

Abstract
Objectives:
Very little is known about mechanisms of idiosyncratic sensitivity to the damaging effects of mercury (Hg); however, there is likely a genetic component. The aim of the present study was to search for genetic variation in genes thought to be involved in Hg metabolism and transport in a group of individuals identified as having elevated Hg sensitivity compared to a normal control group.
Materials and Methods:
Survivors of pink disease (PD; infantile acrodynia) are a population of clinically identifiable individuals who are Hg sensitive. In the present study, single nucleotide polymorphisms in genes thought to be involved in Hg transport and metabolism were compared across two groups: (i) PD survivors (n = 25); and (ii) age- and sex-matched healthy controls (n = 25).
Results:
Analyses revealed significant differences between groups in genotype frequencies for rs662 in the gene encoding paraoxanase 1 (PON1) and rs1801131 in the gene encoding methylenetetrahydrofolate reductase (MTHFR).
Conclusions:
We have identified two genetic polymorphisms associated with increased sensitivity to Hg. Genetic variation in MTHFR and PON1 significantly differentiated a group formerly diagnosed with PD (a condition of Hg hypersensitivity) with age- and gender-matched healthy controls.
Keywords: Genetics, mercury sensitivity, methylenetetrahydrofolate reductase, pink disease, paraoxanase 1
INTRODUCTION
The potent neurotoxicity of mercury (Hg) has been known for centuries, yet it remains in use in a variety of medical, commercial, and industrial practices. Of most concern internationally is the fact that Hg is widely distributed throughout our food chain, air, water, and land at levels that have proven toxic to flora, fauna, and human beings.[1] Hg in all its forms (ionic Hg2+ and Hg+/Hg22+; vapor Hg0 and organic alkyl forms MeHg+, Me2 Hg, EtHg+, and Et2 Hg) has potent neurotoxicity for the brain and particularly for the human fetal brain.[2] The central nervous system is a major target of Hg, and both high- and low-dose exposure can produce significant long-term neurological damage.[3,4] Neurological symptoms attributed to Hg intoxication include ataxia, deafness, psychosis, loss of speech, erethism, and constriction of the visual field.[5] For example, the direct causal link between cerebral palsy and Hg exposure to the developing human fetus was first established by the Swedish Expert Group in 1971[6] and confirmed recently by other researchers.[7,8] For a fetus, Hg brain levels can be significantly higher than in maternal blood, and the developing fetal brain demonstrates high sensitivity to Hg toxicity. Current research has identified that Hg exposure during early development (including in utero) is associated with tic disorders,[9] PD,[10] cerebral palsy, and Hunter–Russell syndrome.[11]
The PD outbreak of the 20th century highlights the potential severity of the reaction (including death) that some children may have to a low level of Hg exposure,[12] and it is a variable, idiosyncratic, and unpredictable individual susceptibility to Hg that determines the reaction.[13] In the early 1900s, the increased availability of new and untested pharmaceuticals led to the exposure of infants to inorganic Hg present as calomel in teething powder. Calomel contained Hg levels low enough to not be poisonous to the average infant; however, for a small subset of the population highly sensitive to Hg, the levels were sufficient to be detrimental and produce symptoms, which would eventually be classified as a disease entity known as PD or infantile acrodynia.[12] New cases of PD essentially ceased in the mid-1950s as calomel was identified as the causal agent and removed from teething products at that time. Some survivors of PD are still living and represent an important cohort of clinically identifiable Hg-sensitive individuals. Given the ubiquitous nature of low level Hg exposure, it remains possible that various adverse health effects are manifesting in Hg-sensitive individuals whom we are unable, at the present time, to identify on the basis of any clinical or genetic testing. Although unproven, various clinical effects have been hypothesised to result from low-level Hg exposure including autism,[13] tic disorders,[9] and Alzheimer's disease.[14] Additionally, the symptoms of an adverse response to Hg have been known to mimic various conditions including Parkinson's disease,[15] amyotrophic lateral sclerosis,[16] and schizophrenia.[17] An understanding of the genetic moderators of Hg impact would facilitate testing of these hypotheses.
The toxicokinetics and clinical effects of Hg exposure depend greatly on the dose, pattern, timing, route of exposure, individual excretion rates, and idiosyncratic sensitivity to Hg.[18] There is a substantial body of evidence to show that much of the variance in Hg sensitivity is determined by genetic factors.[3] The aim of this study was to test for association between genetic variation in genes involved in Hg transport and metabolism and PD.
MATERIALS AND METHODS
Ethical approval for this study was granted by the Deakin University Human Research Ethics Committee.
Participants
Survivors of PD: Potential participants were drawn from the database of PD survivors held by the Australian Pink Disease Support Group (http://www.pinkdisease.org/). A recruitment notice was placed on the group's website as well as emailed to all members who had supplied an email address. The notice stipulated that participants must have received a formal diagnosis of PD as a child and be residing within 200km of Sydney or Melbourne, Australia. Following receipt of informed consent from 25 volunteers, a suitable time was arranged for a qualified nurse to visit their residence. Participants had 2 × 10 ml of blood drawn. The blood samples were then transported to Genetic Repositories Australia whereupon cell lines were created. Extracted deoxyribonucleic acid (DNA) was then supplied to the research team. The PD survivor sample ranged in age from 60 to 75 years (22 females and three males) and had a mean age of 65.7 years (SD = 3.9).
Healthy controls: Age- and sex-matched healthy control DNA (n = 25) was procured from the Baker IDI Heart and Diabetes Institute Biobank. The healthy control sample was drawn from 22 females and 3 males ranging in age from 59 to 75 years with a mean age of 65.9 years (SD = 5.3).
For all samples, DNA concentration was adjusted to 50 ng/μl in TE buffer (10mM Tris-HCl, 1mM ethylenediaminetetraacetic acid (EDTA)•Na2 (pH 8.0)) and stored at −20°C.
Genotyping
Genetic polymorphisms associated with Hg metabolism and toxicity were identified by literature search. Details of the candidate genes associated with increased sensitivity to Hg are shown in Table 1. A total of 16 polymorphisms were genotyped in this study, 13 of them by polymerase chain reaction (PCR) restriction fragment length polymorphism. Presence/absence of GSTM1 and GSTT1 was assessed by multiplex PCR with albumin as positive control. The 15 bp insertion/deletion polymorphism (rs11267756) in SLC7A5 was assessed by PCR and agarose gel electrophoresis.
Table 1.
Candidate genes associated with increased sensitivity to Hg

Standard PCRs were conducted using 100 ng of DNA, 1 U Taq polymerase (Invitrogen, Melbourne, Australia), 200 μM each dNTP, 0.5 μM forward and reverse primer, 20 mM Tris-HCl pH 8.4, 50 mM KCl, and 2 mM MgCl2 in a total volume of 20 μl. The multiplex PCR for GSTM1, GSTT1, and albumin was conducted using 100 ng of DNA, 2 U Taq polymerase (Invitrogen), 400 μM each dNTP, 0.3 μM GSTM1 primers, 0.2 μM GSTT1 primers, 0.15 μM albumin primers, 20 mM Tris-HCl pH 8.4, 50 mM KCl, and 3 mM MgCl2 in a total volume of 20 μl.
Following PCR, 10 μl of each PCR product was purified using QIAquick PCR Purification kits (Qiagen, Melbourne, Australia) and eluted in 20 μl of nuclease-free water.
Restriction digestion of purified PCR products was conducted using 10 μl of purified DNA, 5-20 U restriction enzyme, and 1x corresponding restriction enzyme buffer, made up to a total volume of 20 μl with nuclease-free water.
Following restriction digestion, the samples were separated using agarose gel electrophoresis (1-5%, depending on product size), labeled with SybrSafe (Invitrogen) and visualized under ultraviolet (UV) light. Genotype calling was conducted blindly by two independent reviewers.
RESULTS
Hardy–Weinberg equilibrium and genotype frequencies for PD survivors and control group participants were compared using Fisher's exact test. Analyses were performed using IBM Statistical Package for Social Sciences (SPSS) 21. Genotype frequencies were considered to be different between groups at P < 0.05. The summary results for all polymorphisms are shown in Table 2.
Table 2.
Genotype data for 16 candidate gene polymorphisms in pink disease survivors and matched control subjects
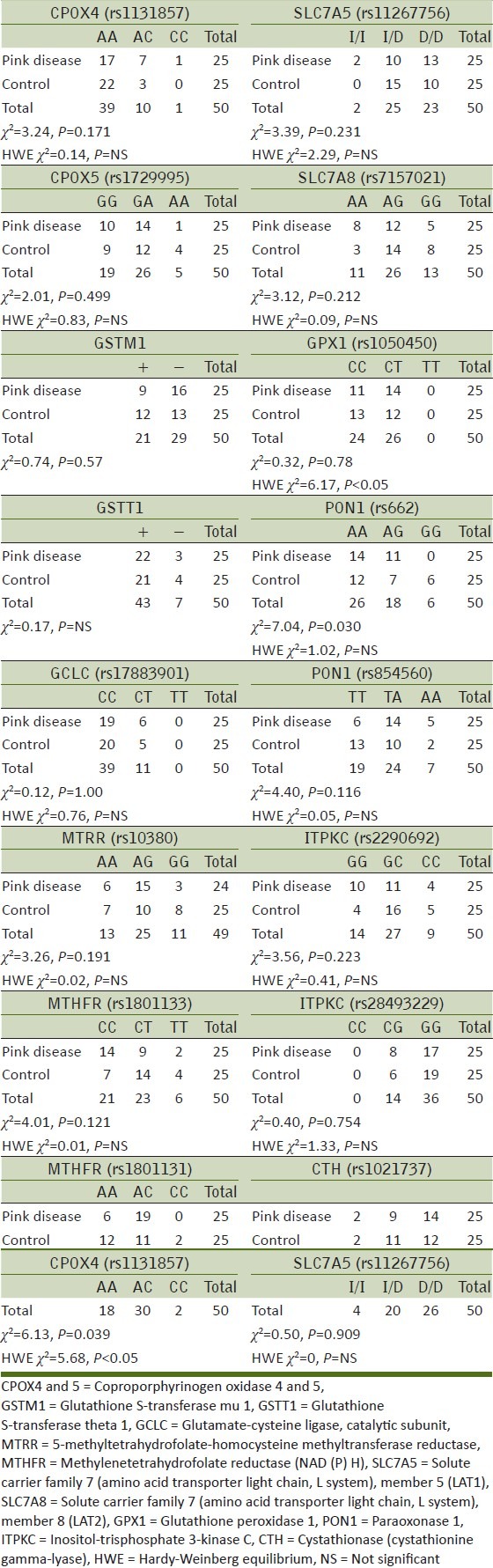
The only significant difference in genotype frequencies between PD survivors and controls were found for methylenetetrahydrofolate reductase (NAD (P) H) (MTHFR) at rs1801131 (χ2 = 6.13, P = 0.039) and for paraoxonase 1 (PON1) at rs662 (χ2 = 7.04, P = 0.030). Representative digests for the various genotypes are shown in Figure 1.
Figure 1.
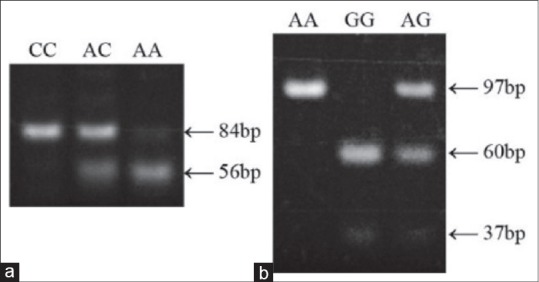
Representative genotyping results for (a) MTHFR rs1801131 and (b) PON1 rs662
Hardy–Weinberg equilibrium was present for all polymorphisms examined, except for rs1801131 in the MTHFR gene (χ2 = 5.68, P < 0.05) and rs1050450 in GPX1 (χ2 = 6.17, P < 0.05).
DISCUSSION
The old toxicology adage attributed to Paracelsus, “the dose makes the poison” is an incomplete and insufficient model of toxicokinetics, with modern perspectives emphasizing the large individual variance in response to any given dose of a toxicant.[18] Therefore, in order to comprehensively understand the etiology of clinical syndromes arising from Hg exposure, it is critical to understand individual sensitivity. Here we report the first study of Hg sensitivity based on genetic variance amongst survivors of PD, a condition which is the result of an idiosyncratic sensitivity to Hg.[12]
In this study, we found polymorphisms in two genes that differentiated PD survivors from age- and gender-matched healthy control subjects. This suggests a possible role in Hg sensitivity. The associated single nucleotide polymorphisms were rs662, which results in a glutamine to arginine substitution at position 192 of PON1, and rs1801131, which causes a glutamic acid to alanine substitution at amino acid 429 of MTHFR. Neither of these polymorphisms have previously been associated with sensitivity to Hg or other heavy metals, although studies have shown that heavy metals, including Hg, can affect PON1 activity levels.[19]
PON1 plays an important role in the hydrolysis and detoxification of several organophosphate insecticides, including diazinon and parathion, the nerve gases sarin and soman, and aromatic esters and lactones. Although no direct role for PON1 in the detoxification of Hg has been demonstrated, it is feasible that it could play a role and that this genetic variation at rs662, which is known to affect enzymatic activity, could affect the ability of the enzyme to protect against the toxic effects of Hg. Previous studies have shown association between genetic polymorphism in PON1 (including rs662) and a range of clinical disorders including coronary artery disease,[20] microvascular complications of diabetes,[21] and macular degeneration.[22] In most cases, these associations are thought to be driven by altered metabolism and levels of oxidized lipids in subjects with genetic variation in PON1.
MTHFR catalyzes the conversion of 5,10-methyle netetrahydrofolate to 5-methyltetrahydrofolate, a cosubstrate for homocysteine remethylation to methionine. This enzyme therefore plays a crucial role in amino acid and folate metabolism. MTHFR deficiency results in homocysteinemia and altered folate metabolism, and it has been studied extensively in the context of a range of diseases. Genetic variation in MTHFR has been reproducibly associated with neural tube defects,[23] vascular disease,[24] cleft lip/palate,[25] depression,[26] and schizophrenia.[27,28] For many of these associations, there appears to be interaction between genetic variation in MTHFR and plasma levels of either homocysteine or folate metabolites (or both), suggesting that MTHFR may act as a modifier of disease risk.
The rs1801131 polymorphism in MTHFR was not in Hardy–Weinberg equilibrium in our participants. This is consistent with published studies showing deviations from Hardy–Weinberg expectations that suggest there may be selective pressures for sequence variation in this gene.[29] Furthermore, a previous study provided data to suggest that mutations in MTHFR may have occurred on a founder haplotype with a selective advantage.[30]
A limitation of the study was the modest sample size of 50. This figure was a function of both funding limitations and challenges in communicating with and recruiting PD survivors. The PD survivor population is diminishing through age-related mortality, with the youngest survivors born in the 1950s. It is a very small, elderly, and unique population. As a result of the sample size, statistical power was, of course, impacted. Although the statistical analyses conducted were conservative and therefore accounted for the low power, it remains important to note the number of comparisons conducted (16) and interpret the results cautiously in light of the possibility of type I error.
Despite these challenges, the PD survivor population remains, to our knowledge, the only identifiable clinical population of individuals that have a demonstrated history of Hg sensitivity. They remain a vitally important population to researchers aiming to further our understanding of Hg sensitivity. The issue is particularly salient as low level Hg exposure is ubiquitous and the potent neurotoxicity of Hg is well established as being especially devastating to the central and peripheral nervous systems of infants and unborn babies.[3]
Beyond this emerging understanding of how Hg may affect individuals at doses known to produce clinical effects, there is less certainty as to predicting who is most at risk, and why, as a result of our lack of understanding of individual difference variables related to Hg sensitivity. This study contributes to our knowledge in this area by identifying two genetic variants that significantly differentiate healthy controls from a clinical population of Hg-sensitive individuals. Of course, there are likely many more genetic variables contributing to Hg sensitivity that await identification.
CONCLUSIONS
In this study, we have identified two genetic polymorphisms associated with increased sensitivity to Hg. Our results showed that genetic variation in MTHFR and PON1 significantly differentiated a group formerly diagnosed with PD (a condition of Hg hypersensitivity) with age- and gender-matched healthy controls. Given the range of diseases in which Hg has been implicated, these findings will facilitate further investigation into the source and mechanisms of Hg sensitivity with far-ranging clinical implications.
Footnotes
Source of Support: This study was funded by SafeMinds, USA.
Conflict of Interest: None declared.
REFERENCES
- 1.Clarkson TW, Magos L. The toxicology of mercury and its chemical compounds. CRC Crit Rev Toxicol. 2006;36:609–62. doi: 10.1080/10408440600845619. [DOI] [PubMed] [Google Scholar]
- 2.Amin-Zaki L, Elhassani S, Majeed MA, Clarkson TW, Doherty RA, Greenwood M. Intra-uterine methylmercury poisoning in Iraq. Pediatrics. 1974;54:587–95. [PubMed] [Google Scholar]
- 3.Tchounwou PB, Ayensu WK, Ninashvili N, Sutton D. Environmental exposure to mercury and its toxicopathologic implications for public health. Environ Toxicol. 2003;18:149–75. doi: 10.1002/tox.10116. [DOI] [PubMed] [Google Scholar]
- 4.Zahir F, Rizwi SJ, Haq SK, Khan RH. Low dose mercury toxicity and human health. Environ Toxicol Pharmacol. 2005;20:351–60. doi: 10.1016/j.etap.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 5.Ceccatelli S, Daré E, Moors M. Methylmercury-induced neurotoxicity and apoptosis. Chem Biol Interact. 2010;188:301–8. doi: 10.1016/j.cbi.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 6.Swedish Expert Group. Methyl mercury in fish: A toxicologic-epidemiologic evaluation of risks: Report from an expert group. Nord Hyg Tidskr Suppl. 1971;4:1–364. [PubMed] [Google Scholar]
- 7.Berlin M, Zalups RK, Fowler BA. Mercury. In: Nordberg GF, Fowler BA, Nordberg M, Friberg LT, editors. Handbook on the Toxicology of Metals. 3rd Edition. Burlington: Academic Press; 2007. pp. 675–729. [Google Scholar]
- 8.Summer K-H, Halbach S, Kappus H, Greim H. Toxicology and risk assessment: A comprehensive introduction. In: Greim H, Snyder R, editors. 1st edition. West Sussex, United Kingdom: John Wiley and Sons; 2008. pp. 276–278. [Google Scholar]
- 9.Verstraeten T, Davis RL, DeStefano F, Lieu TA, Rhodes PH, Black SB, et al. Safety of thimerosal-containing vaccines: A two-phased study of computerized health maintenance organization databases. Pediatrics. 2003;112:1039–48. [PubMed] [Google Scholar]
- 10.Mizukoshi K, Nagaba M, Ohno Y, Ishikawa K, Aoyagi M, Watanabe Y, et al. Neurotological studies upon intoxication by organic mercury compounds. ORL J Otorhinolaryngol Relat Spec. 1975;37:74–87. doi: 10.1159/000275209. [DOI] [PubMed] [Google Scholar]
- 11.Black J. The puzzle of pink disease. J R Soc Med. 1999;92:478–81. doi: 10.1177/014107689909200918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dally A. The rise and fall of pink disease. Soc Hist Med. 1997;10:291–304. doi: 10.1093/shm/10.2.291. [DOI] [PubMed] [Google Scholar]
- 13.Austin D. An epidemiological analysis of the ‘autism as mercury poisoning’ hypothesis. Int J Risk Saf Med. 2008;20:135–42. [Google Scholar]
- 14.Hock C, Drasch G, Golombowski S, Müller-Spahn F, Willershausen-Zönnchen B, Schwarz P, et al. Increased blood mercury levels in patients with Alzheimer's disease. J Neural Transm. 1998;105:59–68. doi: 10.1007/s007020050038. [DOI] [PubMed] [Google Scholar]
- 15.Haddad LM, Shannon MW, Winchester J. 4th edition. Philadelphia: WB Saunders Company; 2007. Clinical management of poisoning and drug overdose; pp. 1111–18. [Google Scholar]
- 16.Adams CR, Ziegler DK, Lin JT. Mercury intoxication simulating amyotrophic lateral sclerosis. JAMA. 1983;250:642–3. [PubMed] [Google Scholar]
- 17.Goetz GG. 3rd ed. St. Louis, MO: Saunders; 2007. Textbook of clinical neurology; p. 46. [Google Scholar]
- 18.Clarkson TW. The toxicology of mercury. Crit Rev Clin Lab Sci. 1997;34:369–403. doi: 10.3109/10408369708998098. [DOI] [PubMed] [Google Scholar]
- 19.Barcelos GR, Grotto D, de Marco KC, Valentini J, Lengert Av, Oliveira AÁ, et al. Polymorphisms in glutathione-related genes modify mercury concentrations and antioxidant status in subjects environmentally exposed to methylmercury. Sci Total Environ. 2013;463:319–25. doi: 10.1016/j.scitotenv.2013.06.029. [DOI] [PubMed] [Google Scholar]
- 20.Hernández AF, Gil F, Leno E, López O, Rodrigo L, Pla A. Interaction between human serum esterases and environmental metal compounds. Neurotoxicology. 2009;30:628–35. doi: 10.1016/j.neuro.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 21.Chen Q, Reis SE, Kammerer CM, McNamara DM, Holubkov R, Sharaf BL, et al. Association between the severity of angiographic coronary artery disease and paraoxonase gene polymorphisms in the National Heart, Lung, And Blood Institute-sponsored Women's Ischemia Syndrome Evaluation (WISE) study. Am J Med Genet. 2003;72:13–22. doi: 10.1086/345312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kao YL, Donaghue K, Chan A, Bennetts B, Knight J, Silink M. Paraoxonase gene cluster is a genetic marker for early microvascular complications in type 1 diabetes. Diabet Med. 2002;19:212–5. doi: 10.1046/j.1464-5491.2002.00660.x. [DOI] [PubMed] [Google Scholar]
- 23.Ou C, Stevenson RE, Brown VK, Schwartz CE, Allen WP, Khoury MJ, et al. 5, 10 Methylenetetrahydrofolate reductase genetic polymorphism as a risk factor for neural tube defects. Am J Med Genet. 1996;63:610–4. doi: 10.1002/(SICI)1096-8628(19960628)63:4<610::AID-AJMG15>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 24.Klerk M, Verhoef P, Clarke R, Blom HJ, Kok FJ, Schouten EG. MTHFR 677C → T polymorphism and risk of coronary heart disease: A meta-analysis. JAMA. 2002;288:2023–31. doi: 10.1001/jama.288.16.2023. [DOI] [PubMed] [Google Scholar]
- 25.Bjelland I, Tell GS, Vollset SE, Refsum H, Ueland PM. Folate, vitamin B12, homocysteine, and the MTHFR 677C → T polymorphism in anxiety and depression: The Hordaland Homocysteine Study. Arch Gen Psychiatry. 2003;60:618–26. doi: 10.1001/archpsyc.60.6.618. [DOI] [PubMed] [Google Scholar]
- 26.Mills JL, Kirke PN, Molloy AM, Burke H, Conley MR, Lee YJ, et al. Methylenetetrahydrofolate reductase thermolabile variant and oral clefts. Am J Med Genet. 1999;86:71–4. [PubMed] [Google Scholar]
- 27.Lewis SJ, Lawlor DA, Smith GD, Araya R, Timpson N, Day IN, et al. The thermolabile variant of MTHFR is associated with depression in the British Women's Heart and Health Study and a meta-analysis. Mol Psychiatry. 2006;11:352–60. doi: 10.1038/sj.mp.4001790. [DOI] [PubMed] [Google Scholar]
- 28.Lewis SJ, Zammit S, Gunnell D, Smith GD. A meta-analysis of the MTHFR C677T polymorphism and schizophrenia risk. Am J Med Genet B Neuropsychiatr Genet. 2005;135B:2–4. doi: 10.1002/ajmg.b.30170. [DOI] [PubMed] [Google Scholar]
- 29.Muntjewerff JW, Hoogendoorn ML, Kahn RS, Sinke RJ, Heijer MD, Kluijtmans LA, et al. Hyperhomocysteinemia, methylenetetrahydrofolate reductase 677TT genotype, and the risk for schizophrenia: A Dutch population based case-control study. Am J Med Genet B Neuropsychiatr Genet. 2005;135B:69–72. doi: 10.1002/ajmg.b.30179. [DOI] [PubMed] [Google Scholar]
- 30.Wilcken B, Bamforth F, Li Z, Zhu H, Ritvanen A, Renlund M, et al. Geographical and ethnic variation of the 677C > T allele of 5, 10 methylenetetrahydrofolate reductase (MTHFR): Findings from over 7000 newborns from 16 areas world wide. J Med Genet. 2003;40:619–25. doi: 10.1136/jmg.40.8.619. [DOI] [PMC free article] [PubMed] [Google Scholar]