

Abstract
Objective
To determine whether super-activation of PPARγ can reprogram human myoblasts into brownlike adipocytes and to establish a new cell model for browning research.
Methods
To enhance the PPARγ signaling, we fused M3, the transactivation domain of MyoD, to PPARγ. PPARγ and M3-PPARγ-lentiviral vectors were used to convert human myoblasts into adipocytes. Brown adipocyte markers of the reprogrammed adipocytes were assessed by qPCR and protein analyses. White adipocytes differentiated from subcutaneous stromal vascular cells and perithyroid brown fat tissues were used as references.
Results
In transient transfection, M3-PPARγ has stronger constitutive activity than PPARγ reporter assay. Although both the transduction of PPARγ and M3-PPARγ induced adipogenesis in myoblasts, M3-PPARγ, compared to PPARγ, drastically induced the brown adipocyte markers of UCP1, CIDEA and PRDM16 by 1,050, 2.4, and 5.0 fold, respectively and increased mitochondria contents by 4 fold. The gene expression levels of the browning makers in PPARγ-reprogrammed adipocytes are comparable to those of in vitro differentiated white adipocytes.
Conclusions
We have found that super-activation of PPARγ can effectively convert human myoblasts into brown-like adipocytes and can be a new approach to derive brown-like adipocytes.
Keywords: brown adipocyte, obesity, human myoblasts, super-active PPARγ, cellular reprogramming
Introduction
Obesity represents a major risk factor, particularly for diabetes type 2 (T2D), dyslipidemias and cardiovascular diseases (1). Obesity develops when energy intake exceeds expenditure. Recent discovery that adult humans have functional brown adipose tissue (BAT) mediated thermogenesis has re-energized interest in targeting BAT bioenergetics to increase energy expenditure for the treatment of obesity and associated diseases (2, 3, 4).
In mammals, two major distinctive types of adipose tissue exist, white (WAT) and brown (BAT) adipose tissue; the former stores energy in the form of triglyceride whereas the latter dissipates energy as heat. Brown adipocytes, the functional component of BAT, are characterized by the presence of multilocular lipid droplets, the abundance of mitochondria and the expression of the cell type-specific uncoupling protein 1(UCP1). Both an increase in the number of brown adipocytes and the induction of UCP1 expression are characteristics for adipocyte “browning” (5). Great advances have been made in understanding the regulation and development of brown adipocytes or BAT in recent years (6, 7) through studies on rodent models. However, because of apparent species differences in non-shivering thermogenesis, it is unclear whether the knowledge derived from the studies using rodent models can be directly extrapolated to humans. For example, systemic β-adrenergic stimulation of thermogenesis is accompanied by brown adipose tissue activity in rodents (8), but not in humans (9).
Since human BAT localizes diffusely in areas of the neck, perithyroid and supraclavicles (10, 11), studies using primary brown adipocytes are scarce, probably due to the technical difficulty in obtaining such cells for research. Recently, human brown or brown-like adipocytes derived from pluripotent stem cells through over-expression of browning factors (12) and differentiation (13) have been developed and will be an excellent cell model for differentiation studies.
However, brown or brown-like adipocytes derived from somatic cells and the knowledge derived from the somatic cell reprogramming may be more applicable to human obesity treatment in the near future. To date, the activation of PPARγ signaling is perhaps the most reproducible regimen to brown murine and human adipocytes in vitro (14, 15, 16, 17), but there are few reports about whether clinical doses of thiazolidinedione will result in meaningful adipocyte browning in humans. Thus, researcher are seeking more effective means to brown human adipocytes. Recently, Hirai et al reported (18, 19) that the addition of the transactivation domain of MyoD (M3) to a transcription factor can drastically enhance its capacity to reprogram fibroblasts into induced pluripotent stem cells or cardiomyocytes. Aiming to address the questions of 1) whether brown-like adipocytes can be derived from direct somatic cell conversion and 2) whether the enhancement or superactivation of PPARγ signaling can promote a deeper browning, we engineered a fusion protein of M3-PPARγ and demonstrated that it has an enhanced transcriptional activity and can effectively convert myoblasts into brown-like adipocytes.
Materials and Methods
Molecular cloning
The M3 fragment was obtained by PCR amplification on the template of M3O (fusion protein of M3 with Oct4), (20)) and cloned into a Gateway pEntr vector (Invitrogen, Carlsbad, CA) to generate pEntr-M3. pEntr M3-PPARγ was constructed by cloning of human PPARγ1 into pEntr-M3 at the 3′-end in frame by Infusion (Clontech, Mountain View, CA). To make the lenti-viral destination vector pSMPUW-CMV-DEST, a fragment of the CMV promoter-ccdB was cloned into the universal lentiviral vector pSMPUW (Cell BioLabs, San Diego, CA). Standard Gateway LR cloning protocol was utilized to generate pLenti-M3-PPARγ, PPARγ and GFP by using LR Clonase II reaction (Invitrogen). cDNA inserts of all clones were verified without mutation by restriction enzyme digestion and DNA sequence analysis.
Reporter Assays
The human HEK293 cells were cultured in 6-well plates in Dulbecco's modified Eagle's medium containing 10% fetal calf serum (Invitrogen), 100 μg/mL penicillin, and 100 μg/mL streptomycin (Invitrogen). Luciferase reporter assays were performed by transfecting 1 μg of PPRE3x-tk-Luc reporter construct, 1 μg of pSMPUW-M3-PPARγ, pSMPUW-PPARγ or pSMPUW-GFP control, and 2 ng of pCMV-Renilla (Promega) into HEK293 cells using LipoD293 (SignaGen Laboratories, Rockville, MD). The cells were cultured in the presence or absence of rosiglitazone (1 μM) for 48 hr after transfection and lysed for luciferase and renilla (for correction) activity assay by using Dual-Luciferase Reporter Assay System (Promega, Madison, WI).
Production of lentivirus
HEK293T cells were used for lentivirus production. To produce lentiviruses, 1.2 μg of the transfer vector (lenti-M3-PPARγ, lenti-PPARγ, or lenti-GFP), 1.2 μg of pCD/NL-BH*DDD (Addgene plasmid 17531) and 0.2 μg of pVSVG (Cell Biolabs) were co-transfected into HEK293T cells using LipoD293 reagent (SignaGen) in a 6-well plate. LipoD293/DNA complex-containing medium was removed and replaced with fresh medium (DMEM/F12 supplemented with 10% FBS and 100 U/ml penicillin-streptomycin) 16 hr post-transfection. Cell medium containing the viral particles was collected twice at 24 and 48 hr after the medium change, passed through a 0.22 μm filter, and either used fresh or stored at 4°C for up to one week for cell infection.
Human studies
The human study protocols were approved by the institutional review board of the University of Maryland. All subjects provided informed consent. To isolate human myoblasts, a percutaneous muscle biopsy was obtained from the lateral portion of the vastus lateralis. The tissue samples were cut into small pieces (∼10 mg), washed in Hanks buffer without calcium or magnesium and digested with a mixture of collagenase IV (1 mg/mL, Sigma-Aldrich, St Louis, MO) and trypsin (Gibco, 0.025%) in PBS for 30 min at 37°C. Tissue pieces and cells were centrifuged for 5 min at 170 × g, resuspended in complete SkBM medium with Singlequots supplements containing rhEGF, fetuin, BSA and GA-1000 minus insulin and hydrocortisone and minus insulin and hydrocortisone (Lonza, Walkersville, MD), 100 U/mL penicillin-streptomycin and 10% FBS and plated on collagen-coated plates. Myoblasts were allowed to grow out of the tissue pieces and were passaged in the incomplete SkBM medium. Human adipose stromal vascular cells (AdSVCs) were isolated as described before (21, 22). Human brown adipose tissues (BAT) were obtained from dissection of the perithyroid adipose tissues during surgery for obesity-related diseases.
Cell culture and differentiation
Human myoblasts were grown in the incomplete SkBM medium until 90-100% confluence in 12-well plates and were infected by exposure to lentiviral supernatant (∼ 4 MOI) and polybrene (0.8 μg/mL, Millipore, Billerica, MA) daily for two days. After the second lentiviral infection, human myoblasts or AdSVCs underwent adipogenesis according to the non-modified adipogenesis protocol (23). Briefly, the cells were cultured in the adipocyte induction medium DMEM/F12 (Gibco) containing d-Biotin (33 nM, Sigma), human insulin (70 nM, Sigma I9278), dexamethasone (100 nM, APP pharmaceuticals, Schaumburg, IL), pantothenate (4 μg/mL, Sigma), human transferrin (10 μg/mL, Calbiochem), 3,3′, 5-triiodo-L-thyronine sodium salt (2 μM, Sigma), rosiglitazone (1μM, Enzo Lifesciences, Farmingdale, NY), isobutylmethylxanthine (IBMX, 0.5 mM, Sigma), 10% FBS, 100U/mL penicillin-streptomycin. After 72 hr, cells were switched to the adipocyte differentiation medium (induction medium without rosiglitazone and IBMX) for additional 8 days or until collection.
RNA extraction, cDNA synthesis and RT-qPCR
Total RNAs were extracted using RNeasy Lipid Tissue Mini Kit (Qiagen, Valencia, CA) with on-column DNase digestion (Qiagen). cDNAs were synthesized using AMV Reverse Transcriptase kit (Promega) from 1μg of total RNA. Quantitative PCR was performed on Light Cycler 480 (Roche, Indianapolis, IN) using primers listed in Table S1. β-Actin was used as a reference gene.
Western blot
Cultured cells were homogenized in lysis buffer containing 50 mM Tris-HCl (pH 6.8) and 2 % SDS. The protein concentration was measured using Pierce TM BCA Protein Assay Kit (Thermo Fisher scientific, Waltham, MA). Lysates were loaded onto NuPAGE® Novex® Bis-Tris gels for electrophoresis. Then proteins were transferred to PVDF membrane. Antibodies used were anti-UCP1 antibody (Cat#ab10983, 1:1000, Abcam, Cambridge, MA), PPARγ (Cat#sc-7196, 1:2000,Santa Cruz Biotechnology, Santa Cruz, CA), MyoD (Cat#TA309755, OriGene, Rockville, MD), anti-β-actin antibody (Sigma) and goat anti-rabbit IgG (H+L) HRP conjugate (Bio-Rad Laboratories, Hercules, CA). The quantitative analysis of UCP1 relative signal was performed using ImageJ software (NIH Image, Bethesda, MD).
Mitotracker staining and quantification
Cells were incubated with 100 nM Mitotracker Red CMXRos (Invitrogen), which contains a mildly thiol-reactive chloromethyl moiety for labeling mitochondria, for 30 min at 37°C at about 100% confluency, then washed twice with PBS before imaging. Confocal imaging was performed at using a Zeiss LSM510 microscope (Carl Zeiss MicroImaging, Inc.), Ten cells of each group were randomly picked for quantification of the red fluorescence signal by ImageJ software.
Statistics
qPCR and Western analyses were conducted in cells from three to four subjects. Statistical analyses were performed using GraphPad Prism 6 (La Jolla, CA). Data are expressed as mean ± SEM. Group comparisons were determined using ANOVA or two-tailed Student's t test, and P < 0.05 was considered significant.
Results
Fusion of M3 to PPARγ enhances PPARγ signaling
Fusion of the M3 transacting domain to transcription factors Oct4 and Mef2 enhances their cellular reprogramming activities (19, 20). We speculated that M3 may enhance the transcriptional activity of PPARγ, and engineered a synthetic protein of M3-PPARγ by fusing M3 to the N-terminus of PPARγ (Fig. 1a). To evaluate the transcriptional activity of M3-PPARγ, we examined the luciferase activities of the promoter reporter (PPRE 3x-Luc) which contains three peroxisomal proliferator responsive elements (PPRE) by transient transfection assays in HEK293 cells. As shown in Fig. 1b, basal promoter activity with the control vector GFP was low in the cells but increased by 17 fold after transfection of PPARγ, which was further augmented by about one fold in the presence of the PPARγ ligand rosiglitazone. Impressively, M3-PPARγ increased the promoter activity by 82.6 and 4.8 fold, respectively, when compared to GFP control and PPARγ. The M3-PPARγ remained responsive to rosiglitazone by increasing the luciferase activity by 177%. These results indicate that M3-PPARγ is constitutively super-active in activating the PPRE reporter and remains responsive to the PPARγ ligand.
Figure 1.
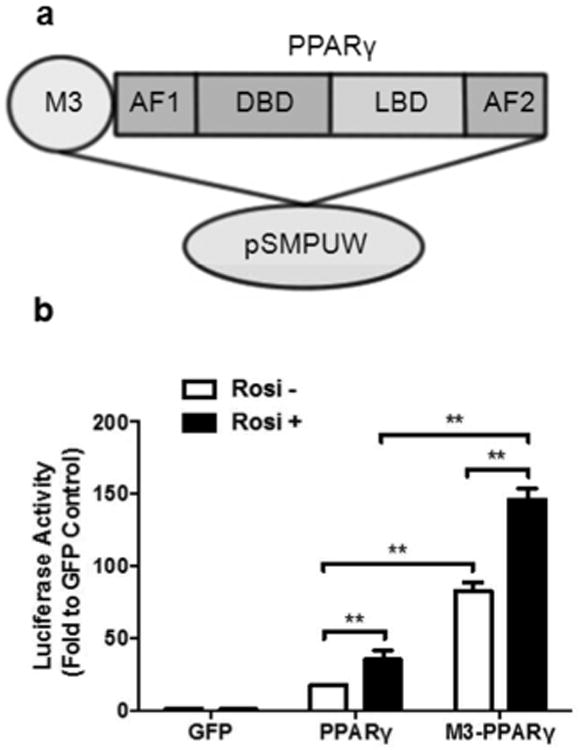
Fusion of M3 transacting domain to PPARγ enhances its transcriptional activity of PPARγ. (a) Illustration of M3 domain to N-terminus of PPARγ, which is comprised of a transactivation domain (AF1), a DNA-binding domain (DBD), a C-terminal ligand-binding domain (LBD) and a ligand-dependent transactivation domain (AF2). The entire construct is cloned into the lentiviral expression vector pSMPUW. (b) Transcriptional activity of M3-PPARγ on PPRE3x-luciferase promoter. HEK293 cells are transfected with the reporter construct and GFP, PPARγ and M3-PPARγ in the presence or absence of rosiglitazone (1 μM), and assayed for luciferase activities. Data are expressed as mean ± SE (n = 3). **: p < 0.01.
M3-PPARγ directly converts myoblasts into brown-like adipocytes
We then generated lentiviruses expressing M3-PPARγ, PPARγ and GFP. The myoblasts transduction rate was nearly 100% after two rounds of the viral infection at an MOI of 4. We confirmed the protein expression of PPARγ and M3-PPARγ at the expected size (Fig. 2a) in myoblasts collected 48 hr post-infection. Next, we tested human primary myoblasts derived from five individual muscle biopsies for adipogenesis in response to the adipogenic cocktail without PPARγ lentivirus infection and found that the basal adipogenic potential of the myoblasts differed among individuals. We then chose the myoblasts from three subjects with the least adipogenic potential to assess the modified PPARγ reprogramming efficiency. Cell reprogramming procedures are illustrated in Fig. 2b. The adipogenesis program was initiated by treatment with the adipogenic cocktail containing insulin, dexamethasone and IBMX. Myoblasts transduced with GFP remained fibroblast-like during the entire adipogenesis protocol. In contrast, cells transduced with PPARγ and M3-PPARγ started to accumulate lipid droplets from day 2 and became fully differentiated adipocytes between days10 and 14. Eventually, more than 90% of PPARγ and M3-PPARγ myoblasts differentiated into adipocytes (Fig. 3).
Figure 2.
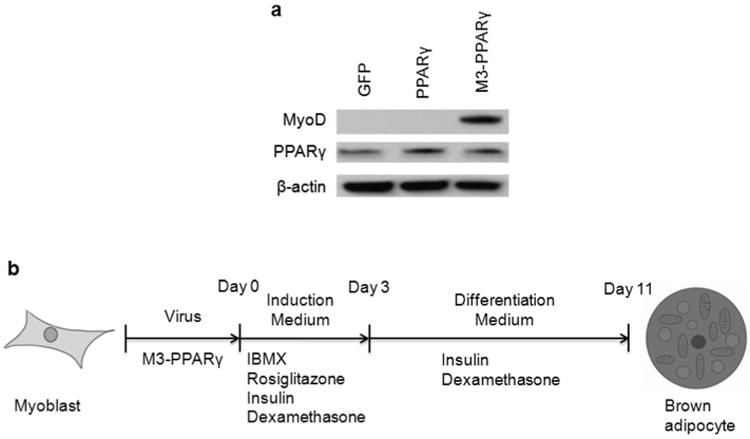
Lentivirus production and experiment scheme. (a) Western blot analysis of PPARγ and M3-PPARγ. Human myoblasts were transduced with lenti-GFP, lenti-PPARγ or lenti-M3-, and protein lysates were collected for Western blot by antibodies against MyoD, PPARγ, and β-actin (as a loading control) 48 h post-transduction. (b) Experimental scheme testing for the differentiation of human myoblasts into brown-like adipocytes. Human myoblasts were transduced with the indicated lentiviruses and cultured in induction medium containing IBMX, rosiglitazone, insulin and dexamethasone for 3 days. Then the cells were cultured in differentiation medium without IBMX and rosiglitazone for 8 days before analysis.
Figure 3.
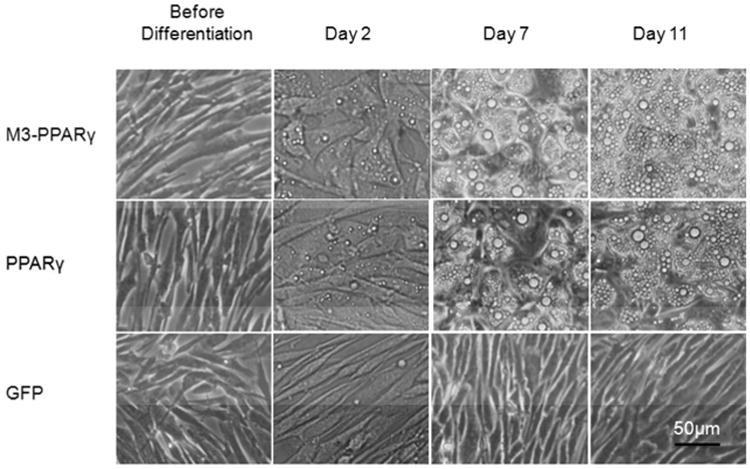
Representative microscopic images of myoblasts before and during adipocyte differentiation. Myoblasts, transduced with lentiviruses of GFP, PPARγ or M3-PPARγ, were examined with phase-contrast microscopy before differentiation and on the indicated days of differentiation. Scale bar: 50 μm for all images.
We next conducted qPCR to selectively measure the expression of genes for adipocytes and myocytes at differentiation day 11 in reference to the in vitro differentiated white adipocytes and BAT. Fatty acid binding protein 4 (FABP4), an adiposity marker, was barely expressed in GFP-expressing myoblasts, but was induced by ∼30,000 fold and 128,000 fold in PPARγ- and M3-PPARγ-reprogrammed cells, respectively. The expression pattern of another adiposity gene CIDEC or FSP27 was similar to that of FABP4. Leptin was induced by 1.5 and 18.6 fold in PPARγ and M3-PPARγ transduced cells, respectively, versus the GFP control (Fig. 4a). Interestingly, the adipogenic transcription factor C/EBPβ was induced by 4 and 9 fold in PPARγ and M3-PPARγ cells where PPARγ transcripts were less induced in M3-PPARγ (132 fold) than PPARγ-transduced cells (387 fold). For selective brown adipocyte genes (24), compared to the GFP control, PPARγ-overexpression induced brown fat-selective genes including CIDEA (46 fold), PRDM16 (2.8 fold) and UCP1 (47 fold). Remarkably, further induction was observed in M3-PPARγ cells for genes of CIDEA (2.4 fold), PRDM16 (5 fold) and UCP1 (1,057 fold) over PPARγ cells (Fig. 4b). Finally, we measured myocyte-specific genes myosin (25) and MyoD (26) and found their expressions were significantly reduced in M3-PPARγ cells by ∼0.6 fold (p <0.05 for both), but were not significantly reduced in PPARγ cells (Fig. 4c), compared to the GFP control. In reference to the white adipocytes differentiated from AdSVCs [i.e. induced white adipocytes (iWA)] and the perithyroid BAT, the expressions of most white adipocyte and adiposity markers in PPARγ and M3-PPARγ-induced adipocytes were higher than iWA except leptin. On the other hand, the expression of brown adipocyte markers UCP1, PRDM16 and CIDEA in the M3-PPARγ-reprogrammed adipocytes was significantly higher than that in iWA and PPARγ-induced adipocytes. As to UCP1 and PRDM16, their expression levels were close to the perithyroid BAT. These data showed that the M3-PPAR γ-reprogrammed adipocytes were expressing high levels of brown adipocyte genes.
Figure 4.
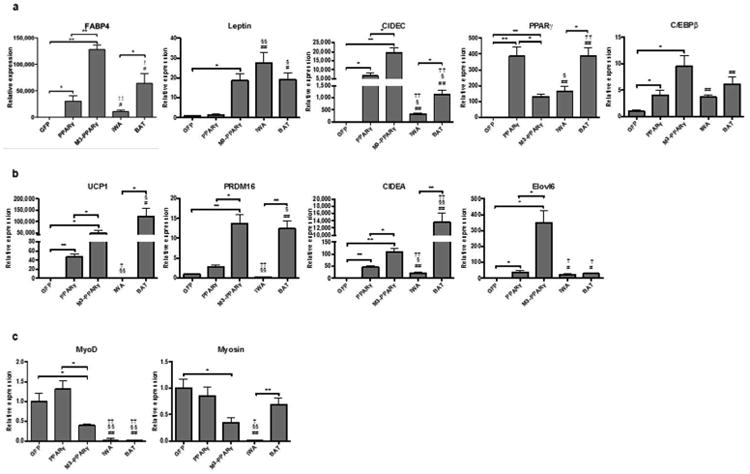
Gene expression analyses of human myoblasts transduced with lentiviruses of GFP, PPARγ or M3-PPARγ after treatment with the adipose differentiation cocktail, compared to the in vitro differentiated white adipocytes (iWA) and the perithyorid BAT. iWA are adipocytes differentiated from subcutaneous AdSVCs and BAT are biopsies of perithyroid brown adipose tissues. Total RNAs were isolated from cells on day 11 of differentiation or BAT. (a) Selective white and adiposity markers. (b) Selective brown adipocyte selective markers (c) Selective myocyte markers. Data are presented as mean ± SEM (n = 3 for myocytes and BAT; n = 4 for iWA); *p < 0.05, **p < 0.01; #p < 0.05, ##p < 0.01 versus GFP; §p < 0.05, §§p < 0.01 versus PPARγ; †p < 0.05, ††p < 0.01 versus M3-PPARγ.
UCP1 is considered to be both the molecular and functional marker of browning. We then conducted Western blotting analysis to determine the UCP1 protein expression. As shown in Fig. 5, UCP1 protein was not detectable in GFP cells, but the protein band became visible in the PPARγ-expressing cells. Strikingly, the protein was increased by 47 fold in M3-PPARγ cells versus PPARγ cells (p < 0.01).
Figure 5.
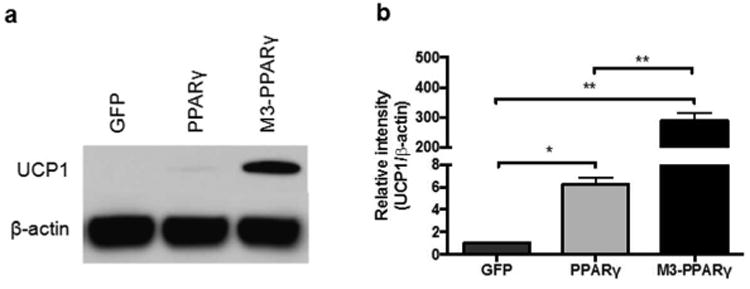
UCP1 protein expression analyses of human myoblasts transduced with GFP, PPARγ and M3-PPARγ 11 days after treatment of adipose differentiation cocktail. (a) Representative image of Western blot of UCP1 protein expression in differentiated cells and (b) quantification in reference to β-actin control. Data are presented as mean ± SEM (n=3); *p < 0.05, **p < 0.01.
Brown adipocytes are rich in mitochondria and the increase in mitochondria content is considered a typical sign of adipocyte browning. We then conducted MitoTracker fluorescence analysis by confocal microscopy in the cells transduced with GFP, PPARγ or M3-PPARγ. As depicted in Fig. 6, the GFP myoblasts remained a myoblast morphology and stained strongly for mitochondria, whereas the staining in the PPARγ-induced adipocytes was reduced by ∼71%. Significantly, the fluorescence intensity was increased by 3 fold in the M3-PPARγ group than the PPARγ group (p < 0.01). This result indicated that the M3-PPARγ induced mitobiogenesis.
Figure 6.
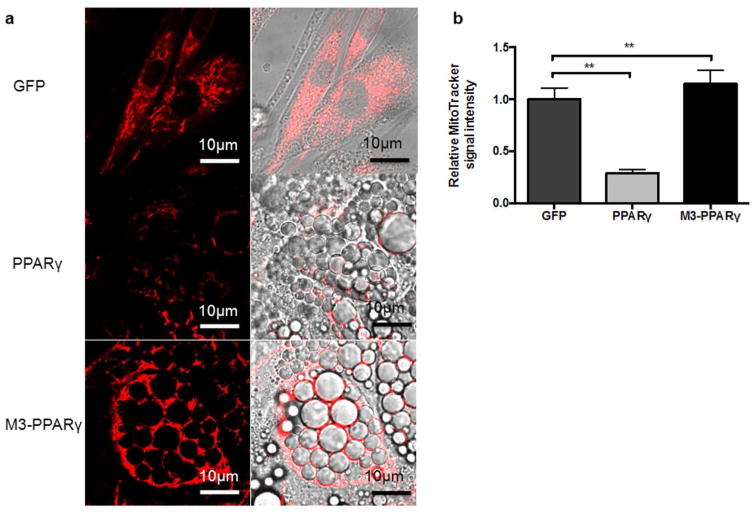
Induction of mitobiogenesis by M3-PPARγ. (a) Representative image of MitoTracker (red) staining of human myoblasts transduced with GFP, PPARγ and M3-PPARγ 11 days after treatment of adipose differentiation. (b) Quantitative fluorescence signal of MitoTracker staining. Data are presented as mean ± SEM; **p < 0.01 versus PPARγ (n = 10). Scale bar: 10 μm for all images.
Discussion
In this study, we show that human myoblasts can be converted into brown-like adipocytes through cellular reprogramming by super-activation of PPARγ signaling. Since the functionalities of the reprogrammed adipocytes have yet to be characterized in more detail, we call our reprogramming-derived, high UCP1-expressing cells brown-like adipocytes. Although M3-PPARγ appeared to induce brown adipocyte gene expression, especially UCP1 to a great extent, it induced white adipose genes, such as leptin, as well. Whether M3-PPARγ selectively promotes browning is not definite at this stage, so we define the M3-PPARγ-mediated, general enhanced PPARγ signaling above the degree of activation by ligand on wild-type PPARγ as “super-activation”. Although brown or brown-like adipocytes have been obtained from human pluripotent stem cells through over-expression of browning factors (12) and differentiation (13), this is the first report, to our knowledge, demonstrating that human brown-like adipocytes can be derived through cellular reprogramming of non-pluripotent somatic cells.
Since the breakthrough discovery that adult skin fibroblasts can be reprogrammed into induced pluripotent stem cells (iPSCs)(27), the great plasticity of cell conversion from one type to another has been well recognized. Moreover, cell lineage conversion occurs amenably between developmentally related types of cells due to a lower epigenetic barrier (28, 29, 30). Since brown adipocytes share common precursor cells with skeletal myocytes (31, 32, 33), we reasoned that human myoblasts would be a type of somatic cells well-suited for the reprogramming study to derive brown-like adipocytes. Although many transcription factors, hormones and growth factors (34, 35, 36, 37) are reported to induce browning in murine model systems, only a few have shown browning activities in human cells. Exceptionally, activation of PPARγ signaling reproducibly induces UCP1 expression in human adipocytes. We thus speculated that super-activation of PPARγ would promote human adipocyte browning and engineered a M3-PPARγ fusion protein, which indeed activated the reporter gene activity by 3 fold more than the wild-type PPARγ (Fig 1). Although both PPARγ and M3-PPARγ induced adipogenesis, the super-active PPARγ drastically stimulated the UCP1gene expression by ∼1,050 fold and the protein expression by ∼47 fold versus the PPARγ group, and induced significant mitobiogenesis. Moreover, using in vitro differentiated white adipocytes and perithyroid BAT as references, we showed that the M3-PPARγ-reprogrammed cells express brown adipocyte markers at the levels much closer to BAT than the white adipocytes do. Collectively, we demonstrate the browning characteristics of the M3-PPARγ-reprogrammed adipocytes at both the molecular and cellular levels.
Although the browning approach taken in the study is artificial, our success in reprogramming human myoblasts into brown-like adipocytes has several implications. Firstly, we demonstrate the feasibility of deriving brown-like cells through cellular reprogramming of somatic cells, providing a new cell model for browning research. Secondly, our study shows that the PPARγ super-activation can significantly promote browning, revealing that human adipocytes have a great capacity for browning. Although PPARγ activation by thiazolidinediones can cause browning of adipocytes in vitro, significant adipocyte browning at clinical doses of the medication has not been noticed in human subjects. Thus, a super-activation of PPARγ may be needed for browning in humans. However, whether an enhancement of the PPARγ signaling pathway would promote further browning was unknown. Fusion of VP16, a viral transacting domain, to PPARγ (38) can enhance PPARγ's transcriptional activity; however, whether it would induce adipocyte browning has not been studied. In this study, we chose the M3 domain based on the following considerations: 1) the M3 domain will more likely maintain its transacting activity in myoblasts where MyoD is expressed natively and 2) the M3 domain appears to possess stronger transacting activity than VP16 for cellular reprogramming (19). The mechanism by which M3-fusion increases PPARγ signaling is not clear. Whether the addition of M3 domain non-specifically increases PPARγ signaling or preferentially activates browning genes is an important question to the understanding of the PPARγ-mediated browning. Presumably, the addition of M3 to the N-terminus of PPARγ may enable PPARγ to recruit more or new co-activators to target genes or to facilitate the chromatin accessibility to co-transcription factors (19). Further investigation may lead to new strategies to selectively activate browning genes.
In summary, we have derived human brown-like adipocytes through cellular reprogramming of somatic cells, providing a new cell model to study adipocyte browning and opening up a new avenue towards the derivation of brown adipocytes.
Supplementary Material
Table S1. Primers Sequences for Q-PCR Analysis
What is already known about this subject?
Adipocyte browning is considered a new anti-obesity strategy and has been extensively studied in rodent systems.
PPARγ agonist treatment induces browning of mouse and human adipocytes in vitro; however, whether enhancement of PPARγ signaling will promote a deeper browning is unknown.
Human somatic cells are known to possess great plasticity and are amenable for conversion through cellular reprogramming.
What does this study add?
Generation of a synthetic molecular M3-PPARγ by fusing of M3, the transacting domain of MyoD, to PPARγ, which is constitutively super-active and remains responsive to its ligand rosiglitazone.
Compared to PPARγ, M3-PPARγ can directly convert human myoblasts into brown-like adipocytes with a drastic UCP1 induction at gene and protein expression levels by ∼ 1,050 and 47 fold, respectively, and increased mitochondria content by 4 fold.
Our studies demonstrate the feasibility of obtaining human brown-like adipocytes through cellular reprogramming from myoblasts, providing a new cell model for research about browning and its mechanisms.
Acknowledgments
We thank Nobuaki Kikyo for the M3-Oct4 construct, Geoffrey Girnun for the PPRE 3x construct, Mi-jeong Lee for advice on UCP1 protein detection, Richard Horenstein for muscle biopsy. This work was partly supported by Maryland Stem Cell Research Fund, NIH grants NORC of Maryland (DK072488), Baltimore Diabetes and Research Teaching Center (P60 DK79637), National Natural Science Foundation of China (Grant award 8137094) and the Geriatric Research, Education and Clinical Center, Baltimore Veterans Affairs Health Care Center.
Funding: Maryland Stem Cell Research Fund, NIH grants to Mid-Atlantic NORC (DK072488) and to Baltimore Diabetes and Research Teaching Center (P60 DK79637), the National Natural Science Foundation of China (Grant award 81370947) and the Geriatric Research, Education and Clinical Center, Baltimore Veterans Affairs Health Care Center, the Clinical Nutrition Research Unit of Maryland (DK072488).
Footnotes
Disclosure: The authors declared no conflict of interest
References
- 1.Flegal KM, Graubard BI, Williamson DF, Gail MH. Cause-specific excess deaths associated with underweight, overweight, and obesity. JAMA. 2007;298:2028–2037. doi: 10.1001/jama.298.17.2028. [DOI] [PubMed] [Google Scholar]
- 2.Nedergaard J, Cannon B. The changed metabolic world with human brown adipose tissue: therapeutic visions. Cell metabolism. 2010;11:268–272. doi: 10.1016/j.cmet.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 3.Boss O, Farmer SR. Recruitment of brown adipose tissue as a therapy for obesity-associated diseases. Front Endocrinol (Lausanne) 2012;3:14. doi: 10.3389/fendo.2012.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ravussin E, Galgani JE. The implication of brown adipose tissue for humans. Annu Rev Nutr. 2011;31:33–47. doi: 10.1146/annurev-nutr-072610-145209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cinti S. The adipose organ at a glance. Dis Model Mech. 2012;5:588–594. doi: 10.1242/dmm.009662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Townsend KL, Tseng YH. Brown fat fuel utilization and thermogenesis. Trends Endocrinol Metab. 2014;25:168–177. doi: 10.1016/j.tem.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dulloo AG. Translational issues in targeting brown adipose tissue thermogenesis for human obesity management. Ann N Y Acad Sci. 2013;1302:1–10. doi: 10.1111/nyas.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 9.Vosselman MJ, van der Lans AA, Brans B, Wierts R, van Baak MA, Schrauwen P, et al. Systemic beta-adrenergic stimulation of thermogenesis is not accompanied by brown adipose tissue activity in humans. Diabetes. 2012;61:3106–3113. doi: 10.2337/db12-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frontini A, Cinti S. Distribution and development of brown adipocytes in the murine and human adipose organ. Cell metabolism. 2010;11:253–256. doi: 10.1016/j.cmet.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 11.Cannon B, Nedergaard J. Yes, even human brown fat is on fire! J Clin Invest. 2012;122:486–489. doi: 10.1172/JCI60941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahfeldt T, Schinzel RT, Lee YK, Hendrickson D, Kaplan A, Lum DH, et al. Programming human pluripotent stem cells into white and brown adipocytes. Nat Cell Biol. 2012;14:209–219. doi: 10.1038/ncb2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishio M, Yoneshiro T, Nakahara M, Suzuki S, Saeki K, Hasegawa M, et al. Production of functional classical brown adipocytes from human pluripotent stem cells using specific hemopoietin cocktail without gene transfer. Cell metabolism. 2012;16:394–406. doi: 10.1016/j.cmet.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 14.Pisani DF, Djedaini M, Beranger GE, Elabd C, Scheideler M, Ailhaud G, et al. Differentiation of Human Adipose-Derived Stem Cells into “Brite” (Brown-in-White) Adipocytes. Front Endocrinol (Lausanne) 2011;2:87. doi: 10.3389/fendo.2011.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elabd C, Chiellini C, Carmona M, Galitzky J, Cochet O, Petersen R, et al. Human multipotent adipose-derived stem cells differentiate into functional brown adipocytes. Stem cells. 2009;27:2753–2760. doi: 10.1002/stem.200. [DOI] [PubMed] [Google Scholar]
- 16.Petrovic N, Walden TB, Shabalina IG, Timmons JA, Cannon B, Nedergaard J. Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J Biol Chem. 2010;285:7153–7164. doi: 10.1074/jbc.M109.053942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ohno H, Shinoda K, Spiegelman BM, Kajimura S. PPARgamma agonists induce a white-to-brown fat conversion through stabilization of PRDM16 protein. Cell metabolism. 2012;15:395–404. doi: 10.1016/j.cmet.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirai H, Katoku-Kikyo N, Karian P, Firpo M, Kikyo N. Efficient iPS cell production with the MyoD transactivation domain in serum-free culture. PloS one. 2012;7:e34149. doi: 10.1371/journal.pone.0034149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirai H, Katoku-Kikyo N, Keirstead SA, Kikyo N. Accelerated direct reprogramming of fibroblasts into cardiomyocyte-like cells with the MyoD transactivation domain. Cardiovasc Res. 2013;100:105–113. doi: 10.1093/cvr/cvt167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirai H, Tani T, Katoku-Kikyo N, Kellner S, Karian P, Firpo M, et al. Radical acceleration of nuclear reprogramming by chromatin remodeling with the transactivation domain of MyoD. Stem cells. 2011;29:1349–1361. doi: 10.1002/stem.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang RZ, Lee MJ, Hu H, Pray J, Wu HB, Hansen BC, et al. Identification of omentin as a novel depot-specific adipokine in human adipose tissue: possible role in modulating insulin action. Am J Physiol Endocrinol Metab. 2006;290:E1253–1261. doi: 10.1152/ajpendo.00572.2004. [DOI] [PubMed] [Google Scholar]
- 22.Yang RZ, Lee MJ, Hu H, Pollin TI, Ryan AS, Nicklas BJ, et al. Acute-Phase Serum Amyloid A: An Inflammatory Adipokine and Potential Link between Obesity and Its Metabolic Complications. PLoS Med. 2006;3:e287. doi: 10.1371/journal.pmed.0030287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee MJ, Wu Y, Fried SK. A modified protocol to maximize differentiation of human preadipocytes and improve metabolic phenotypes. Obesity. 2012;20:2334–2340. doi: 10.1038/oby.2012.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu J, Bostrom P, Sparks LM, Ye L, Choi JH, Giang AH, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 2012;150:366–376. doi: 10.1016/j.cell.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng W, Brooks B. Identification of dynamical correlations within the myosin motor domain by the normal mode analysis of an elastic network model. J Mol Biol. 2005;346:745–759. doi: 10.1016/j.jmb.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 26.Weintraub H. The MyoD family and myogenesis: redundancy, networks, and thresholds. Cell. 1993;75:1241–1244. doi: 10.1016/0092-8674(93)90610-3. [DOI] [PubMed] [Google Scholar]
- 27.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 28.Sancho-Martinez I, Baek SH, Izpisua Belmonte JC. Lineage conversion methodologies meet the reprogramming toolbox. Nat Cell Biol. 2012;14:892–899. doi: 10.1038/ncb2567. [DOI] [PubMed] [Google Scholar]
- 29.Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bar-Nur O, Russ HA, Efrat S, Benvenisty N. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell. 2011;9:17–23. doi: 10.1016/j.stem.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 31.Atit R, Sgaier SK, Mohamed OA, Taketo MM, Dufort D, Joyner AL, et al. Beta-catenin activation is necessary and sufficient to specify the dorsal dermal fate in the mouse. Dev Biol. 2006;296:164–176. doi: 10.1016/j.ydbio.2006.04.449. [DOI] [PubMed] [Google Scholar]
- 32.Timmons JA, Wennmalm K, Larsson O, Walden TB, Lassmann T, Petrovic N, et al. Myogenic gene expression signature establishes that brown and white adipocytes originate from distinct cell lineages. Proc Natl Acad Sci U S A. 2007;104:4401–4406. doi: 10.1073/pnas.0610615104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961–967. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bordicchia M, Liu D, Amri EZ, Ailhaud G, Dessi-Fulgheri P, Zhang C, et al. Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. J Clin Invest. 2012;122:1022–1036. doi: 10.1172/JCI59701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tseng YH, Kokkotou E, Schulz TJ, Huang TL, Winnay JN, Taniguchi CM, et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature. 2008;454:1000–1004. doi: 10.1038/nature07221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koncarevic A, Kajimura S, Cornwall-Brady M, Andreucci A, Pullen A, Sako D, et al. A novel therapeutic approach to treating obesity through modulation of TGFbeta signaling. Endocrinology. 2012;153:3133–3146. doi: 10.1210/en.2012-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bostrom P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature. 2012;481:463–468. doi: 10.1038/nature10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Y, Lazar MA. Differential gene regulation by PPARgamma agonist and constitutively active PPARgamma2. Mol Endocrinol. 2002;16:1040–1048. doi: 10.1210/mend.16.5.0825. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers Sequences for Q-PCR Analysis