future study, we will examine if NKX2.5+ or GATA4+ cells are immature committed cardiomyocytes by coimmunostaining with antibodies against some markers for cardiomyocytes such as contractile proteins.
Proliferating CPCs Exhibited Differentiation Defect.
Overall cell proliferation was reduced in the heart of −/−mz (M−Z−) E13.5 embryos in comparison with +/− (M+Z+), −/+ (M−Z+), or −/−z (M+Z−) E13.5 embryos (Fig. 4). To find out which cell types constitute the remaining proliferating cells in the heart of −/−mz (M−Z−) E13.5 embryos, we performed EdU labeling in combination with immunostaining with antibodies against NKX2.5 or c-KIT (Fig. 5 D and E and SI Appendix, Figs. S10 and S11). We found that the fraction of EdU+ cells that were NKX2.5+ was reduced in the heart of −/−mz (M−Z−) E13.5 embryos compared with −/+ (M−Z+) E13.5 embryos (Fig. 5 D and F). By contrast, the fraction of EdU+ cells that were c-KIT+ was increased in the heart of −/−mz (M−Z−) E13.5 embryos versus Zfp57−/+ (M−Z+) E13.5 embryos (Fig. 5 E and G). Thus, proliferating c-KIT+ cells, i.e., presumably proliferating CPCs, accumulated, whereas proliferating NKX2.5+ cells, i.e., presumably immature committed cardiomyocytes, decreased without ZFP57.
NKX2.5+ Cells Exhibited Proliferation Defect.
We also found the fraction of NKX2.5+ cells that were EdU+ was reduced in the heart of −/−mz (M−Z−) E13.5 embryos compared with −/+ (M−Z+) E13.5 embryos (SI Appendix, Fig. S10A). When quantified, it was significantly reduced in the whole heart (SI Appendix, Fig. S10B). If measured separately, it was significantly reduced in the ventricle and ventricular septum but not in the atria (SI Appendix, Fig. S10C). These results suggest that NKX2.5+ cells exhibited reduced proliferation without ZFP57.
c-KIT+ Cells Displayed Increased Proliferation.
Interestingly, we found c-KIT+ cells had increased proliferation in the heart of −/−mz (M−Z−) E13.5 embryos compared with −/+ (M−Z+) E13.5 embryos (SI Appendix, Fig. S11A). When quantified in the whole heart, the fraction of c-KIT+ cells that were EdU+ was significantly increased in −/−mz (M−Z−) E13.5 embryos in comparison with −/+ (M−Z+) E13.5 embryos (SI Appendix, Fig. S11B). When quantified separately, it was significantly increased in the ventricle and ventricular septum but not in the atria (SI Appendix, Fig. S11C). These results suggest that c-KIT+ cells exhibited increased cell proliferation without ZFP57.
Discussion
Maternal and zygotic Zfp57 play redundant roles in the survival of mouse embryos (13). In this study, we found that Zfp57 mutants exhibited multiple cardiac defects, with the maternal-zygotic mutant displaying higher penetrance and more severe phenotypes than the zygotic mutant. Indeed, we found expression of some key players in cardiac development such as Hey2, Nkx2.5, and Gata4 was only severely down-regulated in the Zfp57 maternal-zygotic mutant E13.5 embryos, less so in E10.5 embryos, but not in Zfp57 zygotic mutant or heterozygous embryos with or without maternal Zfp57 (Fig. 3 and SI Appendix, Figs. S5, S6, and S8). These results suggest that maternal Zfp57, together with zygotic Zfp57, contributes to cardiac development. Maternal and zygotic Zfp57 play redundant roles in normal cardiac septation and ventricular wall thickness. This serves as an example of maternal function that is involved in mammalian organ development. Surprisingly, zygotic Zfp57 appears to be necessary and sufficient for Bmp10 expression and ventricular trabeculation in the embryonic heart (Figs. 1E and 3). Therefore, maternal and zygotic Zfp57 are partially redundant in cardiac development, with maternal Zfp57 functioning in some but not all processes.
ZFP57 is required for the maintenance of the DNA methylation imprint at a large number of imprinted regions in both mouse and humans (13, 17). Maternal and zygotic Zfp57 are partially redundant in its maintenance (13). DNA methylation at the imprinting control regions is essential for proper expression of the imprinted genes (14, 16, 42). Previously, we hypothesized that maternal Zfp57 may exert its effect on the late stage of mouse embryonic development through its roles in the maintenance of the DNA methylation imprint in early mouse embryos (43). In this case, maternal Zfp57, together with zygotic Zfp57, maintains the DNA methylation imprint at some target imprinted regions that control expression of certain imprinted genes required for cardiac development in mouse embryos. For example, expression of the imprinted Dlk1 gene is dependent on DNA methylation at the IG-DMR, the imprinting control region of the Dlk1-Dio3 imprinted region (22, 44). In this study, we found both the DNA methylation imprint at the IG-DMR and expression of Dlk1 were lost in the embryonic heart without ZFP57 (Fig. 2 B and C and SI Appendix, Fig. S4 A and B). Furthermore, we found that loss of ZFP57 causes cardiac defects that are reminiscent of various knockout mice in the NOTCH signaling pathway (Fig. 6). Further analysis revealed that NOTCH1 activation was inhibited in the heart of Zfp57 mutant embryos. Expression of Hey2, Nkx2.5, and Bmp10, three target genes of NOTCH1, was down-regulated in the heart without ZFP57 confirming that NOTCH signaling was attenuated in the heart of Zfp57 mutants. Together these results support our hypothesis that maternal and zygotic Zfp57 influence cardiac development through regulating DNA methylation imprint and expression of the imprinted genes that function in NOTCH signaling.
Fig. 6.
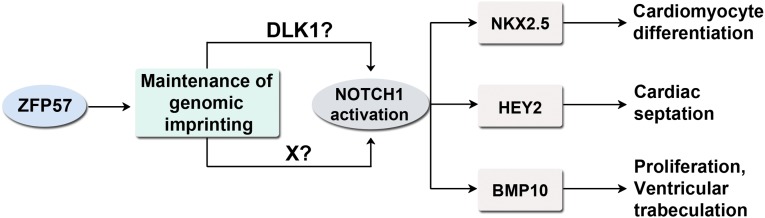
A schematic model is proposed for the probable molecular mechanisms of ZFP57 in Notch signaling and cardiac development. ZFP57 maintains genomic imprinting and it is required for the proper expression of its target imprinted genes including Dlk1. DLK1 and/or other target gene product of ZFP57 (indicated by factor X here) may activate NOTCH1 to generate N1ICD that can stimulate transcription of the NOTCH1 target genes in the embryonic heart. NOTCH1 activation could be either DLK1 dependent or DLK1 independent. NKX2.5, HEY2, and BMP10 are three downstream targets of NOTCH1 that control cardiomyocyte differentiation, cardiac septation, proliferation, and ventricular trabeculation, respectively.
The imprinted Dlk1 gene is highly expressed during embryogenesis (45–49). It is also called Pref-1, a factor involved in adipogenesis (20, 21, 50). Some previous studies had implicated DLK1 and its homologs as positive regulators in LIN-12/NOTCH signaling (25, 51, 52), whereas other studies had shown the opposite or no effect of DLK1 on NOTCH signaling (26, 27, 50, 53–56). These seemingly contradictory results could be due to different experimental systems used in different studies. Another possible explanation for these discrepancies could be caused by the complex expression pattern and structure of the imprinted Dlk1 gene (47, 49). It is reported that there are various isoforms of Dlk1 transcripts that produce membrane-bound or secreted DLK1 proteins (13, 24, 49, 53, 57–59). The membrane-bound form of DLK1 can also be cleaved to generate soluble peptides (58). These different isoforms of DLK1 proteins could exhibit opposite effects in cell signaling and development (45, 55, 60, 61). Therefore, DLK1 may act as a positive or negative regulator of NOTCH signaling under different conditions. The cumulative effect of various DLK1 proteins may also result in no effect on NOTCH signaling under certain circumstances.
Most N1ICD+ cells were DLK1+ in the heart of heterozygous, −/+ (M−Z+), embryos, whereas the N1ICD+DLK1+ cells were almost absent in the maternal-zygotic mutant, −/−mz (M−Z−), embryos (Fig. 2 F and G). We propose here that DLK1 may be involved in NOTCH1 activation in cardiac development (Fig. 6). Intriguingly, a small portion of N1ICD+ cells were DLK1−, indicative of DLK1-independent NOTCH1 activation, that remained constant in the heart of heterozygous, −/+ (M−Z+), or maternal-zygotic mutant, −/−mz (M−Z−), embryos (Fig. 2 F and G). These results suggest that DLK1 probably promotes N1ICD generation and DLK1-mediated NOTCH1 activation may account for most N1ICD in the heart of heterozygous −/+ (M−Z+) embryos. This may be one of the first instances implicating DLK1 as a positive regulator in NOTCH signaling in mammalian heart development.
Intriguingly, DLK1 and N1ICD were present in the same cells in the embryonic heart. We hypothesize that DLK1 may act as a cofactor such as a coreceptor, rather than a noncanonical ligand, for NOTCH1 and other NOTCH receptors. Alternatively, DLK1 may activate NOTCH signaling by recycling and/or presenting NOTCH receptor proteins for activation by canonical DSL ligands. It is also possible that DLK1 functions as a ligand for NOTCH1 and other NOTCH receptors within the same cells to activate NOTCH receptors through an autocrine mechanism (29). These hypotheses need to be tested in the future by investigating the specific interactions of DLK1 and NOTCH receptors.
It is also plausible that ZFP57 activates NOTCH1 via a DLK1-independent pathway (via factor X in Fig. 6), with DLK1 being just a bystander in the NOTCH1 receptor activation process. Based on current data, we could not distinguish these possibilities. However, there have been a number of papers suggesting a probable role of DLK1 in LIN-12/NOTCH signaling regardless of whether it is a positive or negative regulator (25–27, 51, 52, 56). Therefore, we favor our current model that DLK1, probably together with other target genes of ZFP57 (e.g., factor X in Fig. 6), is involved in NOTCH1 activation.
Loss of ZFP57 caused accumulation of c-KIT+ cells and decrease of NKX2.5+ or GATA4+ cells in the heart. Although we need to use antibodies against some markers of cardiomyocytes for coimmunostaining to confirm these cell identities, we think NKX2.5+ or GATA4+ cells are likely immature committed cardiomyocytes that are decreased in the embryonic heart of Zfp57 mutant embryos. These results implicating cell differentiation defects are consistent with attenuation of NOTCH signaling in the heart without ZFP57. Indeed, LIN-12/NOTCH signaling is essential for cell-fate specification (8). This differentiation block, together with cell proliferation defect, may result in most cardiac defects observed in Zfp57 mutants (Fig. 6).
Both mouse and human ZFP57 proteins maintain genomic imprinting (13, 17). Interestingly, some human patients with ZFP57 mutations exhibited partially penetrant ASD, VSD, and tetralogy of Fallot (TOF) (17), resembling those present in the Zfp57 zygotic mutant (Fig. 1). Based on this observation, we hypothesize that human ZFP57 mutations may also compromise NOTCH signaling.
Materials and Methods
Mouse Breeding and Timed Pregnancy Mating.
The animal protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the Icahn School of Medicine at Mount Sinai. All mouse strains were on a mixed genetic background of 129Sv/Ev and Black Swiss origins. Zfp57 mutant mice containing the deleted null allele of Zfp57 were used here for timed pregnancy mating (13). Noon of the day when vaginal plug was found was counted as the half-day pregnancy (E0.5). Zfp57 zygotic mutant, −/−z (M+Z−), embryos lacking just the zygotic Zfp57 and heterozygous, +/− (M+Z+), embryos with both maternal and zygotic Zfp57 were generated from the cross between heterozygous (+/−) female mice and homozygous (−/−) male mice or from the cross between heterozygous (+/−) female mice and heterozygous (+/−) male mice (SI Appendix, Fig. S1). Similarly, Zfp57 maternal-zygotic mutant, −/−mz (M−Z−), embryos without maternal or zygotic Zfp57, and heterozygous, −/+ (M−Z+), embryos lacking maternal Zfp57, were generated from the cross between homozygous (−/−) female mice and heterozygous (+/−) male mice (SI Appendix, Fig. S1).
Histological Analysis and Quantification.
Embryos or heart samples isolated from pregnant mice were fixed in 4% (wt/vol) of paraformaldehyde (PFA) solution overnight and embedded in paraffin. Transverse serial sections of 8 µm in thickness were stained with hematoxalin and eosin (H&E). Ventricular wall (vw) thickness and trabecular area were measured by ImageJ software on comparable sections through the atrioventricular canal region for five separate embryos for each genotype. For vw, four measurements were made in the selected areas of the left and right ventricles of each embryo. Total average trabecular area was quantified in the left and right ventricles of each embryo.
RNA in Situ Hybridization with Digoxigenin Labeling.
Sense and antisense probes were made of a DIG RNA labeling kit (Roche). Embryos fixed in 4% (wt/vol) of PFA solution for 24 h were dehydrated in either 30% (wt/vol) or 2 M of sucrose solution before being embedded in OCT compound (Sakura). Transverse serial cryosections of 10 µm in thickness obtained from these embryos were incubated with sense or antisense digoxigenin-labeled RNA probe at 65 °C overnight. Alkaline phosphatase (AP) substrate was added to the sections for detection after incubation with AP-coupled antidigoxigenin antibody.
Immunohistochemical Analysis and Quantification.
Samples were fixed in 4% (wt/vol) of PFA solution for 4 h to overnight at 4 °C and embedded in paraffin or OCT. Transverse serial sections of 8 µm in thickness were obtained for immunostaining. Sodium citrate antigen retrieval was applied to paraffin sections. For NOTCH1 or N1ICD immunostaining, paraffin sections or cryosections were blocked with 5% (vol/vol) goat serum for 1 h at room temperature (RT) followed by incubation with rabbit monoclonal antibodies against NOTCH1 (1:100, Cell Signaling 3608) or N1ICD (1:100, Cell Signaling 4147) at 4 °C overnight. After incubation with the primary antibodies, these sections were incubated with biotinylated goat anti-rabbit IgG (1:300, Vector BA1000) secondary antibodies for 1 h at RT followed by the signal amplification procedure with TSA Plus Cy3 System (Perkin-Elmer) that was performed according to the manufacturer’s suggested protocol. For c-KIT, NKX2.5, or GATA4 immunostaining, cryosections were blocked with 5% (vol/vol) donkey serum for 1 h at RT followed by incubation with goat anti–c-KIT (1:300, R&D AF1356), goat anti-NKX2.5 (1:300, Santa Cruz SC8697), or goat anti-GATA4 (1:600, Santa Cruz SC1237) antibodies. After incubation with the primary antibodies, these sections were incubated with biotinylated donkey anti-goat IgG (1:300, Millipore AP180B) secondary antibodies for 1 h at RT followed by the signal amplification procedure with TSA Plus Cy3 System. For cleaved Caspase-3 immunostaining, paraffin sections were blocked with 5% (vol/vol) of goat serum for 1 h at RT followed by incubation with rabbit anti-cleaved Caspase-3 (1:500, Cell Signaling 9664) antibody at 4 °C overnight. After incubation with the primary antibodies, these sections were incubated with biotinylated goat anti-rabbit IgG (1:300, Vector BA1000) secondary antibodies for 1 h at RT followed by signal amplification with TSA Plus Cy3 System. For DLK1 immunostaining, cryosections were incubated with rat anti-DLK1 antibody (Enzo Life Sciences PF105B) at 4 °C overnight and then stained with FITC-labeled goat anti-rat IgG (1:300, Southern Biotech 3030-02) secondary antibodies for 1 h at RT. All stained sections were imaged under a Axioplan2IE microscope with Apotome (Zeiss). Immunofluorescent cells were counted on comparable sections with ImageJ software for at least three separate embryos for each genotype.
EdU Incorporation Assay.
Pregnant female mice were injected with 25 μg of EdU (Molecular Probes) per gram of body weight 1 h before embryos were collected for fixation. The embryos were fixed in 4% (wt/vol) of PFA for 4 h at 4 °C and embedded in OCT. Transverse serial sections of 8 µm in thickness obtained from these embryos were stained with Click-iT Imaging Kit (Molecular Probes).
Statistical Analysis.
Statistical analyses were performed using Microsoft Excel. All results are reported as mean ± SEM of at least three independent samples. Student’s t test was used for statistical comparisons between two groups of data. The results with **P < 0.01 and *P < 0.05 were considered statistically significant in Student’s t test. One-way analysis of variance (ANOVA) was performed for statistical comparisons of more than two groups of data, followed by the post hoc independent sample t test with Bonferroni correction. The results with *P < 0.05 were considered statistically significant in ANOVA.
Supplementary Material
Acknowledgments
The authors thank Drs. James Bieker, Bruce Gelb, and Paul Wassarman of Icahn School of Medicine at Mount Sinai for their critical reading of the manuscript. Microscopy was performed at the Microscopy CORE facility located at the Icahn School of Medicine at Mount Sinai. This work was supported by National Institutes of Health (NIH) Grant GM093335, NYSTEM Contract C026434, and American Heart Association Grant 09SDG2400151 (to X.L.). Y.S. and D.E.C. were partly supported by NIH T32 Cancer Biology or Developmental Biology Training Grants, respectively.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1415541112/-/DCSupplemental.
References
- 1.Zaidi S, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013;498(7453):220–223. doi: 10.1038/nature12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin CJ, Lin CY, Chen CH, Zhou B, Chang CP. Partitioning the heart: Mechanisms of cardiac septation and valve development. Development. 2012;139(18):3277–3299. doi: 10.1242/dev.063495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bruneau BG. The developmental genetics of congenital heart disease. Nature. 2008;451(7181):943–948. doi: 10.1038/nature06801. [DOI] [PubMed] [Google Scholar]
- 4.Fahed AC, Gelb BD, Seidman JG, Seidman CE. Genetics of congenital heart disease: The glass half empty. Circ Res. 2013;112(4):707–720. doi: 10.1161/CIRCRESAHA.112.300853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greenwald I, Kovall R. Notch signaling: Genetics and structure. WormBook. 2013:1–28. doi: 10.1895/wormbook.1.10.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kopan R. Notch signaling. Cold Spring Harb Perspect Biol. 2012;4(10) doi: 10.1101/cshperspect.a011213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Artavanis-Tsakonas S, Muskavitch MA. Notch: The past, the present, and the future. Curr Top Dev Biol. 2010;92:1–29. doi: 10.1016/S0070-2153(10)92001-2. [DOI] [PubMed] [Google Scholar]
- 8.Greenwald I. LIN-12/Notch signaling: Lessons from worms and flies. Genes Dev. 1998;12(12):1751–1762. doi: 10.1101/gad.12.12.1751. [DOI] [PubMed] [Google Scholar]
- 9.MacGrogan D, Nus M, de la Pompa JL. Notch signaling in cardiac development and disease. Curr Top Dev Biol. 2010;92:333–365. doi: 10.1016/S0070-2153(10)92011-5. [DOI] [PubMed] [Google Scholar]
- 10.Jain R, Rentschler S, Epstein JA. Notch and cardiac outflow tract development. Ann N Y Acad Sci. 2010;1188:184–190. doi: 10.1111/j.1749-6632.2009.05099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gridley T. Notch signaling in the vasculature. Curr Top Dev Biol. 2010;92:277–309. doi: 10.1016/S0070-2153(10)92009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boni A, et al. Notch1 regulates the fate of cardiac progenitor cells. Proc Natl Acad Sci USA. 2008;105(40):15529–15534. doi: 10.1073/pnas.0808357105. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Li X, et al. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev Cell. 2008;15(4):547–557. doi: 10.1016/j.devcel.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartolomei MS, Ferguson-Smith AC. Mammalian genomic imprinting. Cold Spring Harb Perspect Biol. 2011;3(7) doi: 10.1101/cshperspect.a002592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X. Genomic imprinting is a parental effect established in mammalian germ cells. Curr Top Dev Biol. 2013;102:35–59. doi: 10.1016/B978-0-12-416024-8.00002-7. [DOI] [PubMed] [Google Scholar]
- 16.Barlow DP. Genomic imprinting: A mammalian epigenetic discovery model. Annu Rev Genet. 2011;45:379–403. doi: 10.1146/annurev-genet-110410-132459. [DOI] [PubMed] [Google Scholar]
- 17.Mackay DJ, et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet. 2008;40(8):949–951. doi: 10.1038/ng.187. [DOI] [PubMed] [Google Scholar]
- 18.Conway SJ, Kruzynska-Frejtag A, Kneer PL, Machnicki M, Koushik SV. What cardiovascular defect does my prenatal mouse mutant have, and why? Genesis. 2003;35(1):1–21. doi: 10.1002/gene.10152. [DOI] [PubMed] [Google Scholar]
- 19.Chen H, et al. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development. 2004;131(9):2219–2231. doi: 10.1242/dev.01094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laborda J, Sausville EA, Hoffman T, Notario V. dlk, a putative mammalian homeotic gene differentially expressed in small cell lung carcinoma and neuroendocrine tumor cell line. J Biol Chem. 1993;268(6):3817–3820. [PubMed] [Google Scholar]
- 21.Smas CM, Sul HS. Pref-1, a protein containing EGF-like repeats, inhibits adipocyte differentiation. Cell. 1993;73(4):725–734. doi: 10.1016/0092-8674(93)90252-l. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt JV, Matteson PG, Jones BK, Guan XJ, Tilghman SM. The Dlk1 and Gtl2 genes are linked and reciprocally imprinted. Genes Dev. 2000;14(16):1997–2002. [PMC free article] [PubMed] [Google Scholar]
- 23.Takada S, et al. Delta-like and gtl2 are reciprocally expressed, differentially methylated linked imprinted genes on mouse chromosome 12. Curr Biol. 2000;10(18):1135–1138. doi: 10.1016/s0960-9822(00)00704-1. [DOI] [PubMed] [Google Scholar]
- 24.Kopan R, Ilagan MX. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell. 2009;137(2):216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Komatsu H, et al. OSM-11 facilitates LIN-12 Notch signaling during Caenorhabditis elegans vulval development. PLoS Biol. 2008;6(8):e196. doi: 10.1371/journal.pbio.0060196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baladrón V, et al. dlk acts as a negative regulator of Notch1 activation through interactions with specific EGF-like repeats. Exp Cell Res. 2005;303(2):343–359. doi: 10.1016/j.yexcr.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 27.Bray SJ, Takada S, Harrison E, Shen SC, Ferguson-Smith AC. The atypical mammalian ligand Delta-like homologue 1 (Dlk1) can regulate Notch signalling in Drosophila. BMC Dev Biol. 2008;8:11. doi: 10.1186/1471-213X-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fortini ME. Notch signaling: The core pathway and its posttranslational regulation. Dev Cell. 2009;16(5):633–647. doi: 10.1016/j.devcel.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 29.D’Souza B, Meloty-Kapella L, Weinmaster G. Canonical and non-canonical Notch ligands. Curr Top Dev Biol. 2010;92:73–129. doi: 10.1016/S0070-2153(10)92003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Struhl G, Greenwald I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature. 1999;398(6727):522–525. doi: 10.1038/19091. [DOI] [PubMed] [Google Scholar]
- 31.Fischer A, Schumacher N, Maier M, Sendtner M, Gessler M. The Notch target genes Hey1 and Hey2 are required for embryonic vascular development. Genes Dev. 2004;18(8):901–911. doi: 10.1101/gad.291004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Donovan J, Kordylewska A, Jan YN, Utset MF. Tetralogy of fallot and other congenital heart defects in Hey2 mutant mice. Curr Biol. 2002;12(18):1605–1610. doi: 10.1016/s0960-9822(02)01149-1. [DOI] [PubMed] [Google Scholar]
- 33.Gessler M, et al. Mouse gridlock: No aortic coarctation or deficiency, but fatal cardiac defects in Hey2 -/- mice. Curr Biol. 2002;12(18):1601–1604. doi: 10.1016/s0960-9822(02)01150-8. [DOI] [PubMed] [Google Scholar]
- 34.Sakata Y, et al. Ventricular septal defect and cardiomyopathy in mice lacking the transcription factor CHF1/Hey2. Proc Natl Acad Sci USA. 2002;99(25):16197–16202. doi: 10.1073/pnas.252648999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCulley DJ, Black BL. Transcription factor pathways and congenital heart disease. Curr Top Dev Biol. 2012;100:253–277. doi: 10.1016/B978-0-12-387786-4.00008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grego-Bessa J, et al. Notch signaling is essential for ventricular chamber development. Dev Cell. 2007;12(3):415–429. doi: 10.1016/j.devcel.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moens CB, Stanton BR, Parada LF, Rossant J. Defects in heart and lung development in compound heterozygotes for two different targeted mutations at the N-myc locus. Development. 1993;119(2):485–499. doi: 10.1242/dev.119.2.485. [DOI] [PubMed] [Google Scholar]
- 38.Charron J, et al. Embryonic lethality in mice homozygous for a targeted disruption of the N-myc gene. Genes Dev. 1992;6(12A):2248–2257. doi: 10.1101/gad.6.12a.2248. [DOI] [PubMed] [Google Scholar]
- 39.Muller-Sieburg CE, Sieburg HB, Bernitz JM, Cattarossi G. Stem cell heterogeneity: Implications for aging and regenerative medicine. Blood. 2012;119(17):3900–3907. doi: 10.1182/blood-2011-12-376749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boisset JC, et al. In vivo imaging of haematopoietic cells emerging from the mouse aortic endothelium. Nature. 2010;464(7285):116–120. doi: 10.1038/nature08764. [DOI] [PubMed] [Google Scholar]
- 41.Scott IC. Life before Nkx2.5: Cardiovascular progenitor cells: Embryonic origins and development. Curr Top Dev Biol. 2012;100:1–31. doi: 10.1016/B978-0-12-387786-4.00001-4. [DOI] [PubMed] [Google Scholar]
- 42.Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366(6453):362–365. doi: 10.1038/366362a0. [DOI] [PubMed] [Google Scholar]
- 43.Li X. Extending the maternal-zygotic effect with genomic imprinting. Mol Hum Reprod. 2010;16(9):695–703. doi: 10.1093/molehr/gaq028. [DOI] [PubMed] [Google Scholar]
- 44.Lin SP, et al. Asymmetric regulation of imprinting on the maternal and paternal chromosomes at the Dlk1-Gtl2 imprinted cluster on mouse chromosome 12. Nat Genet. 2003;35(1):97–102. doi: 10.1038/ng1233. [DOI] [PubMed] [Google Scholar]
- 45.Falix FA, Aronson DC, Lamers WH, Gaemers IC. Possible roles of DLK1 in the Notch pathway during development and disease. Biochim Biophys Acta. 2012;1822(6):988–995. doi: 10.1016/j.bbadis.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 46.Appelbe OK, Yevtodiyenko A, Muniz-Talavera H, Schmidt JV. Conditional deletions refine the embryonic requirement for Dlk1. Mech Dev. 2013;130(2-3):143–159. doi: 10.1016/j.mod.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rogers ED, Ramalie JR, McMurray EN, Schmidt JV. Localizing transcriptional regulatory elements at the mouse Dlk1 locus. PLoS ONE. 2012;7(5):e36483. doi: 10.1371/journal.pone.0036483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.da Rocha ST, et al. Restricted co-expression of Dlk1 and the reciprocally imprinted non-coding RNA, Gtl2: implications for cis-acting control. Dev Biol. 2007;306(2):810–823. doi: 10.1016/j.ydbio.2007.02.043. [DOI] [PubMed] [Google Scholar]
- 49.Miller AJ, Cole SE. Multiple Dlk1 splice variants are expressed during early mouse embryogenesis. Int J Dev Biol. 2014;58(1):65–70. doi: 10.1387/ijdb.130316sc. [DOI] [PubMed] [Google Scholar]
- 50.Sul HS. Minireview: Pref-1: Role in adipogenesis and mesenchymal cell fate. Mol Endocrinol. 2009;23(11):1717–1725. doi: 10.1210/me.2009-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Singh K, et al. C. elegans Notch signaling regulates adult chemosensory response and larval molting quiescence. Curr Biol. 2011;21(10):825–834. doi: 10.1016/j.cub.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li L, et al. DLK1 promotes lung cancer cell invasion through upregulation of MMP9 expression depending on Notch signaling. PLoS ONE. 2014;9(3):e91509. doi: 10.1371/journal.pone.0091509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ferrón SR, et al. Postnatal loss of Dlk1 imprinting in stem cells and niche astrocytes regulates neurogenesis. Nature. 2011;475(7356):381–385. doi: 10.1038/nature10229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Müller D, et al. Dlk1 promotes a fast motor neuron biophysical signature required for peak force execution. Science. 2014;343(6176):1264–1266. doi: 10.1126/science.1246448. [DOI] [PubMed] [Google Scholar]
- 55.Andersen DC, et al. Dual role of delta-like 1 homolog (DLK1) in skeletal muscle development and adult muscle regeneration. Development. 2013;140(18):3743–3753. doi: 10.1242/dev.095810. [DOI] [PubMed] [Google Scholar]
- 56.Schober A, et al. MicroRNA-126-5p promotes endothelial proliferation and limits atherosclerosis by suppressing Dlk1. Nat Med. 2014;20(4):368–376. doi: 10.1038/nm.3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smas CM, Chen L, Zhao L, Latasa MJ, Sul HS. Transcriptional repression of pref-1 by glucocorticoids promotes 3T3-L1 adipocyte differentiation. J Biol Chem. 1999;274(18):12632–12641. doi: 10.1074/jbc.274.18.12632. [DOI] [PubMed] [Google Scholar]
- 58.Smas CM, Chen L, Sul HS. Cleavage of membrane-associated pref-1 generates a soluble inhibitor of adipocyte differentiation. Mol Cell Biol. 1997;17(2):977–988. doi: 10.1128/mcb.17.2.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Garcés C, Ruiz-Hidalgo MJ, Bonvini E, Goldstein J, Laborda J. Adipocyte differentiation is modulated by secreted delta-like (dlk) variants and requires the expression of membrane-associated dlk. Differentiation. 1999;64(2):103–114. doi: 10.1046/j.1432-0436.1999.6420103.x. [DOI] [PubMed] [Google Scholar]
- 60.Shin S, Suh Y, Zerby HN, Lee K. Membrane-bound delta-like 1 homolog (Dlk1) promotes while soluble Dlk1 inhibits myogenesis in C2C12 cells. FEBS Lett. 2014;588(7):1100–1108. doi: 10.1016/j.febslet.2014.02.027. [DOI] [PubMed] [Google Scholar]
- 61.Traustadottir GA, Kosmina R, Sheikh SP, Jensen CH, Andersen DC. Preadipocytes proliferate and differentiate under the guidance of Delta-like 1 homolog (DLK1) Adipocyte. 2013;2(4):272–275. doi: 10.4161/adip.24994. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.