

Abstract
Photoaffinity labeling (PAL) using a chemical probe to covalently bind its target in response to activation by light has become a frequently used tool in drug discovery for identifying new drug targets and molecular interactions, and for probing the location and structure of binding sites. Methods to identify the specific target proteins of hit molecules from phenotypic screens are highly valuable in early drug discovery. In this review, we summarize the principles of PAL including probe design and experimental techniques for in vitro and live cell investigations. We emphasize the need to optimize and validate probes and highlight examples of the successful application of PAL across multiple disease areas.
The use of photoaffinity labeling (PAL) in medicinal chemistry and drug discovery has recently come to fruition [1]. PAL is a powerful technique used for the study of protein–ligand interactions, where it can identify unknown targets of ligands, assist in the elucidation of protein structures, functions and conformational changes as well as identify novel or alternative binding sites in proteins [2]. In the current review, we will discuss the general principles of photoaffinity labeling concerning photoaffinity probe design and experimental strategies and the performance of the different photoaffinity groups available. We then focus on examples of the successful application of PAL in relation to the identification of molecular targets of small molecules, discovery of off-target interactions and the classification and structural elucidation of binding sites. The majority of examples presented here were published within the past 15 years, and greater emphasis has been given to recent examples illustrative of the general approaches.
Photoaffinity probe design
PAL is the use of a chemical probe that can covalently bind to its target in response to activation by light [3]. This is made possible by the incorporation of a photoreactive group within an otherwise reversibly binding probe compound. On irradiation with a specific wavelength of light, the photogroup forms a reactive intermediate that rapidly reacts with and binds to the nearest molecule, which ideally will be the target protein. Frank Westheimer first introduced the concept of photoaffinity labeling in the early 1960s, using acylation to incorporate an aliphatic diazo group into the enzyme chymotrypsin, which formed an intramolecular crosslink on photolysis [4].
The ideal traits of a photoaffinity probe include stability in the dark at a range of pHs, a high degree of similarity to the parent compound with comparable activity and affinity levels, and little steric interference to binding. The optimal probe also requires activation at wavelengths that do minimal damage to biological molecules, but still generate highly reactive intermediates, capable of reacting with many bond types to form stable adducts. The newly formed bond must also remain intact and not be destroyed by the isolation or detection methodology [5]. All these characteristics can be hard to optimize simultaneously in a single molecule, so it is often the case that no photoaffinity probe is ideal. The effort required in optimizing the probe may be comparable to that in optimizing an early drug lead for potency, selectivity and physicochemical properties [6].
The general design of photoaffinity probes involves the incorporation of three important functionalities; an affinity/specificity unit, in other words, the small molecule of interest, a photoreactive moiety (e.g., trifluoromethylphenyl diazirine) and an identification/reporter tag (e.g., biotin) (Figure 1). The specificity unit is responsible for reversible binding to target proteins. The photoreactive moiety allows photo-inducible permanent attachment to targets, and the identification component is vital for the detection and isolation of probe–protein adducts. The identification tag can be a fluorescent dye, a radioisotope or a partner for a specific binding event (e.g., biotin–avidin). The length of the linker/spacer groups between functionalities is a key component in photoaffinity probes [7]. Too short a linker may lead to the probe cross-linking with itself while too long a linker may place the photoreactive group at too great a distance to capture the target protein efficiently. The photogroup can either be placed on a linker or can be directly incorporated into the reversible binding pharmacophore (Figure 1A & B). Extensive structure–activity relationships (SAR) are often required to produce the best possible probe. Suitable points on the parent compound are needed to add the other functionalities required. Depending on the desired probe structure and function, two separate modifiable sites may be needed.
Figure 1. General designs for photoaffinity probes.

(A) Design for a PAL probe where the photogroup, pharmacophore and reporter tags are remote from one another and connected by linkers. (B) Design for a PAL probe where the photogroup is directly incorporated within the pharmacophore, but both are remote from the reporter tag. (C) Design for a two-component PAL probe where the pharmacophore and photogroup are in a separate molecule to the reporter tag. Alkyne and azide functionality on the two halves allows a conjugation step through 1,3-cycloaddition reaction (click chemistry).
Finding a productive site on a parent ligand from which to build the rest of the probe and optimizing the photoreactivity of the molecule are potentially substantial tasks. The identification of an attachment site on a parent ligand that does not disrupt binding is made easier if the ligand originates from a screening set of related analogs from which SAR for the desired cellular bioactivity can be deduced. Linker attachment can be quickly focused on the areas where diverse substituents are tolerated and a useful linker may be identified through synthesis of fewer than ten analogs. However, this is predicated on the occurrence of perhaps 10–50 or more, useful and diversely substituted analogs in the initial screening set. If the parent reversibly binding probe is a singleton, as is often the case for natural product hits in phenotypic screens, then this SAR will need to be developed by new analog synthesis. It is also important to note that the fully assembled probes, with long linkers, photoaffinity group and identification tag incorporated are seldom cell permeable. Thus activity in cell-based assays may only be discernible when truncated linkers are attached, for example, short polyethylene glycol chains, and the degree to which completion of the probe structure may compromise binding to the target cannot be readily assessed.
In some cases, it is possible to use structural biology information about known targets of a ligand to guide the design of a probe to identify other targets in the same protein class. For example, ATP-site kinase inhibitors may be assumed to have broadly similar binding modes across the protein superfamily, and vectors to solvent identified from a protein crystal structure of the ligand complexed with one target could provide a preferred starting point for linker attachment for a probe to identify other kinase targets [8].
Optimizing the reaction of the photoaffinity probe, a product of the linker length between specificity unit and photophore and the irradiation conditions, is made easier if capture of a known target of the probe can be used as a surrogate control for specific binding. However, as many interesting bioactive molecules from cell-based screens do not have known targets, experimental protocols that use competition between the photoaffinity probe and the unmodified ligand to confirm specific interactions have come to be seen as essential in the deconvolution of putative targets identified through PAL [9].
Cell-permeable photoaffinity probes are very useful tools as they allow the capture of protein targets in live cells. The nature of most reporter tags and the commonly used polyethylene glycol linker chains generally makes photoaffinity probes large and cell impermeable. The principle means to overcome this is conjugation via click chemistry that allows the introduction of the reporter tag at a later stage [10]. Click chemistry is often used in conjunction with affinity probes, photoaffinity probes and fluorescent probes as well as in structure-based design and combinatorial chemistry. The most successful examples of click chemistry in target identification strategies make use of the Huisgen 1,3-dipolar cycloaddition of alkynes and azides to form 1,2,3-triazoles. A review discussing the use of clickable probes has recently been published [11]. The photoaffinity probe is generally split in two; one half contains the affinity unit, photogroup and an alkyne handle (Figure 1C). The other half has a terminal azide on a linker chain connected to a reporter tag. Alternatively, the azide and the alkyne can switch positions. The component containing the pharmacophore and photogroup can enter cells, and upon irradiation should covalently capture its target proteins in cell. Following cell lysis, a copper catalyst, reducing agent and the azide-containing reporter can be added, allowing the click chemistry reaction to occur and linking the captured proteins to a detectable tag through triazole formation.
Photoaffinity groups
The main photoreactive groups used for PAL are phenylazides, phenyldiazirines and benzophenones, where the reactive intermediates formed on irradiation with specific wavelengths of light are a nitrene, a carbene and a diradical, respectively (Figure 2A). The half-life of the generated reactive species is shorter than the dissociation of the ligand–protein complex. Ideally, the photoaffinity group should be stable in daylight and preferably, its activation energy should be greater than the absorption wavelength of proteins to avoid damaging biomolecules. Proteins absorb UV light at 280 and 200 nm, where absorbance at 280 nm is due to the aromatic amino acids, tryptophan, tyrosine and phenylalanine, while peptide bonds are responsible for absorbance at 200 nm. Excess absorption of UV light by proteins leads to the formation of an electronically excited state molecule, which is then capable of undergoing degradative chemical transformations. Other functionalities that have been used for PAL include diazo groups, diazocarbonyls, enones, sulfur radicals, halogenated substrates, nitrobenzenes, diazonium salts as well as alkyl derivatives of azides and diazirines. Fleming has comprehensively reviewed the variety of different chemical reagents used in PAL [12]. Sakurai et al. recently reported a study comparing the main photoreactive groups and assessing their reactivity [13].
Figure 2. The major photoaffinity functional groups and their reactive intermediates.

(A) Phenylazides, phenyldiazirines and benzophenones undergo photolysis on irradiation with specific light wavelengths to produce reactive nitrene, carbene and diradical intermediates, respectively. (B) Modified PAL reagents with improved stability of the reactive intermediates.
Phenylazides are frequently used in PAL because they are easily synthesized and are commercially available [3]. On the other hand, the shorter wavelengths required to excite these compounds can cause damage to biological molecules and the nitrene intermediate formed decreases photoaffinity yields compared with carbenes [14]. This may be because nitrenes are less reactive than carbenes. Nitrenes can also rearrange to form benzazirines and dehydroazepines/ketenimines as undesired side products [3,15]. Another reported disadvantage is that azides can be reduced to amine by thiols [16]. However, this reaction is most efficient in alkaline media rather than the physiological pH range in which most photoaffinity labeling experiments with cell lysates take place. In order to improve photoaffinity suitability, substituted arylazides such as tetrafluorophenylazide (Figure 2B) have been developed and are reported to prevent rearrangement of nitrenes to ketenimines. Substituents ortho to the azide group are generally avoided because of undesired intramolecular cyclizations that occur after photolysis [11].
Benzophenones form a reactive triplet diradical when irradiated with light and are activated by a long wavelength, which lowers the risk of damaging biomolecules. Benzophenones are also beneficial due to their inertness to solvents and easy preparation. A disadvantage to their use is that a longer irradiation period is often required which can increase nonspecific labeling [17]. The benzophenone group is also a rather bulky group that can affect the interaction between the affinity pharmacophore and the target protein, this steric hindrance can also lead to increased nonspecific labeling and capture [3].
Aryldiazirines are the most commonly used photoaffinity group, particularly the trifluoromethyl derivative, which is favored due to its excellent chemical stability (Figure 2B). Aryldiazirines are highly resistant toward a number of factors such as temperature, nucleophiles, acidic and basic conditions as well as oxidizing and reducing agents. They require a high wavelength (350–355 nm) to be activated which reduces the potential damage to biological molecules. The carbene intermediate is extremely reactive and has a short half-life, which is beneficial as it rapidly forms covalent cross-links to biomolecules. However, due to this high reactivity, carbenes are often quickly quenched by water; this can be a disadvantage in terms of photoaffinity yields, but also an advantage as it minimizes nonspecific binding. When irradiated with light, the diazirine is converted to a singlet-state carbene, however around 30% is converted into the diazo compound shown in Figure 3. This compound can form the singlet carbene yet the process of conversion is relatively slow. As the reactive intermediates for all photogroups are high energy, there are many reactions which can occur depending on the environment they are in, as illustrated for carbenes in Figure 3. Das has authored an extensive overview of aliphatic diazirines in PAL [5] while Dubinsky et al. have highlighted recent advances in the use of diazirine photogroups [14].
Figure 3. Productive and nonproductive reactions of carbenes.

The carbene species generated by photolysis of trifluoromethylphenyldiazirine (red) undergoes many possible reactions to give productive capture of targets (blue) or destructive side reactions (green).
ISC: Intersystem crossing.
Reporter/identification tags
To facilitate the successful isolation of photolabeled biomolecules, reporter tags are incorporated either directly or indirectly in the photoprobe. A technique is required to measure, detect and observe tagged proteins regardless of the method by which they are captured.
Radioactive isotopes (radiolabels) such as 125I and 3H (tritium) are frequently used as reporter tags for PAL. The main disadvantage of radiolabels is that they do not provide a direct means to isolate labeled proteins from the rest of the proteome, and identification of targets can only be based on gel-based readouts. Consequently, radio-labels are preferentially used for profiling known targets. Other disadvantages can include the fast degradation of the radiolabel due to short half-lives, and the special handling needed for the reagents. There are advantages to using this type of reporter group; radiolabels are small and therefore cause minimal change to the structure of the parent compound. Incorporation of a radiolabel can be synthetically tractable and allows easy detection with high sensitivity. Radiolabels are chemically stable and allow the assessment of in vivo activity [18].
Affinity tags are most commonly used in PAL, where biotin is the most popular choice due to its extremely high affinity toward avidin. Antibody epitope tags can also be used. Affinity tags allow easier enrichment and isolation of labeled proteins, and are detected by relevant antibodies specific for the tag. Epitope tags are beneficial as they can easily be eluted from antibody resins; this step is more difficult for biotin due to its strong binding to avidin. Affinity tags are usually large and non-cell permeable, and they may cause steric interference between the specificity unit and the target protein if incorrectly placed [15].
Fluorophores are frequently used for activity-based proteomics [19], and this group of reporter tags has the largest variety available. Commonly used classes include fluorescein, BODIPY (dipyrromethene boron difluoride), rhodamine, cyanine dyes (Cy-5 and Cy-3) and NBD (nitrobenz-2-oxa-1,3-diazole). Each class has its own advantages and disadvantages; the general advantage is that the tags are hydrophobic and can generally penetrate cell membranes. Fluorescein and rhodamine are inexpensive compared with the other alternatives; however they suffer from photobleaching which is a major drawback to their use. BODIPY and the cyanine dyes exhibit high absorption coefficients, narrow absorption peaks and high quantum yields providing clearer detection and identification.
Recent developments in this area of PAL have included the use of photoaffinity probes where a staple isotope label is incorporated within the molecule as well as the traditional affinity unit, photoreactive moiety and reporter tag, although it can also directly replace the reporter tag. The inclusion of mixed isotopes can aid the identification of photo-labeled proteins through MS, as demonstrated by Lamos et al. [20], who developed a deuterated benzophenone. The authors incorporated this photophore into a probe for cyclosporin (CsA) and used it to capture its known target, CypA. The probe was incubated in a mixture of proteins including CypA, and following UV radiation and western blot analysis, a band was clearly seen at 22 kDa matching the molecular weight of CypA. Labeled proteins were purified using avidin resin and, after proteolysis, eluted proteins were analyzed by MS. The 11 Da mass differences due to the deuterium isotopes allowed the recognition of labeled peptides and the authors were able to distinguish clearly between labeled and nonlabeled peptides. Song et al. [21] have also used this approach, as well as fluorous tags to enrich and identify captured proteins. Sadaghiani et al. discussed in the detail the advantages and disadvantages of the different reporter groups for a variety of proteomics methods [18].
General experimental workflows
In most protocols, cells or cell lysates are treated with photoaffinity probes and time is allowed for the probes to associate to their targets. Samples are then irradiated with a specific wavelength of light to activate the photophore and covalently cross-link with proximate biomolecules. If performed on live cells, samples are lysed and then undergo click chemistry conjugation with the reporter tag moiety. Labeled and tagged proteins are separated from the rest of the proteome by affinity purification using the reporter/affinity tag. Analysis using SDS-PAGE allows the range of labeled proteins to be visualized using the equipment and reagents specific for the reporter tag. Proteins are excised from the gel and undergo multiple protein digests to produce peptide fragments. These fragments can be sequenced using MS to determine the identity of the isolated proteins (Figure 4). It is also possible to derive the amino acid sequence the photoaffinity probe has reacted with, which may provide extra information about the binding site.
Figure 4. Overview of a typical PAL protocol.


Nonspecific cross-linking can be a limitation in PAL. It is often the case that photoaffinity probes cross-link to highly abundant or ‘sticky’ proteins. To distinguish between background labeling and specific cross-linking, several control experiments can be applied. The amount of labeling which occurs in the absence of a photoaffinity probe and without UV radiation should be assessed. It is also especially useful to conduct a competition experiment, which should highlight more specific cross-linking to proteins. It is often the case that the parent unmodified compound has greater affinity toward its target than the photoaffinity probe. The competition experiments are performed by incubating samples with an excess of the parent compound or another photostable competitor. Specific labeling of proteins can be recognized by a dose-dependent decrease of signal from the reporter tag in the presence of the unlabeled ligand. Taunton and MacKinnon [9] have provided a useful general protocol for PAL, which can be adapted to a variety of systems and photaffinity probe designs.
Molecular target identification
Target deconvolution is an important part of drug discovery and development [11]. Accurate identification of a drug/small molecule’s range of targets is key to maximizing therapeutic potential and minimizing toxicity [22]. Phenotypic screens are widely used to discover new bioactive molecules that elicit desirable responses in cells; this involves a disease-relevant cell-based assay, usually focused on particular cell-signaling pathways or complex behaviors, which allows the simultaneous modulation of multiple molecular targets. SAR studies may then be conducted to increase compound potency, prior to target deconvolution and target validation [23]. There are a variety of techniques which can be applied to identify the molecular targets of small molecules that have been found to cause distinct phenotypic changes. Several thorough reviews cover the wider range of target identification techniques [17,24–26].
In general terms, target deconvolution can be split into two categories; affinity-based methods and expression cloning-based methods. A fundamental aspect, however, of all the technologies used for target identification is the affinity of the small molecule to its target protein. The strength of the association between a reversibly binding ligand and its target(s) determines whether the interaction will persist through experimental procedures for target detection and/or isolation. Photoaffinity probes can overcome this by permanently capturing targets with weak affinity through the formation of the covalent attachment. However, the confidence with which a specific target can be identified over nonspecific binding events generally increases with the affinity of the probe. If the affinity of the probe is low, then the likelihood that nonspecific associations between the small molecule and other proteins are also detected increases, driven for example by the hydrophobic effect. The output of the photoaffinity experiment may become dominated by weak but nonfunctional binding interactions to abundant cellular proteins. It is therefore useful to consider the potency of the parent probe molecule in the initial biological assay and how this may reflect the affinity for specific targets.
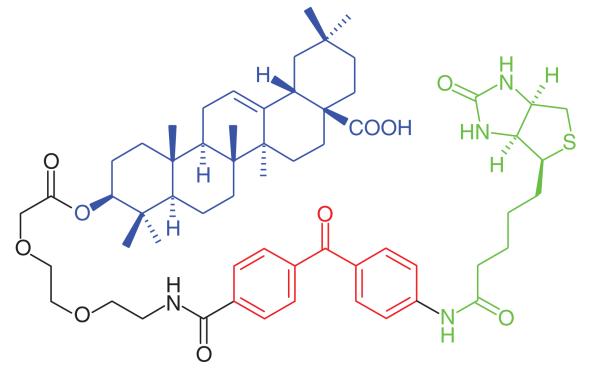
One recent use of effective photoaffinity labeling was the identification of the target proteins of oleanolic acid. Oleanolic acid demonstrates a variety of biological activities in areas including cancer, HIV and hyperglycemia [27]. Zhang et al. [28] successfully synthesized two types of oleanolic acid photoaffinity probes (Table 1, Entry 1): one containing an identification tag, photogroup (benzophenone) and oleanolic acid bonded together by separate linkers and the other being a ‘tag free’ probe with a terminal azide group available to react with a biotin-derivatized alkyne. From previous SAR studies, modification of the hydroxyl group of oleanolic acid was identified to have little effect on its activity, and was therefore deemed a suitable attachment point for the probe. One known target of oleanolic acid is the phosphorylase RMGPa, so in order to determine that the probes retained activity, an enzyme inhibition assay against RMGPa was carried out. Surprisingly, the smaller ‘tag free’ probe exhibited the poorest activity (fivefold less potent than oleanolic acid), compared with the full probe which was only twofold less potent. The full-length probe was incubated in the soluble proteosome from HepG2 cells and two proteins of 40–50 kDa were tagged and captured. However, definitive target identification has not yet been reported.
Table 1.
Examples of photoaffinity probes used to identify the targets of ligands or to characterize ligand-binding sites.
| Entry | Sample | Design†, PAG | Target(s)‡ | Photoaffinity probe(s) | Ref. |
|---|---|---|---|---|---|
| 1 | HepG2 liver cancer cell lysate | A, Benzophenone | n.a. |
|
[28] |
|
| |||||
| 2 | HeLa cervical cancer cells | A, Phenyl diazirine | SAP130 of SF3b |
|
[29] |
|
| |||||
| 3 | Nuclear extracts from a cell line derived from a FRDA patient | C, Benzophenone | HDAC3 |
|
[30] |
|
| |||||
| 4 | T47D breast cancer cell lysate | B, Phenyl azide | TIP47 |
|
[31] |
|
| |||||
| 5 | HCT116 colon cancer cells | C, Phenyl diazirine | MDH2 |
|
[32] |
|
| |||||
| 6 | PC-3 prostate cancer cells | C, Benzophenone | n.a. |
|
[33] |
|
| |||||
| 7 | Nuclear fraction of DU145 prostate cancer cells | A, Benzophenone | n.a. |
|
[34] |
|
| |||||
| 8 | Endoplasmic reticulum microsome fraction | C, Diazirine§ | Sec61 protein complex |
|
[35] |
|
| |||||
| 9 | Mouse liver mitochondrial fraction | A, Phenyl azide | Mpc 1 and Mpc 2 |
|
[36] |
|
| |||||
| 10 | Mouse erythroleukemia (MEL) cells | B, Phenyl azide | Ribosomal protein S3 |

|
[37] |
|
| |||||
| 11 | K562 leukemia cells and cell lysate; HepG2 live cancer cells and cell lysate | C, Diazirine | PKA,PIM-3, PKN2 |
|
[8] |
|
| |||||
| 12 | Microsomal fraction | B, Benzophenone | hmEH, ES10, L-FABP |

|
[38] |
|
| |||||
| 13 | PC-3 cells | C, Diazirine# | BCAR3, LDHA, LTA4H, PRDX3, RPS3, RPS6KA1 |

|
[39] |
|
| |||||
| 14 | Rat liver peroxisomes | A, Phenyl diazirine | MFE2 |
|
[40] |
|
| |||||
| 15 | HEK293 embryonic kidney cells | B, Phenyl azide | GPR54 |
 Amino acids in red were replaced with cysteine and then modified to introduce an azide photophore as shown below. Each probe contained one modified amino acid;*starred amino acids represent probes which cross-linked successfully |
[41] |
|
| |||||
| 16 | Membrane fraction from Saccharomyces cerevisiae | B, Benzophenone | Rce1p |
|
[42] |
|
| |||||
| 17 | HeLa cervical cancer cell membrane fractions | C, Benzophenone | PS1 in the γ-secretase protein complex |
|
[43] |
|
| |||||
| 18 | HeLa cervical cancer cell lysate | B, Benzophenone | PS1 of the γ-secretase protein complex |
|
[44] |
|
| |||||
| 19 | Solubilized nAChR-rich membranes isolated from Torpedo californica | B, Phenyl diazirine | nAChR |

|
[45] |
|
| |||||
| 20 | HT1080 fibrosarcoma cell lysate | B, Benzophenone | α-tubulin and β-tubulin |
|
[46] |
|
| |||||
| 21 | HEK293 embryonic cancer cells | C, Benzophenone | Nat10 and Naa50 |
|
[47] |
Photoaffinity probe design classified in accordance with Figure 1.
Color code: Red: Photogroups; Blue: Pharmacophores; Green: Reporter tags; Black: Linker groups.
Representative probe chosen to illustrate the approach from a combinatorial set of diazirines.
n.a.: Not available; PAG: Photoaffinity group.
Pladienolides are natural products that exhibit anti-tumor activity in a variety of in vivo and in vitro systems but until recently, uncertainty remained about their protein target(s). Kotake et al. [29] prepared three types of pladienolide-derived probes; a 3H-labeled analog, a fluorescence-tagged (BODIPY-FL) compound and a photoaffinity probe (Table 1, Entry 2). These molecules were constructed based on the results of previous SAR. HeLa cells were treated with the tritiated probe, and the subcellular distribution of the radioactive signal was evaluated. This revealed that the nuclear fraction exhibited the highest radioactivity, strongly suggesting that pladienolides have a high affinity toward a nuclear protein. In order to limit the number of possible targets, immunoprecipitation experiments were performed using specific antibodies for a range of nuclear proteins on the nuclear fraction from the tritiated probe-treated HeLa cells. The radioactivity was detected in six of the co-precipitates, five of which were formed using antibodies known to recognize components of the ribonuclear protein U2 snRNP, which has a key splicing apparatus function at the 3′ site of mRNA. The main target of these natural products was identified by treating fractionated HeLa cells with the photoaffinity probe. An immunoprecipitate of the nuclear fraction was formed using an anti-SAP155 antibody, and the photoaffinity probe was activated by UV creating a covalent bond between the probe and its target. Western blotting with streptavidin horseradish peroxidise revealed a significant band of around 140 kDa. This was identified as a spliceosome-associated protein using LC–MS/MS peptide sequencing. Overall, the results showed that pladienolides have affinity to SAP130 of the SF3b complex, and can therefore inhibit mRNA splicing. This work demonstrated that mRNA splicing machinery may be a viable antitumor drug target.
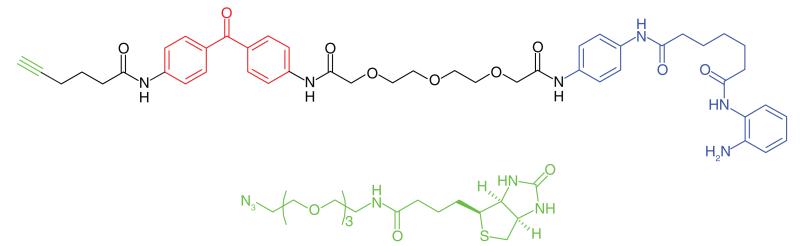
HDAC inhibitors are drugs with therapeutic potential against diseases such as Huntington’s [48], Friedreich’s ataxia (FRDA) [49] and cancer [50]. The chemotype, pimelic diphenylamide, was discovered by Xu et al. [30] to exhibit good activity in a mouse model of FRDA. Photoaffinity probes were developed (Table 1, Entry 3) based upon lead compounds in the series. The authors opted for the use of a benzophenone photogroup and a click conjugated biotin reporter tag for streptavidin enrichment. The studies showed that HDAC3 was the primary molecular target for pimelic diphenylamide, and subsequent work highlighted its role in gene silencing in FRDA. Modifications to the lead compound shifted the specificity from HDAC3 in favor of HDAC1–2. This supported the hypothesis that HDAC3 inhibition was responsible for the observed phenotype, as the desired activity was not seen when HDAC1 or HDAC2 were inhibited.
Apoptosis is a frequent mode of action for many anticancer drugs and targeting of irregular apoptotic traits in cancerous cells can lead to potent and selective anticancer agents. However, drugs designed to induce apoptosis often suffer from the inability to distinguish between malignant and nonmalignant cells. 3,5-Diaryl-oxadiazoles have been shown to be capable of causing apoptosis in tumor cells. Jessen et al. [31] built on SAR studies to develop a photoaffinity probe of the lead compound MX-126374 (Table 1, Entry 4). The authors chose an azide photophore and tritium as the reporter tag. Subsequent target ID experiments using 2D-PAGE gel electrophoresis and MS sequencing revealed the primary target of MX-126374 to be TIP47, which has been implicated in IGF-IIR signaling as it binds directly to the receptor itself. The identification of this molecular target for the induction of apoptosis supports further investigation of manipulating IGF-IIR signaling as a cancer treatment.
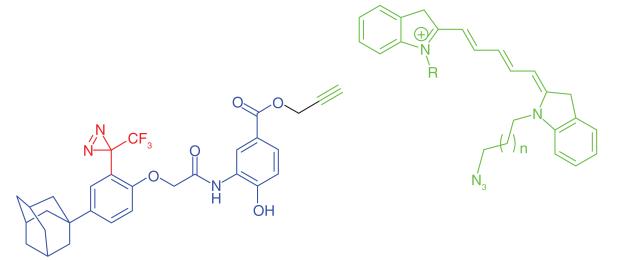
The transcription factor HIF promotes angiogenesis and metastasis and is a potential target for cancer therapy. The small molecule LW6 inhibits HIF1α accumulation and consequential HIF transcriptional activity; however the molecular targets responsible were unknown. Lee et al. [32] designed and synthesize photoaffinity probes of LW6 based on prior SAR studies (Table 1, Entry 5), which were placed in HCT116 cells, irradiated and click conjugated with a fluorescent reporter tag. The proteins were separated by SDS-PAGE and visualized by in-gel fluorescence scanning, giving a prominent band around 37 kDa. Following trypsin digest and sequencing of the peptides by MS, this protein was identified as MDH2, a mitochondrial enzyme involved in the tricarboxylic acid cycle. Further studies showed LW6 interacted with and inhibited MDH2, and also that a known inhibitor of MDH2 potently inhibited HIF transcriptional activity.
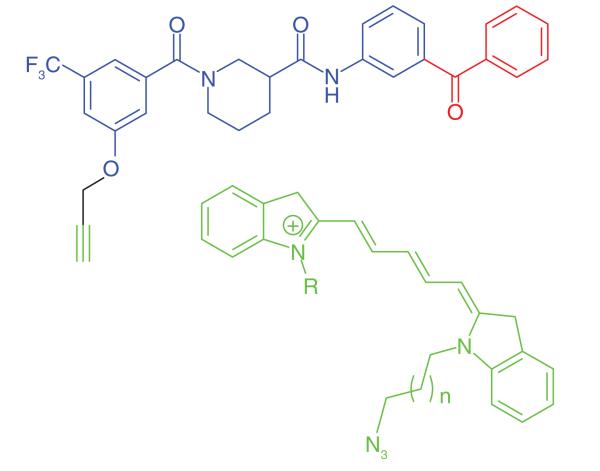
A similar strategy was implemented by Bell et al. [33] to determine the molecular targets for CCG-1423, which was discovered to be an inhibitor of the Rho signaling pathway. Photoaffinity probes using the benzophenone group and click conjugation to a fluorescent tag (Cy5.5) (Table 1, Entry 6) were used to identify a protein of 24 kDa, where labeling of this protein was clearly competed off by an excess of unlabeled CCG-1423, providing strong evidence that the protein was a bona fide molecular target. However, further studies are needed to identify the protein and assess its relation to Rho signaling.
Kotoku et al. [34] discovered a novel bioactive compound, furospinosulin-1, from a cell-based screen assessing a range of natural products and analogs. The small molecule of interest was isolated from the Indonesian marine sponge Dactylospongia elegans, and was found to be a dose-dependent, hypoxia-selective growth inhibitor of DU145 prostate cancer cells. Unlike many hypoxia-targeting inhibitors, furospinosulin-1 did not interfere with HIF1α accumulation, but did deplete expression of IGF-2, prompting the authors to hypothesize the compound was acting by a novel mode of action. To deconvolute the mechanism of action, four photoaffinity probes were synthesized (Table 1, Entry 7), with different linker lengths between the specificity unit and the photophore (benzophenone). In all cases, biotin was used as the reporter tag. Probes with shorter linkers between the pharmacophore and photogroup were found to retain the best activity and remained selective for hypoxic conditions over normoxia. The probes were evaluated using an electrophoretic mobility shift assay and shown to disrupt the binding of nuclear proteins to DNA, supporting the hypothesis that furospinosulin-1 disrupts the formation of a complex of nuclear proteins possibly involved in transcription, but direct target confirmation has not yet been reported.
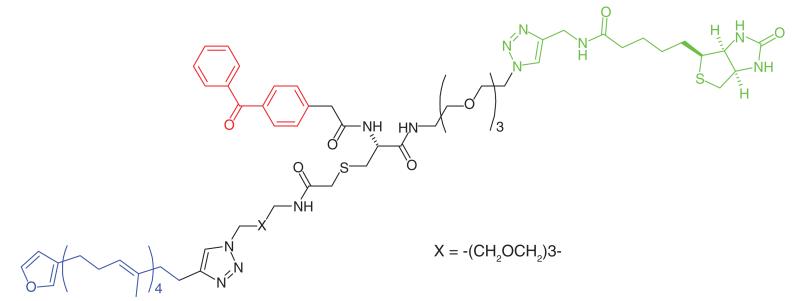
Cyclodepsipeptides are inhibitors of VCAM expression that function by preventing the co-translational translocation of VCAM into the endoplasmic reticulum. Inhibition of VCAM has been highlighted as a potential therapeutic option for cancer [51] and arthritis [52]. The fungal cyclodepsipeptide HUN-7293 was used by MacKinnon et al. [35] to identify the molecular target of this chemical class. The authors incorporated photo-leucine into the molecule (Table 1, Entry 8) as well as a propargyl handle at a separate site to allow conjugation to a rhodamine reporter. The molecular target of this chemotype was found to be the Sec61 complex, a heterotrimeric membrane protein that forms the structural architecture of the translocation channel in the ER membrane. This result supported previous data from biochemical experiments showing that cyclodepsipeptides act at the ER membrane [53]. This example illustrates the ability of PAL to capture low abundance proteins, as Sec61α identified in this experiment comprised less than 1% of the total ER protein content. It was clearly a highly labeled protein when analyzed using relevant antibodies, but was not a major band when the SDS-PAGE gel was Coomasie stained, supporting the specificity of protein capture and that lower abundance proteins are detectable among a complex mixture of proteins. Other authors have reported the capture of proteins present at femtomole quantities from complex mixtures [54].
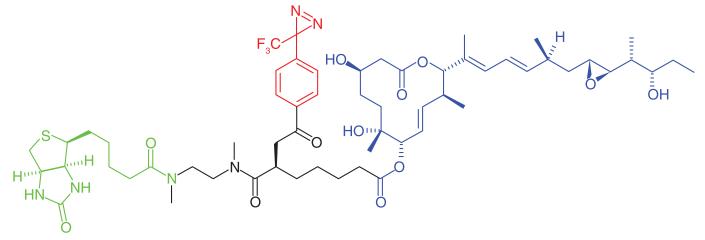
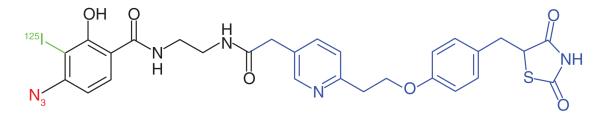
Thiazolidinediones (TZDs) are treatments for Type 2 diabetes [55]. The compounds are insulin sensitizers and known agonists of nuclear receptor PPARγ [56]. However, there are studies that hypothesize that another mode of action involving mitochondrial membrane proteins may also be responsible for the observed insulin sensitization. TZD analogs have been shown not to bind to PPARγ under physiological conditions [57]. Colca et al. [58] used a radiolabeled photoaffinity analog of pioglitazone (PNU-91323) to investigate the alternate mode of action of TZDs (Table 1, Entry 9). Intact and solubilized mitochondrial fractions were treated with the probe and a previously unidentified mitochondrial protein complex of 17 kDa was discovered. The authors performed further PAL experiments using the same probe (now called MSDC-1101) [36] to characterize the complex, and found that the photoaffinity probe bound to two distinct mitochondrial proteins; Mpc1 and Mpc2. Additional studies showed that RNAi knockdown of these proteins in Drosophila increased glucose production, and that photolabeling of the Mpc proteins could be inhibited by the presence of a known pyruvate inhibitor, confirming that TZDs are a ligand for these proteins. Modulation of mitochondrial pyruvate carrier proteins for therapeutic benefit in metabolic diseases is currently being investigated [59].
An older example of target identification using PAL was the discovery of ribosomal protein S3 as a potential target for hybrid polar cytodifferentiation (HPC) agents such as suberoylanilide hydroxamic acid (SAHA) [37]. SAHA was developed as an HDAC inhibitor [60] and is known as the marketed drug vorinostat [61]. HPC agents are capable of inducing terminal differentiation or apoptosis of cancerous cells [62]. Webb et al. developed a tritiated photoaffinity analog of SAHA (Table 1, Entry 10) containing a photoreactive azide. PAL was performed in murine erythroleukemia cell lysates, proteins were separated by SDS-PAGE and labeled proteins were identified by fluorography. A specific band was seen at approximately 30 kDa, which was excised from the gel and identified as ribosomal protein S3 by sequencing. Inhibition of this protein may be responsible in part for the antitumor activity displayed by HPC drugs, as individual ribosomal proteins are involved in mediating several cellular activities including proliferation and neoplastic alterations [63].
Identification of off-target interactions in cells
The majority of small molecule off-target interactions are discovered by in vitro screening assays. For example, biochemical assays for GPCRs, ion channels, kinases, nuclear receptors and transporters are all frequently used to detect off-target pharmacologies in the early stages of drug discovery. The productivity of these methods relies on the in vitro activity of small molecules being translated in vivo, as well as the in vivo function of proteins being preserved in vitro. A major limitation in this approach is that the composition of the screening panels limits the off-target interactions that can be detected. PAL provides an opportunity to identify the targets of molecules of interest in cells without the need to make prior choices of the targets to be screened, and where the proteins are at their natural relative abundance and in the presence of endogenous ligands and co-factors. This can be complimentary to the in vitro screening described above and informs on which in vitro interactions translate into cells. A caveat is that the position of the attachment of the linker group to the specificity unit affects the range of targets that can be identified. Preserving the known pharmacophore for interaction with a given protein structural family would permit other proteins in the same class to be detected, for example, as described below for kinases [8]. The ability to detect other targets outside of this family may be compromised if the attachment point disrupts a pharmacophore essential for the interaction, a problem when there is no prior knowledge of the off-targets expected. Multiple PAL probes constructed from the same affinity unit with different linker attachment points may be helpful to ensure that all possible interactions of the ligand are probed.
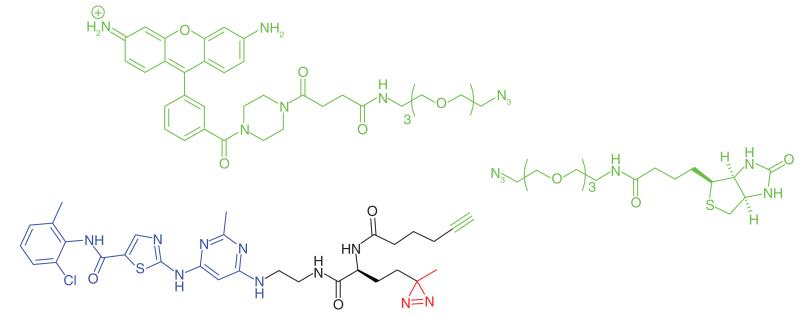
Many protein kinase inhibitors do not show a high specificity profile and understanding how the range of kinases or other targets inhibited contributes to biological activity is important. To achieve a more comprehensive insight into the off-target interactions of dasatinib (a dual Src/Abl inhibitor, IC50 Src 3.6 nM, IC50 Abl 8.8 nM) in vivo, Shi et al. [8] developed cell-permeable photoaffinity probes from dasatinib which could profile the proteome for off-target interactions (Table 1, Entry 11). A diazirine group was incorporated directly into the pharmacophore of dasatinib in place of a hydroxyethylpiperazinyl moiety, which had been shown to be replaceable without incurring significant loss of activity. An alkyne handle was added to conjugate to reporter tags using click chemistry. The activities of the probes were similar to that of the parent compound. Pull-down and target ID experiments were performed in both live cells and cell lysates. In addition to the known interactions of dasatinib with Abl and Src family tyrosine kinases, a number of serine/threonine kinase interactions were discovered and validated, most notably PKA (IC50 4.8 μM), PIM-3 (IC50 8.2 μM) and PKN2 (IC50 11.5 μM), albeit at a greatly reduced potency compared with the targeted kinase activities.
Tamoxifen is a selective ER modulator (SERM) used to treat ER-positive breast cancer in pre- and postmenopausal women [64]. There are proteins and protein complexes which possess antiestrogen binding sites (AEBS) that have high affinities for triphenylethylenic compounds like tamoxifen. These proteins were thought to be responsible for some of the other biological effects of tamoxifen, such as changes in serum lipid composition. To identify AEBS-containing proteins that directly interacted with tamoxifen, Mésange et al. [38] developed a tritiated photaffinity probe, 4-(2-morpholinoethoxy)benzophenone ([3H]MBoPE) (Table 1, Entry 12) which did not bind to ER but retained activity against hmEH, a known AEBS-containing protein identified from a previous PAL experiment [65]. Labeling of this protein by [3H]MBoPE was clearly competed by increasing concentrations of tamoxifen. [3H]MBoPE was incubated in solubilized microsomal proteins, and following irradiation, subsequent separation, isolation and MS sequencing; three proteins were identified. These were the previously known target, hmEH and two novel targets; ES10 and L-FABP. Both of these proteins are involved in lipid metabolism and their discovery as molecular targets of tamoxifen suggested they may account for the mode of action by which tamoxifen affects serum lipids.
An interesting study of PAL was performed by Kambe et al. [39], where the authors synthesized an assorted library of 1,5-disubstituted tetrazole photoaffinity probes using the Ugi-azide multicomponent condensation reaction (Table 1, Entry 13). The diazirine photophore and an alkyne handle were incorporated into each member of the library via either the carbonyl (aldehyde or ketone) or the amine substrates. PC-3 prostate cancer cells were treated with each library member, irradiated with UV light, lysed and labeled proteins were coupled to a rhodamine azide reporter using click chemistry. Proteins were separated and detected using SDS-PAGE and in-gel fluorescence scanning. The authors discovered a wide range of UV-dependent protein labeling, with specificity trends exhibited by most probes and protein targets. Interestingly, a small subset of proteins was labeled in the absence of UV irradiation, suggestive of nonspecific labeling, and highlighting the importance of relevant controls. The selectivity profile of one probe included a high number of proteins labeled by others in the probe library. There was limited evidence that this probe was responsible for unique labeling events, therefore it served as a nonspecific control to highlight more specific labeling events by the rest of the library. The very diverse targets for a selection of six probes were identified and the abundance of each labeled protein was compared with that labeled by the nonspecific probe; when the abundance was at least threefold greater, the protein was deemed to be a specific target. This study highlights the potential problem of promiscuous background labeling and how it can be accounted for to identify more specific probe targets.
Determination of binding site location & structure
Enzymes, multifunctional proteins and receptors can have multiple ligand-binding sites, many of which have been characterized, while others are known of but their location and arrangement are not. It is often the case that ligand-binding sites are found within a multisub-unit protein complex. Binding sites located across subunit interfaces are potentially hard to locate and characterize and PAL can be applied to these, as demonstrated for binding sites in the GABAAR and nAChR complexes [45,66–68], provided the complexes survive lysis protocols or if the experiment is conducted in intact cells using a cell permeable probe. Sites formed from both subunits at an interface can also be detected [46]. Once specific subunits within complexes are identified as the targets of PAL probes and their cognate reversible binding ligands, competition experiments can discern where other classes of inhibitor use the same or different binding sites within the complex [43,68]. PAL has been used for the elucidation of binding sites for approximately 30 years [69], and many new binding sites are still being discovered using this technique.
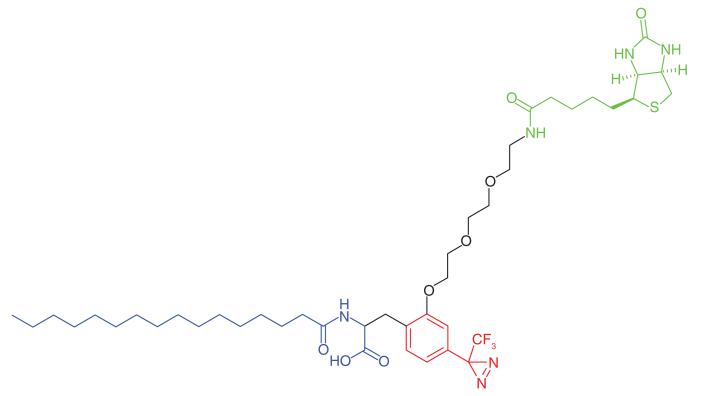
Peroxisomes, membrane-enclosed subcellular organelles with a high content of oxidative enzymes, play a key role in various biosynthetic pathways. A diazirine analog of palmitic acid (Table 1, Entry 14) was synthesized by Kashiwayama et al. [40], and used to probe potential binding sites of purified rat liver peroxisomes. An 80 kDa protein was reproducibly labeled, and the efficiency of labeling was decreased under competing conditions. The protein was found to be MFE2, a β-oxidation peroxisome enzyme. MS analysis disclosed that residues Arg251 and Trp249 were labeled by the probe, and from the crystal structure of MFE2, the authors deduced that these residues were present at the top of a hydrophobic pocket leading to the catalytic site, and proposed that this hydrophobic region is important for substrate interaction and orientation toward the active site.
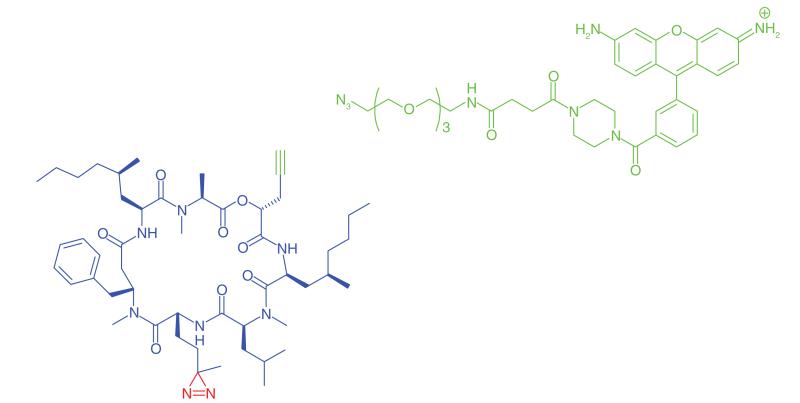
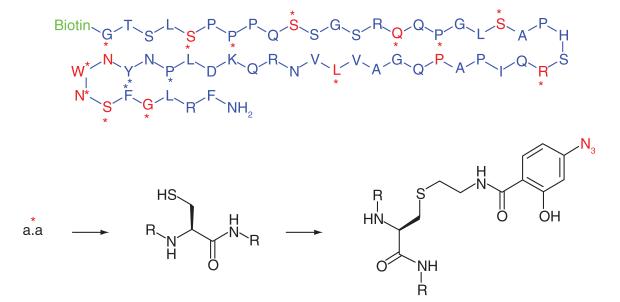
Kisspeptins are human proteins that are the endogenous ligands for the GPCR, GPR54. The Kisspeptin-GPR54 signaling axis is critically involved in the secretion of the hormone GnRH, and therefore GPR54 agonists have potential to treat hormonal secretion diseases. To better exploit this therapeutic opportunity, greater knowledge of the GPR54 ligand binding residues is required and for this reason Misu et al. [41] developed kisspeptin peptide photoaffinity probes (Table 1, Entry 15). The C-terminal region was known to be essential to GPR54 binding, therefore the biotin reporter tag was connected at the N-terminus. Sixteen photoaffinity probes were synthesized based on the Kp-54 peptide sequence, where on each probe only one individual residue was replaced with a cysteine which was later modified to incorporate the azide photogroup. Only six probes covalently bound to GPR54 and all had modified amino acids within the same 18 amino acid sequence, suggesting this short sequence plays a key part in the interaction between kisspeptins and GPR54.
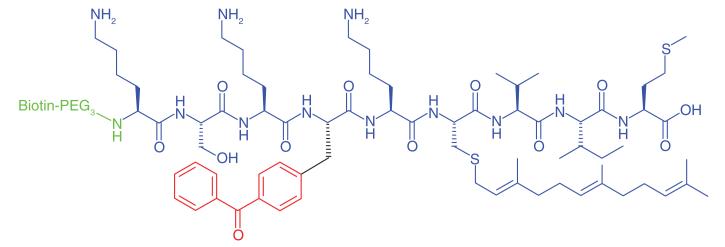
The Ras signaling transduction cascade is an important human cellular pathway involved in cell proliferation. Aberrations within the signaling axis are strongly implicated in cancer [70]. The protease Rce1p directly interacts with and modifies Ras and other CAAX (C: Cysteine, A: Aliphatic amino acid, X: One of several amino acids) proteins by endroproteolytic trimming. Rce1p itself is localized in the ER membrane; however the topology of the enzyme’s catalytic residues is unknown. Kyro et al. [42] synthesized a range of photoaffinity peptide substrates based on part of the C-terminal peptide sequence of K-Ras4b. Rigorous investigations identified the best probe design for labeling of Rce1p. Probes containing a 2-aminobenzoyl fluorophore, dinitrophenyl quencher and a benzophenonemodified isoprenoid to mimic the isoprenyl group Rce1p recognizes were compared with probes which had biotin added to the N-terminal domain, replacing the fluorophore. Probes were also synthesized where the photogroup was added as a separate moiety to the isoprenyl group and reporter tag. Optimal labeling was achieved when the photophore was incorporated onto the peptide chain rather than onto the isoprenoid (Table 1, Entry 16). The authors discovered that labeling of Rce1p also occurred with peptides which were not part of the amino acid sequence which Rce1p recognizes, which may be a consideration for future design of selective inhibitors. A similar approach by Deng et al. [71] used a probe derived from a small peptide which recognized Abl kinase in its active conformation, where PAL captured and isolated Abl locked in that conformation.
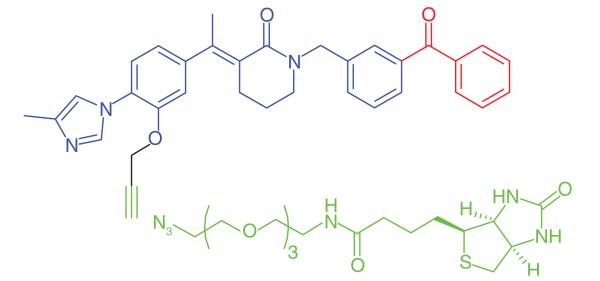
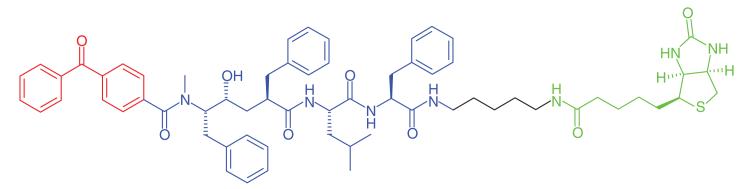
The excess production and deposition of the Aβ peptide is proposed as a root cause of Alzheimer’s disease [72]. γ-Secretase is an intramembrane aspartyl protease, which cleaves transmembrane proteins, the main substrate being the amyloid precursor protein. Proteolytic cleaving of the amyloid precursor protein by both β- and γ-secretases produces short (17–42 amino acids) peptide chains, of which Aβ40 and Aβ42 are believed to be the most neurotoxic. γ-Secretase is a multisubunit protein complex, and its activity is dependent upon the correct assembly of four individual proteins: PS1 or PS2, PEN-2, NCT and APH-1 [73]. γ-Secretase modulators (GSMs) that have been developed as potential treatments for Alzheimer’s disease reduce the production of the pathogenic Aβ42, but do not inhibit the total amount of Aβ formation [74]. The γ-secretase protein complex is a challenging target for drug discovery and various PAL studies have been performed to gain a greater understanding of the binding site of GSMs. Pozdnyakov et al. [43] synthesized a photoaffinity probe (E2012-BPyne) (Table 1, Entry 17) derived from an imidazole GSM (E2012) equipped with a benzophenone and an alkyne handle, which was shown to lower production of Aβ40 and Aβ42 peptides with similar efficacy to the parent compound. The probe was incubated in live HeLa cells in the presence or absence of the parent GSM, cross-linked to targets by UV irradiation, tagged by click chemistry with biotin-azide and isolated by affinity purification. Proteins were separated and western blot analysis was used to distinguish between the various components of γ-secretase. The authors identified PS1-NTF (N-terminal fragment) as the target for E2012. Labeling of PS1-NTF was not observed in the presence of large excess of E2012, and labeling of the other subunits was not seen, suggesting that this is a highly specific interaction. Interestingly, an earlier PAL study was performed by Li et al. [44] on the same target to identify an active site for γ-secretase inhibitors. The authors used a potent inhibitor to design a probe (Table 1, Entry 18) to label the protein complex. The experiment, performed on HeLa cell lysates, resulted in the probe labeling PS1. These results support the hypothesis that the PS1 component is critical for γ-secretase activity.
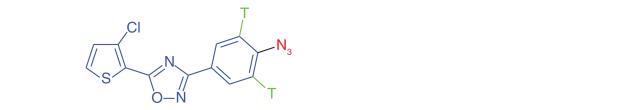
Barbiturates are a large chemical class with sedative and anesthetic effects. They are potent inhibitors of GABAARs and excitatory nicotinic nAChRs [75]. Most barbiturates inhibit these receptors by possessing a high affinity toward the ion channel centralized within the proteins. PAL studies identified distinct binding sites for allosteric modulators of Torpedo nAChR [69,76], particularly within the ion channel itself, but ligand sites have also been found at the γ-α subunit interface and within the δ subunit helix. Hamouda et al. [45] used a tritiated photoaffinity analog of pentobarbital ([3H] mTFD-MPAB) (Table 1, Entry 19) to probe for barbiturate binding sites on Torpedo nAChR. The probe had previously been shown to bind to α1β3γ2 GABAA complexes [66]. [3H]mTFD-MPAB bound to Torpedo nAChR at two separate sites, one within the ion channel, identified by labeling of amino acids within the M2 helices of nAChR subunits. The other site was located at the γ-α subunit interface, recognized by labeling of residue γMet299 of the γM3 helix. The authors discovered that binding preferentially occurred within the ion channel when the receptor was in desensitized state, while lower affinity binding occurred at the subunit interface where other photoaffinity analogs of nAChR modulators had also been shown to bind [67]. Other successful uses of PAL to explore key interacting residues include the work of McKernan et al. [68] who used [3H]flunitrazepam to map the contribution of His102on the α-subunit of GABAARs to the binding of multiple benzodiazepine site ligands.
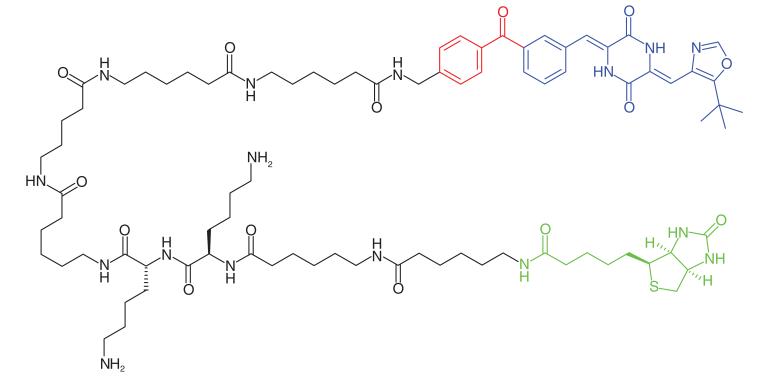
Plinabulin is a microtubule depolymerization agent currently in Phase II clinical trials [77]. Yamazaki et al. [46] synthesized a photoaffinity probe (Table 1, Entry 20) to determine the binding site of plinabulin on microtubules. The authors discovered that a very long linker between the reporter tag and pharmacophore enhanced detection. The anticancer agent bound in a region between α- and β-tubulins, as both subunits were covalently labeled. The binding site is distinct from that of the microtubule inhibitor colchicine, where it was originally thought to bind.
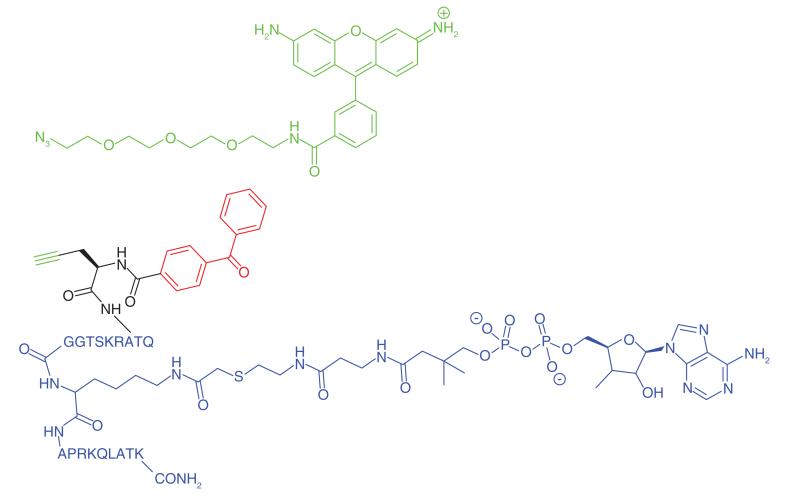
Acetylation and deacetylation of lysine residues by KATs and KDACs, respectively, has a significant impact on the regulation of metabolism and gene transcription. KATs and KDACs are being explored as drug targets for tissue-specific cancers [78]. KAT activity is highly dependent on interactions with protein partners and post-translational modifications, which are challenging to replicate in conventional in vitro assays. Furthermore while there are four major families of KATS, there is little sequence homology between them as well as a number of orphan KATs. Characterization of acetylation-mediated signaling would provide greater understanding and aid inhibitor development. Montgomery et al. [47] developed photoaffinity probes (Table 1, Entry 21) to profile three families of KATs. Probes were based upon known bi-substrate inhibitors which connect coenzyme A with the amino group of lysine-containing peptides to target both cofactor and ligand-binding domains of KATs [79]. A benzophenone and an alkyne handle were added at the C-terminal end of the peptide chain. The selected probes exhibited similar inhibitory and selectivity profiles as their parent compounds, and could effectively label recombinant KATs, where labeling efficiency decreased in the presence of small molecule KAT inhibitors. HEK293 cells were treated with probes, and following irradiation and click-conjugation to a fluorophore showed the labelling of a number of KATs, particularly those belonging to the pCAF family. Further PAL studies were performed to identify novel KAT activities in cancer proteomes. HeLa cell lysates were treated with the photoaffinity probes and the authors identified two specific KATS, Naa50 and Nat10, neither of which belong to a specific family, which could play important roles in protein acetylation in cancer cells.
Future perspective
Many examples of PAL have been published within the past 3 years, underlining the increasing importance of the method to current medicinal chemistry and drug discovery. The reports highlighted in this review show the wide spectrum of probe designs, photophores and reporter tags that can be successfully employed for PAL. It is clear that the technique is applicable to a broad range of small molecules and protein targets relevant to diverse diseases. In the majority of studies, considerable optimization of the photoaffinity probe is carried out, including experiments to prove the ability of the probes to capture known or surrogate targets and to maximize labeling efficiency. The incorporation of relevant competition experiments using unlabeled probes into PAL protocols is often critical for achieving definitive target identification. Despite the widespread use of PAL, it is notable that the experiments do not always lead to complete target identification and validation, as evidenced in some of the reports discussed above.
In the studies surveyed here, the most frequently used photophore was benzophenone. This is surprising given the potential benefits of using the smaller azide or diazirine groups, and may be due in part to the less complex synthetic methodology required for benzophenone incorporation. In some cases benzophenone was the logical choice of label given the structure of the pharmacophore, but it might be expected that diazirines will become the dominant photogroup in the future as synthetic methods are developed for introduction of the minimal alkyldiazirine functionality directly into probe scaffolds [5]. There may be concerns about the chemical stability of photophores to organic reactions as well as light exposure and photoreactive groups have tended to be incorporated into probes at a late stage of synthesis. There have been significant efforts devoted to finding new synthetic routes to the photophores; for example, there are now several methods to install diazirine groups. It is established that an assortment of reactions can be performed on phenyl trifluoromethyl diazirine, with successful examples of Friedel-Crafts acylation, nitration, iodination and subsequent transformations of the products described. This synthetic repertoire will allow earlier incorporation of the photogroup or large-scale synthesis of an intermediate which could be applied to the generation of libraries [2,14].
For other features of probe structure, biotin continues to be the most used reporter tag, with fluorophores and radiolabels applied in equal measure in the minority of cases. Recently introduced DNA-programmed photoaffinity labeling apportions the pharmacophore to one single strand of DNA and the photophore to a complimentary DNA strand, potentially allowing for multiplexed experimental protocols [80,81]. Click chemistry-based PAL is increasingly popular and is described in approximately half of the examples surveyed here. The ability to covalently capture biomolecules in vivo using cell-permeable probes extends the opportunities for target identification to biomolecules that do not usually survive lysis conditions with an intact function or folding state, such as integral membrane proteins. Cell permeable probes are also particularly relevant when targeting multisubunit protein complexes, or proteins which heavily rely upon co-regulators and post-translational modifications to promote the productive binding conformation.
Increasingly, PAL experiments are carried out with multiple probes and combined with other activity-based profiling. Thus a fluorescent or radiolabeled probe without an affinity tag can be used to image the target location in live cells or cell compartment fractions while a more conventional photoaffinity probe is employed for the target capture and isolation. Together, these methods can help sort more rapidly through the candidate targets identified in the PAL experiment.
Probes designed for specific proteins have been used to identify other sites which recognize the same pharmacophore and/or have sequence homology to the original target. For example, this has been demonstrated with peptide probes incorporating amino acid recognition sequences. It is likely that this technique will become more frequently used to identify the full range of biologically relevant targets for individual tools and drugs. On the other hand, generic photoaffinity probes designed to bind widely to classes of enzymes or receptors can be used to profile the diversity and expression of the protein class. Importantly, photoaffinity probes can often be located to single residues within their targets through mass spectrometric sequencing of the labeled peptide fragments following proteolytic digest, providing information on the detailed structure of the interaction of the ligand with the binding site.
Key terms.
Biotin
Vitamin (B7) that is present in small amounts in all living cells. The valeric acid side of chain of biotin can be chemically modified to incorporate various reactive groups to facilitate the addition of a biotin tag to other molecules.
Biotin–Avidin complex
Strongest known ligand–protein noncovalent interaction (Kd=10−15 M). Avidin and streptavidin have the ability to bind up to four biotin molecules. Noncovalent bond formation between the protein and the ligand is rapid and stable toward a variety of factors including pH, temperature and some denaturing agents, making this interaction very useful in protein purification and identification strategies.
Click chemistry
Class of organic reactions that have been defined as modular, wide in scope, high yielding, stereospecific, easy to perform, conducted in easily removable solvents and producing by-products which can be removed without chromatography. Examples include nucleophilic ring opening of epoxides and aziridines, Michael additions and cycloadditions.
Carbenes
Divalent carbon intermediates, where the carbene carbon is covalently bonded to two adjacent groups, and possesses two nonbonding electrons. Carbenes are classified as either singlets or triplets depending on their electronic structure. Singlet carbenes are spin-paired where the two electrons occupy the same orbital, leaving an empty orbital on the carbon. They exhibit both nucleophilic and electrophilic properties. Triplet carbenes exhibit radical-like reactivity, and possess two unpaired electrons in separate orbitals.
Cell lysates
Fluid containing the contents of lysed cells. Cell lysis is the process of breaking down cell walls to release the contents and is achieved by various means including treatment with detergents or salts.
Epitopes
Parts of a molecule that can be recognized and bound by an antibody. Epitopes can be composed of sugars, lipids or amino acids, with the latter being the most common.
Fluorophores
Fluorescent chemical compounds that can re-emit light upon light excitation.
Western blot
Analytical technique used to detect specific proteins. Proteins are separated by their molecular weight through electrophoresis on a polyacrylamide gel in the presence of detergent (e.g., SDS). Proteins are transferred from the gel to a membrane (either nitrocellulose or polyvinylidene difluoride) by a process known as electroblotting. Once the proteins are on the membrane, they can be detected by specific antibodies and/or by fluorescent scanning. Proteins may also be visualized on the gel using relevant stains or by fluorescent scanning.
2D-PAGE
A 2D gel-based method to separate proteins by size and charge, using isoelectric focusing and SDS-PAGE.
Executive summary.
Photoaffinity labeling (PAL) involves the covalent labeling of a protein by a small molecule and has benefits in a variety of applications in medicinal chemistry, most especially target identification for hits from phenotypic screens.
PAL can label low abundance and low affinity proteins.
PAL can be applied productively at multiple stages in drug discovery including discovering new molecular targets, identifying off-target or unwanted interactions and characterizing binding sites to advance new inhibitor design.
Click chemistry combined with PAL enables the identification of targets from live cells by providing cell-permeable affinity probes that can be conjugated in separate steps to tags that enable target isolation.
Synthetic routes for photogroups have improved, particularly in diazirine syntheses where synthetic strategies are now less complex.
Acknowledgments
The authors acknowledge financial support for this work from the Wellcome Trust (studentship to ES) and The Institute of Cancer Research.
Footnotes
Financial & competing interests disclosure The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
• of interest; •• of considerable interest
- 1.Lapinsky DJ. Tandem photoaffinity labeling-bioorthogonal conjugation in medicinal chemistry. Bioorg. Med. Chem. 2012;20:6237–6247. doi: 10.1016/j.bmc.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 2.Hashimoto M, Hatanaka Y. Recent progress in diazirine-based photoaffinity labeling. Eur. J. Org. Chem. 2008;15:2513–2523. [Google Scholar]
- 3.Sadakane Y, Hatanaka Y. Photochemical fishing approaches for identifying target proteins and elucidating the structure of a ligand-binding region using carbene-generated photoreactive probes. Anal. Sci. 2006;22:209–218. doi: 10.2116/analsci.22.209. [DOI] [PubMed] [Google Scholar]
- 4.Singh A, Thornton ER, Westheimer FH. The photolysis of diazoacetylchymotrypsin. J. Biochem. 1962;237:3006–3008. [PubMed] [Google Scholar]
- 5.Das J. Aliphatic diazirines as photoaffinity probes for proteins: recent developments. Chem. Rev. 2011;111:4405–4417. doi: 10.1021/cr1002722. [DOI] [PubMed] [Google Scholar]
- 6.Hughes JP, Rees S, Kalinjian SB, Philpott KL. Principles of early drug discovery. Br. J. Pharmacol. 2011;162:1239–1249. doi: 10.1111/j.1476-5381.2010.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han SY, Choi SH, Kim MH, et al. Design and synthesis of novel photoaffinity reagents for labeling VEGF receptor tyrosine kinases. Tetrahedron Lett. 2006;47:2915–1919. [Google Scholar]
- 8.Shi H, Zhang CJ, Chen GYJ, Yao SQ. Cell-based proteome profiling of potential dasatinib targets by use of affinity-based probes. J. Am. Chem. Soc. 2012;134:3001–3014. doi: 10.1021/ja208518u. [DOI] [PubMed] [Google Scholar]
- 9.MacKinnon AL, Taunton J. Target identification by diazirine photo-cross-linking and click chemistry. Curr. Protoc. Chem. Biol. 2009;1:55–73. doi: 10.1002/9780470559277.ch090167. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Provides comprehensive and practical guidelines to perform photoaffinity labeling (PAL) experiments
- 10.Kolb HC, Sharpless KB. The growing impact of click chemistry on drug discovery. Drug Discov. Today. 2003;8:1128–1137. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh B, Jones LH. Target validation using in-cell small molecule clickable imaging probes. Med. Chem. Commun. 2013;5:247–254. [Google Scholar]
- 12.Fleming SA. Chemical reagents in photoaffinity labelling. Tetrahedron. 1995;51:12479–12520. [Google Scholar]
- 13.Sakurai K, Ozawa S, Yamada R, Yasui T, Mizuno S. Comparison of the reactivity of carbohydrate photoaffinity probes with different photoreactive groups. Chem. Bio. Chem. 2014;15:1399–1403. doi: 10.1002/cbic.201402051. [DOI] [PubMed] [Google Scholar]
- 14.Dubinsky L, Krom BP, Meijler MM. Diazirine based photoaffinity labelling. Bioorg. Med. Chem. 2012;20:554–570. doi: 10.1016/j.bmc.2011.06.066. [DOI] [PubMed] [Google Scholar]
- 15.Geurink PP, Prely ML, van der Marel AG, Bischoff R, Overkleeft HS. Photoaffinity labeling in activity-based protein profiling. Top. Curr. Chem. 2012;324:85–114. doi: 10.1007/128_2011_286. [DOI] [PubMed] [Google Scholar]
- 16.Staros JV, Bayley H, Standring DN, Knowles JR. Reduction of aryl azides by thiols: implications for the use of photoaffinity reagents. Biochem. Biophys. Res. Commun. 1978;80:568–572. doi: 10.1016/0006-291x(78)91606-6. [DOI] [PubMed] [Google Scholar]
- 17.Terstappen GC, Schlüpen C, Raggiaschi R, Gaviraghi G. Target deconvolution strategies in drug discovery. Nat. Rev. Drug Discov. 2007;6:891–903. doi: 10.1038/nrd2410. [DOI] [PubMed] [Google Scholar]
- 18.Sadaghiani AM, Verhelst SHL, Bogyo M. Tagging and detection strategies for activity-based proteomics. Curr. Opin. Chem. Biol. 2007;11:20–28. doi: 10.1016/j.cbpa.2006.11.030. [DOI] [PubMed] [Google Scholar]; • Comprehensive review of the identification tags available in proteomics and their uses.
- 19.Liu J, Liu C, He W. Fluorophores and their applications as molecular probes in living cells. Curr. Org. Chem. 2013;17:564–579. [Google Scholar]
- 20.Lamos SM, Krusemark CJ, McGee CJ, et al. Mixed isotope photoaffinity reagents for identification of small-molecule targets by mass spectrometry. Angew. Chem. Int. Ed. 2006;45:4329–4333. doi: 10.1002/anie.200600743. [DOI] [PubMed] [Google Scholar]
- 21.Song Z, Huang W, Zhang Q. Isotope-coded, fluorous photoaffinity labelling reagents. Chem. Commun. 2012;48:3339–3341. doi: 10.1039/c2cc00027j. [DOI] [PubMed] [Google Scholar]
- 22.Rix U, Superti-Furga G. Target profiling of small molecules by chemical proteomics. Nat. Chem. Biol. 2009;5:616–624. doi: 10.1038/nchembio.216. [DOI] [PubMed] [Google Scholar]
- 23.Cong F, Cheung AK, Huang SMA. Chemical genetics-based target identification in drug discovery. Annu. Rev. Pharmacol. Toxicol. 2012;52:57–78. doi: 10.1146/annurev-pharmtox-010611-134639. [DOI] [PubMed] [Google Scholar]
- 24.Kawatani M, Osada H. Affinity-based target identification for bioactive small molecules. Med. Chem. Commun. 2013;5:277–287. [Google Scholar]
- 25.Sleno L, Emili A. Proteomic methods for drug target discovery. Curr. Opin. Chem. Biol. 2008;12:46–54. doi: 10.1016/j.cbpa.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 26.Lomenick B, Olsen RW, Huang J. Identification of direct protein targets of small molecules. ACS Chem. Biol. 2011;6:34–46. doi: 10.1021/cb100294v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dzubak P, Hajduch M, Vydra D, et al. Pharmacological activities of natural triterpenoids and their therapeutic implications. Nat. Prod. Rep. 2006;22:394–411. doi: 10.1039/b515312n. [DOI] [PubMed] [Google Scholar]
- 28.Zhang L, Zhang Y, Dong J, Liu J, Zhang L, Sun S. Design and synthesis of novel photoaffinity probes for study of the target proteins of oleanolic acid. Bioorg. Med. Chem. Lett. 2012;22:1036–1039. doi: 10.1016/j.bmcl.2011.11.123. [DOI] [PubMed] [Google Scholar]
- 29.Kotake Y, Sagane K, Owa T, et al. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol. 2007;3:570–575. doi: 10.1038/nchembio.2007.16. [DOI] [PubMed] [Google Scholar]; •• Demonstrates the power of multiple orthogonal probes (fluorescence and radiolabeled affinity probes and a photoaffinity probe). to aid rapid target deconvolution.
- 30.Xu C, Soragni E, Chou JC, et al. Chemical probes identify a role for histone deacetylase 3 in Friedrich’s ataxia gene silencing. Chem. Biol. 2009;16:980–989. doi: 10.1016/j.chembiol.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jessen KA, English NM, Wang JY, et al. The discovery and mechanism of action of novel tumor-selective and apoptosis-inducing 3,5-diaryl-1,2,4-oxadiazole series using a chemical genetics approach. Mol. Cancer Ther. 2005;4:761–771. doi: 10.1158/1535-7163.MCT-04-0333. [DOI] [PubMed] [Google Scholar]
- 32.Lee K, Ban HS, Naik R, et al. Identification of malate dehydrogenase 2 as a target protein of the HIF-1 inhibitor LW6 using chemical probes. Angew. Chem. Int. Ed. 2013;52:10286–10289. doi: 10.1002/anie.201304987. [DOI] [PubMed] [Google Scholar]; • Example of target capture and identification from live cells using click chemistry and efficient incorporation of a minimal photophore into the affinity pharmacophore.
- 33.Bell JL, Haak AJ, Wade SM, Sun Y, Neubig RR, Larsen SD. Design and synthesis of tag-free photoprobes for the identification of the molecular target for CCG-1423, a novel inhibitor of the Rho/MKL1/SRF signaling pathway. Beilstein J. Org. Chem. 2013;9:966–973. doi: 10.3762/bjoc.9.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kotoku N, Nakata C, Kawachi T, et al. Synthesis and evaluation of effective photoaffinity probe molecule of furospinosulin-1, a hypoxia-selective growth inhibitor. Bioorg. Med. Chem. 2014;22:2102–2112. doi: 10.1016/j.bmc.2014.02.026. [DOI] [PubMed] [Google Scholar]
- 35.Mackinnon AL, Garrison JL, Hegde R, Taunton J. Photo-leucine incorporation reveals target of cyclodepsipeptide inhibitor of cotranslational translocation. J. Am. Chem. Soc. 2007;129:14560–14561. doi: 10.1021/ja076250y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Colca JR, McDonald WG, Cavey GS, et al. Identification of a mitochondrial target of thiazolidinedione insulin sensitizers (mTOT) – relationship to newly identified mitochondrial pyruvate carrier proteins. PLoS ONE. 2013;8:e61551. doi: 10.1371/journal.pone.0061551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Webb Y, Zhou X, Ngo L, et al. Photoaffinity labeling and mass spectrometry identify ribosomal protein S3 as a potential target for hybrid polar cytodifferentiation agents. J. Biol. Chem. 1999;274:14280–14287. doi: 10.1074/jbc.274.20.14280. [DOI] [PubMed] [Google Scholar]
- 38.Mésange F, Sebbar M, Capedevielle J, et al. Identification of two tamoxifen target proteins by photolabeling with 4-(2-morpholinoethoxy) benzophenone. Bioconjugate Chem. 2002;13:766–772. doi: 10.1021/bc015588t. [DOI] [PubMed] [Google Scholar]; • Highlights the use of PAL to uncover new mechanisms of action for a marketed drug.
- 39.Kambe T, Correia BE, Niphakis MJ, Cravatt BF. Mapping the protein interaction landscape for fully functionalised small-molecule probes in human cells. J. Am. Chem. Soc. 2014;136:10777–10782. doi: 10.1021/ja505517t. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Shows the diversity of targets that can be captured by PAL and informs on the problems of nonspecific binding and how to address this.
- 40.Kashiwayama Y, Tomohiro T, Narita K, et al. Identification of a substrate-binding site in a peroxisomal β-oxidation enzyme by photoaffinity labeling with a novel palmitoyl derivative. J. Biol. Chem. 2010;285:26315–26325. doi: 10.1074/jbc.M110.104547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Misu R, Oishi S, Setsuda S, et al. Characterization of the receptor binding residues of kisspeptins by positional scanning using peptide photoaffinity probes. Bioorg. Med. Chem. Lett. 2013;23:2628–2631. doi: 10.1016/j.bmcl.2013.02.098. [DOI] [PubMed] [Google Scholar]
- 42.Kyro K, Manandhar SP, Mullen D, Schmidt WK, Distefano MD. Photoaffinity labelling of Ras converting enzyme using peptide substrates that incorporate benzoylphenylalanine (Bpa) residues: improved labeling and structural implications. Bioorg. Med. Chem. 2011;19:7559–7569. doi: 10.1016/j.bmc.2011.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pozdnyakov N, Murrey HE, Crump CJ, et al. γ-Secretasemodulator (GSM) photoaffinity probes reveal distinct allosteric binding sites on presenilin. J. Biol. Chem. 2013;288:9710–9720. doi: 10.1074/jbc.M112.398602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li YM, Xu M, Lai MT, et al. Photoactivated-γ-secretase inhibitors directed to the active site covalently label presenilin 1. Nature. 2004;405:689–694. doi: 10.1038/35015085. [DOI] [PubMed] [Google Scholar]
- 45.Hamouda AK, Stewart DS, Chiara DC, Savechenkov PY, Bruzik KS, Cohen JC. Identifying barbiturate binding sites in a nicotinic acetylcholine receptor with [3H] m-trifluoromethyldiazirinemephobarbital a photoreactive barbiturate. Mol. Pharmacol. 2014;85:735–746. doi: 10.1124/mol.113.090985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamazaki Y, Sumikura M, Hidaka K, et al. Anti-microtubule ‘plinabulin’ chemical probe KPU-244 labeled both α- and β- tubulin. Bioorg. Med. Chem. 2010;18:3169–3174. doi: 10.1016/j.bmc.2010.03.037. [DOI] [PubMed] [Google Scholar]
- 47.Montgomery DC, Sorum AW, Meier JL. Chemoproteomic profiling of lysine acetyltransferases highlights an expanded landscape of catalytic acetylation. J. Am. Chem. Soc. 2014;136:8669–8676. doi: 10.1021/ja502372j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomas EA, Coppola G, Desplats PA, et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc. Natl Acad. Sci. USA. 2008;105:15564–15569. doi: 10.1073/pnas.0804249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herman D, Jenssen K, Burnett R, Soragni E, Perlman SL, Gottesfeld JM. Histone deacetylase inhibitors reverse gene splicing in Friedrich’s ataxia. Nat. Chem. Biol. 2006;2:551–558. doi: 10.1038/nchembio815. [DOI] [PubMed] [Google Scholar]
- 50.Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007;25:84–90. doi: 10.1038/nbt1272. [DOI] [PubMed] [Google Scholar]
- 51.Chen QJ, Massaqué J. Molecular pathways: VCAM-1 as a potential therapeutic target in metastasis. Clin. Cancer Res. 2012;18:5520–5525. doi: 10.1158/1078-0432.CCR-11-2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carter RA, Campbell IK, O’Donnel KL, Wicks IP. Vascular cell adhesion molecule-1 (VCAM-1) blockade in collagen-induced arthritis reduces joint involvement and alters B cell trafficking. Clin. Exp. Immunol. 2002;128:44–51. doi: 10.1046/j.1365-2249.2002.01794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Besemer J, Harant H, Wang S, et al. Selective inhibition of cotranslational translocation of vascular cell adhesion molecule 1. Nature. 2005;436:290–293. doi: 10.1038/nature03670. [DOI] [PubMed] [Google Scholar]
- 54.Nury C, Czarny B, Cassar-Lajeunesse E, Georgiadis D, Bregant S, Dive V. A pan photoaffinity probe for detecting active forms of matrix metalloproteinases. Chembiochem. 2013;14:107–114. doi: 10.1002/cbic.201200583. [DOI] [PubMed] [Google Scholar]
- 55.Diamant M, Heine RJ. Thiazolidinediones in type 2 diabetes mellitus: current clinical evidence. Drugs. 2003;63:1373–1405. doi: 10.2165/00003495-200363130-00004. [DOI] [PubMed] [Google Scholar]
- 56.Mansour M, Schwartz D, Judd R, et al. Thiazolidinediones/PPARγ agonists and fatty acid synthase inhibitors as an experimental combination therapy for prostate cancer. Int. J. Oncol. 2011;38:537–546. doi: 10.3892/ijo.2010.877. [DOI] [PubMed] [Google Scholar]
- 57.Chen Z, Vigueira PA, Chambers KT, et al. Insulin resistance and metabolic derangements in obese mice are ameliorated by a novel peroxisome proliferator activated receptor γ-sparing thiazolidinedione. J. Biol. Chem. 2012;287:235237–23548. doi: 10.1074/jbc.M112.363960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Colca JR, McDonald WG, Waldon D, et al. Identification of a novel mitochondrial protein (“mitoNEET”) cross-linked specifically by a thiazolidinedione photoprobe. Am. J. Physiol. Endocrinol. Metab. 2004;286:E252–E260. doi: 10.1152/ajpendo.00424.2003. [DOI] [PubMed] [Google Scholar]
- 59.Divakaruni AS, Wiley SE, Rogers GW, et al. Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc. Natl Acad. Sci. USA. 2013;110:5422–5427. doi: 10.1073/pnas.1303360110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Richon VM, Emililani S, Verdin E, et al. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc. Natl Acad. Sci. USA. 1998;95:3003–3007. doi: 10.1073/pnas.95.6.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grant S, Easley C, Kirkpatrick P. Vorinostat. Nat. Rev. Drug Discov. 2007;6:21–22. doi: 10.1038/nrd2227. [DOI] [PubMed] [Google Scholar]
- 62.Richon VM, Russo P, Venta-Perez G, Cordon-Cardo C, Rifkind RA, Marks PA. Two cytodifferentiation agent-induced pathways, differentiation and apoptosis, are distinguished by the expression of human papillomavirus 16 E7 in human bladder carcinoma cells. Cancer Res. 1997;57:2789–2798. [PubMed] [Google Scholar]
- 63.Shenoy N, Kessel R, Bhagat TD, et al. Alterations in the ribosomal machinery in cancer and hematologic disorders. J. Hematol. Oncol. 2012;5:32. doi: 10.1186/1756-8722-5-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Riggs BL, Hartmann LC. Selective estrogen-receptor modulators – mechanism of action and application to clinical practice. N. Engl. J. Med. 2003;348:618–629. doi: 10.1056/NEJMra022219. [DOI] [PubMed] [Google Scholar]
- 65.Mésange F, Sebbar M, Capedevielle J, et al. Microsomal epoxide hydrolase of rat liver is a subunit of the anti-oestrogen binding site. Biochem. J. 1998;334:107–112. doi: 10.1042/bj3340107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chiara DC, Jayakar SS, Zhou X, et al. Specificity of intersubunit general anesthetic-binding sites in the transmembrane domain of the human a1b3g2 g-aminobutyric acid type A (GABAA) receptor. J. Biol. Chem. 2013;288:19343–19357. doi: 10.1074/jbc.M113.479725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hamouda AK, Stewart DS, Husain SS, Cohen JB. Multiple transmembrane binding sites for p-trifluoromethyldiazirinyletomidate, a photoreactive Torpedo nicotinic acetylcholine receptor allosteric inhibitor. J. Biol. Chem. 2011;286:20466–20477. doi: 10.1074/jbc.M111.219071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McKernan RM, Farrar S, Collins I, et al. Photoaffinity labeling of the benzodiazepine binding site of α1β3γ2 γ-aminobutyric acidA receptors with flunitrazepam identifies a subset of ligands that interact directly with His102 of the α subunit and predicts orientation of these within the benzodiazepine pharmacophore. Mol. Pharmacol. 1998;54:33–43. doi: 10.1124/mol.54.1.33. [DOI] [PubMed] [Google Scholar]
- 69.Hamouda AK, Jayakar SS. Photoaffinity labeling of nicotinic receptors: diversity of drug binding sites. J. Mol. Neurosci. 2014;53:480–486. doi: 10.1007/s12031-013-0150-1. [DOI] [PubMed] [Google Scholar]
- 70.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 71.Deng Y, Couch BA, Koleske AJ, Turk BE. A peptide photoaffinity probe specific for the active conformation of the Abl tyrosine kinase. Chem. Bio. Chem. 2012;13:2510–2512. doi: 10.1002/cbic.201200560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Murphy MP, LeVine H., III Alzheimer’s disease and the β-amyloid peptide. J. Alzheimers. Dis. 2010;19:311–323. doi: 10.3233/JAD-2010-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.De Strooper B, Iwatsubo T, Wolfe MS. Presenilins and γ-secretase: structure, function and role in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012;2:a006304. doi: 10.1101/cshperspect.a006304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tate B, McKee TD, Loureiro RMB, et al. Modulation of gamma-secretase for the treatment of Alzheimer’s disease. Int. J. Alzheimers. Dis. 2012;2012:210756. doi: 10.1155/2012/210756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arias HR, McCardy EA, Gallagher MJ, Blanton MP. Interaction of barbiturate analogs with the Torpedo californica nicotinic acetylcholine receptor ion channel. Mol. Pharmacol. 2001;60:497–506. [PubMed] [Google Scholar]
- 76.Jayakar SS, Dailey WP, Eckenhoff RG, Cohen JB. Identification of propofol binding sites in a nicotinic acetylcholine receptor allosteric inhibitor. J. Biol. Chem. 2013;288:6178–6189. doi: 10.1074/jbc.M112.435909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Singh AV, Bandi M, Raje N, et al. A novel vascular disrupting agent plinabulin triggers JNK-mediated apoptosis and inhibits angiogenesis in multiple myeloma cells. Blood. 2011;117:5692–5700. doi: 10.1182/blood-2010-12-323857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang XJ. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acid Res. 2004;32:959–976. doi: 10.1093/nar/gkh252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lau OD, Kundu TK, Soccio RE, et al. HATs off: selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Mol. Cell. 2000;5:589–595. doi: 10.1016/s1097-2765(00)80452-9. [DOI] [PubMed] [Google Scholar]
- 80.Li G, Liu Y, Liu Y, Chen L, Wu S, Liu Y, Li X. Photoaffinity labeling of small-molecule-binding proteins by DNA-templated chemistry. Angew. Chem., Intl. Ed. 2013;52:9544–9549. doi: 10.1002/anie.201302161. [DOI] [PubMed] [Google Scholar]
- 81.Li G, Liu Y, Yu X, Li X. Multivalent photoaffinity probe for labeling small molecule binding proteins. Bioconj. Chem. 2014;25:1172–1180. doi: 10.1021/bc500195w. [DOI] [PubMed] [Google Scholar]