

Abstract
AIM
To identify and understand the relationship between co-expression pattern and clinic traits in uveal melanoma, weighted gene co-expression network analysis (WGCNA) is applied to investigate the gene expression levels and patient clinic features. Uveal melanoma is the most common primary eye tumor in adults. Although many studies have identified some important genes and pathways that were relevant to progress of uveal melanoma, the relationship between co-expression and clinic traits in systems level of uveal melanoma is unclear yet. We employ WGCNA to investigate the relationship underlying molecular and phenotype in this study.
METHODS
Gene expression profile of uveal melanoma and patient clinic traits were collected from the Gene Expression Omnibus (GEO) database. The gene co-expression is calculated by WGCNA that is the R package software. The package is used to analyze the correlation between pairs of expression levels of genes. The function of the genes were annotated by gene ontology (GO).
RESULTS
In this study, we identified four co-expression modules significantly correlated with clinic traits. Module blue positively correlated with radiotherapy treatment. Module purple positively correlates with tumor location (sclera) and negatively correlates with patient age. Module red positively correlates with sclera and negatively correlates with thickness of tumor. Module black positively correlates with the largest tumor diameter (LTD). Additionally, we identified the hug gene (top connectivity with other genes) in each module. The hub gene RPS15A, PTGDS, CD53 and MSI2 might play a vital role in progress of uveal melanoma.
CONCLUSION
From WGCNA analysis and hub gene calculation, we identified RPS15A, PTGDS, CD53 and MSI2 might be target or diagnosis for uveal melanoma.
Keywords: weighted gene co-expression network analysis, microarray data, gene ontology
INTRODUCTION
Uveal melanoma is an aggressive cancer and can cause a blind and death from metastasis. About 50% of uveal melanoma patients develop metastases within ten years from diagnosis and their median survival is 5 to 7mo after confirming case of metastatic lesions[1]. Although the improvements in diagnosis and the development of more effective local therapies for primary tumors, the rate of metastatic death and poorly prognosis remains unchanged[2]. Previous study utilized the individual differential expression levels of gene or protein and clinic phenotype to understand the mechanism of uveal melanoma[3]–[5]. With the development of proteomic technology and microarray assay, the recent study turned to perform high through-put method to analyze the proteome and genome of uveal melanoma[6]–[8]. Although many studies identified many markers for progress of uveal melanoma, the relationship between expression levels of genes and clinic traits were unclear yet. The data of microarray would provide more information to study. The high through-put data shows global view for understanding the cells or issue. As far, some microarray data of uveal melanoma has been collected by Gene Expression Omnibus (GEO) database[9]. We could construct a biological network in systems level from GEO database.
In this study, GSE27831 microarray data that contains 29 samples are downloaded from GEO database. For investigating the relationships between expression levels of genes and clinic traits, we applied Weighted Gene Co-Expression Network Analysis (WGCNA)[10],[11]. That is a technique that has uncovered patterns of gene co-activity correlated to clinic traits and corresponding to functional pathways[12].
The result shows that the WGCNA identified seven modules and four modules significantly correlated with clinic traits. From network connectivity calculation, the hub gene in each module has been identified. We show that RPS15A, PTGDS, CD53 and MSI2 were hug genes and might play a vital role in clinic diagnose. In particular, RPS15A is closely associated with radiotherapy and MSI2 is closely associated with largest tumor diameter (LTD). These two clinic traits are important to tumor diagnose and therapy.
MATERIALS AND METHODS
Dataset Collected
Expression profiles of mRNA for uveal melanoma samples were collected from GSE27831 (from NCBI GEO database http://www.ncbi.nlm.nih.gov/geo/)[8]. The microarray was used for screenings of GeneChip HumanGenome U133plus2 arrays (Affymetrix, Santa Clara, CA, USA). The mRNA dataset contain 29 unique samples from uveal melanoma patients. Data were preprocessed following the RMA procedure of Console software normalization (http://www.affymetrix.com/). The clinicopathological characteristics of patient samples are downloaded from the study by Gangemi et al[8]. The table of clinicopathological characteristics of patient samples is listed in Table 1.
Table 1. Clinicopathological characteristics of uveal melanoma patient samples.
| Accession No. | Gender | Age | Thickness | LTD (mm) | DFS months | Sclera | Met | Treatment | mda-9 |
| GSM685471 | M | 78 | 7 | 6 | 67 | Y | N | None | L |
| GSM685474 | F | 61 | 10 | 14 | 55 | N | N | Proton | L |
| GSM685475 | F | 74 | 5 | 10 | 41 | Na | N | Proton | H |
| GSM685522 | M | 70 | 16 | 17 | 55 | N | N | None | H |
| GSM686961 | M | 60 | 9 | 11 | 52 | N | N | None | L |
| GSM687001 | F | 82 | 3 | 2 | 20 | Y | N | Proton | L |
| GSM686962 | M | 74 | 7 | 17 | 40 | N | N | Proton | H |
| GSM686963 | M | 33 | 7.56 | 15 | 15 | Y | N | None | L |
| GSM686984 | F | 65 | NA | 15 | 36 | Y | N | None | H |
| GSM686987 | F | 76 | 5.3 | 12 | 44 | Y | N | None | L |
| GSM686986 | M | 59 | 5.2 | 14 | 43 | Y | N | None | L |
| GSM686990 | M | 51 | NA | 16 | 40 | Y | N | None | L |
| GSM685650 | M | 85 | 12 | 16 | 48 | Y | N | None | H |
| GSM685651 | M | 48 | 8 | 12 | 57 | N | N | None | L |
| GSM685470 | F | 77 | 7 | 9 | 54 | N | N | None | L |
| GSM686991 | M | 62 | 4 | 20 | 41 | Y | N | None | L |
| GSM687002 | F | 55 | 11 | 13 | 48 | N | N | None | L |
| GSM687004 | F | 71 | NA | 16 | 42 | Y | N | Proton | L |
| GSM685473 | M | 84 | 14 | 25 | 21 | Y | Y | None | H |
| GSM685523 | M | 74 | 6 | 13 | 33 | N | Y | Proton | H |
| GSM685601 | M | 64 | 15 | 23 | 31 | Y | Y | None | H |
| GSM686985 | F | 69 | 4.9 | 16 | 17 | Y | Y | None | H |
| GSM686988 | M | 51 | 10.5 | 15 | 19 | Y | Y | None | H |
| GSM686989 | M | 80 | 16 | 12 | 19 | N | Y | None | L |
| GSM685603 | M | 61 | 6 | 11 | 31 | N | Y | None | H |
| GSM685652 | F | 69 | 13 | 12 | 25 | N | Y | None | H |
| GSM685602 | F | 74 | 7 | 9 | 18 | N | Y | None | H |
| GSM685472 | M | 66 | 6 | 14 | 17 | N | Y | None | L |
| GSM687003 | F | 42 | 10 | 12 | 51 | N | Y | None | H |
DFS: Disease free survival; NA: Not available; LTD: Largest tumor diameter; Met: Metastasis; mda-9: Expression of mda-9; L: Low expression; H: High expression.
Construct Co-expression Network
The WGCNA is employed to identify the co-expression modules[10],[12],[13]. WGCNA is implemented in the R software package (http://www.r-project.org/). Co-expression methodology is typically used for studying relationship between gene expression levels. WGCNA start from the level of thousands of genes, identifies modules of co-expressed genes, and relates these modules to clinical variables and gene ontology (GO) information. Modules are defined in an unbiased fashion and initially denoted by colors. Grey denotes background genes outside of modules. Highly connective module genes are represented and summarized by their first principal component, and it has been called the module eigengene or ME[11].
The data set used for network construction consisted of 29 Affymetrix HG-U133plus2 microarrays surveying gene expression with 54 675 probe sets. For computational reasons, network analysis was limited 4000 probe sets with greater variance (note: although some genes are represented by multiple probe sets and other probe sets are not fully annotated, for consistency we refer to probe sets as “genes” through this study). When evaluating the significance of the module correlations of WGCNA, we corrected the P-values for the number of modules and the number of tested phenotypic traits. The network analysis was applied to uveal melanoma data set, a signed weighted network adjacency matrix is defined as:
Where xi and xj represent the expression value of probes which are numeric vector whose entries report the β values across the individuals. Note that the adjacency aij is a number between 0 and 1 that is a monotonically increasing function of the correlation coefficient. The power b is a soft-thersholding parameter that can be used to emphasize high positive correlations at the expense of low correlations. A major advantage of weighted correlation networks is that they are highly robust with regard to the choice of b[10].
In co-expression network, the genes represent the nodes and the aij represent the edges. The value of aij represents the strength connectivity of the edges.
Gene Ontology Enrichment
The annotations and functions of proteins were obtained from DAVID Bioinformatics Resources 6.7 (http://david.abcc.ncifcrf.gov/home.jsp)[14],[15]. GO terms assigned a Benjamini-Hochberg adjusted P-value of less than 0.05 by DAVID were deemed to be enriched over the background gene set.
Modules Membership Measure and Statistic Analysis Module-trait associations were estimated using the correlation between the module eigengene and the phenotype (clinic traits), which allows easy identification of expression set (module) highly correlated to the phenotype. For each expression profile, Gene Significance (GS) was calculated as the absolute value of the correlation between expression profile and each trait; module membership (MM) was defined as the correlation of expression profile and each module eigengene. MM, also known as eigengene-based (eigengene: One of a set of right singular vectors of a genes x samples matrix that tabulates, e.g. the mRNA or gene expression of the genes across the samples) connectivity, is a measure of intramoduler connectivity. MM is defined as:
MM(i)=cor(x(i), ME)
Where x(i) represents the expression profile of ith gene and ME represents the eigengene (first principal component) of the given module. We used the MM measure to select module genes for a GO enrichment analysis. For studying more significant association of MM, the threshold of P<0.1 was considered as significance value.
RESULTS
Constructing Gene Co-expression Networks in Uveal Melanoma
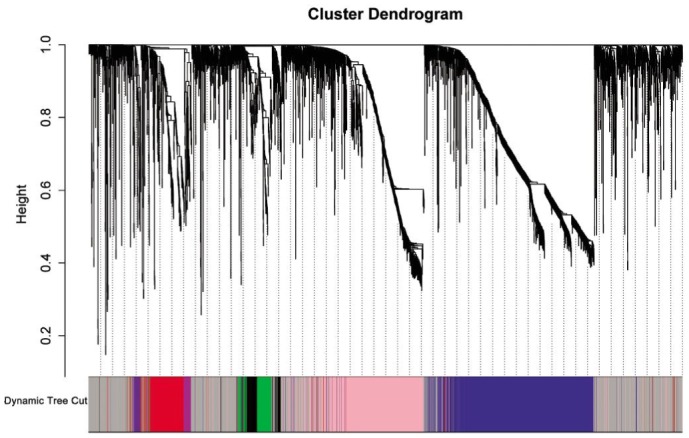
We constructed gene expression networks from microarray data consisting of 29 uveal melanoma samples. All possible pairwise correlations were calculated for 4000 genes in uveal melanoma and converted into measures of connection strength by taking their absolute values and raising them to a power, β[10]. To identify modules of coexpressed genes, we searched for genes or high “topological overlap” (TO). We calculated TO and clustered genes on this basis for uveal melanoma. The results show that identifying seven distinct gene co-expression modules in uveal melanoma (Figure 1).
Figure1. Network analysis of gene expression in uveal melanoma identifies distinct modules of co-expression genes. The dendrogram produced by average linkage hierarchical clustering of 4000 genes based on TO.
Gene Co-expression Modules Correspond to Clinic Traits
The clinic traits are utilized another uveal melanoma study and the dataset was provided by GEO database. The clinic traits were listed in Table 1. And we removed some information that was useless in this study.
Sets of genes (modules) with common expression patterns that were associated with particular traits were identified based on the correlation between ME and clinic traits (Figure 2). We identified four modules that significantly associated with uveal melanoma clinic traits (P<0.1). Modules blue was correlated positively to treatment [ME(blue): treatment r=0.44, P=0.02]. Module purple was correlated positively to sclera and negatively to age [ME(purple): age r=-0.42, P=0.02; sclera r=0.37, P=0.05]. Module red was correlated positively to sclera and negatively to thickness [ME(red): sclera r=0.32, P=0.09; thickness r=-0.32, P=0.09]. Module black was correlated positively to largest diameter [ME(black): largest diameter r=0.36, P=0.06].
Figure 2. Correlation matrix of module eigengene values obtained for uvealmalanoma and clinic traits.

WGCNA groups mRNAs into modules based on patterns of their co-expression. Each of the modules was labelled with a unique color as an identifier. Eight modules were identified; each module eigengene was tested for correlation with clinic traits. Within each cell, upper values are correlation coefficients between module eigengene and the traits; lower values are the correspondent P-value.
Gene Ontology Enrichment of Modules Associated with Clinic Traits
The GO BP FAT function of DAVID to determine GO biological process enriched in the genes in all modules. Genes expressed in each dataset were set as the background measurement for DAVID. The Table 2 showed the GO analysis of modules that associated with clinic traits. The table showed the top 5 of GO biological process.
Table 2. List of the top GO terms in the most significant DAVID functional clusters for each network module.
| Module | Top terms | No. of genes in ME | P | FDR |
| Blue | GO:0006414: translational elongation | 1030 | 1.48e-48 | 2.54E-95 |
| GO:0006412: translation | 2.88e-97 | 4.94e-49 | ||
| GO:0006091: generation of precursor metabolites and energy | 1.59e-37 | 2.72e-34 | ||
| GO:0006119: oxidative phosphorylation | 3.51e-36 | 6.02e-33 | ||
| GO:0022900: electron transport chain | 4.51e-36 | 7.73e-26 | ||
| Purple | GO:0030036: actin cytoskeleton organization | 49 | 0.001889 | 2.673499 |
| GO:0030029: actin filament-based process | 0.002387 | 3.366931 | ||
| GO:0007010: cytoskeleton organization | 0.003335 | 4.674312 | ||
| GO:0006936: muscle contraction | 0.005533 | 7.642884 | ||
| GO:0055002: striated muscle cell development | 0.006677 | 9.154233 | ||
| Red | GO:0009611: response to wounding | 290 | 1.96E-20 | 3.32E-17 |
| GO:0006954: inflammatory response | 7.91E-17 | 1.89E-13 | ||
| GO:0006952: defense response | 1.90E-16 | 3.77E-13 | ||
| GO:0002504: antigen processing and presentation of peptide or polysaccharide antigen via MHC class II | 8.16E-13 | 1.38E-09 | ||
| GO:0006955: immune response | 1.40E-12 | 2.37E-09 | ||
| Black | GO:0006355:regulation of transcription, DNA-dependent | 119 | 1.10E-04 | 0.172753 |
| GO:0010629:negative regulation of gene expression | 1.40E-04 | 0.220258 | ||
| GO:0051252:regulation of RNA metabolic process | 1.54E-04 | 0.242911 | ||
| GO:0016481:negative regulation of transcription | 2.92E-04 | 0.459812 | ||
| GO:0045449:regulation of transcription | 4.00E-04 | 0.629367 |
FDR: False discovery rate; ME: Module eigengene.
The GO results showed that the different modules were corresponded to different biological processes. From the Figure 2, we found that some traits have no correlated with any modules, such as gender, age, DFS, metastasis and expression levels of mda-9. In this study, we analyzed the four modules and tried to investigate the hub genes in each module network.
Inner Module Structure and Key Genes
To determine genes that are centrally located in the modules that are associated with clinic traits, we calculated the intramodular connectivity. The intramodular connectivity (k-within) was calculated for each gene by summing the connection strengths with other module genes and dividing this number by the maximum intramodular connectivity. Genes with high intramodular connectivity are informally referred to as intramodular hub genes. The hub genes in the four modules were listed in Table 3.
Table 3. The hub genes of four modules.
| Module | Hub gene names | Uniprot accession | Description | Association traits | Intramodule connectivity (k) |
| Blue | RPS15A | P62244 | 40S ribosomal protein S15a | Radiotherapy treatment | 284.57 |
| Purple | PTGDS | P41222 | Prostaglandin-H2 D-isomerase | Age and Sclera | 7.88 |
| Red | CD53 | P19397 | Leukocyte surface antigen CD53 | Thickness and sclera | 40.68 |
| Black | MSI2 | Q96DH6 | RNA-binding protein Musashi homolog 2 | Largest diameter | 16.73 |
The modules that closely associated with clinic traits might be very useful for uveal melanoma therapy and diagnose. The hub genes might provide signature target for further prognosis.
DISCUSSION
In this study we utilize gene expression data to identify genes involved in uveal melanoma. The WGCNA was employed to explore the relationship between uveal melanoma transcriptome and clinic traits. WGCNA has many advantages over traditional methods for differential expression analysis, including a focus on co-expression patterns thereby allowing for identification of biologically-relevant modules consisting of related genes, detection of hub genes that may eventually serve as targets for therapeutic modulation, and reducing data allowing for direct association analysis with disease-related variables. We identified 4 co-expression modules (gene networks) that relate to clinic traits status[16]–[20].
Previous study found that expression levels of mda-9 relate with survival time and uveal melanoma metastasis. Unfortunately, we could not found any modules associated with expression levels of mda-9, metastasis and survival times. However, this study provided four modules correlation with other traits and would find novel therapy targets or prognosis targets for uveal melanoma.
The Table 2 shows the GO enrichment of the genes in modules. The module blue is associated with treatment. The treatment of this study is radiotherapy which utilizes proton beam. Therefore, the GO enrichment of module blue might reflect the irradiation response to uveal melanoma. The top 5 biological processes are mainly involved in translation and energy metabolism. Furthermore, the hub gene of module blue is RPS15A which is one of subunits of ribosome protein. In our previous study, we also found that the expression levels of energy metabolism genes were closely associated with radiation[6],[7],[21]. RPS15A is a highly conserved protein that promotes mRNA/ribosome interactions early in translation. Recent evidence showed that RPS15A could stimulate growth in yeast, plant and human lung carcinoma[22]. Another study indicates that RPS15A may play a key role in hepatic cancer cell growth[23]. Module blue contains many ribosome proteins which mainly were involved in protein synthesis. Module blue positively correlating with radiotherapy treatment indicates that irradiation promote up-regulated of translations and energy metabolism genes. RPS15A is a hub gene that strongly connects with other genes. So, RPS15A might be an important target of radiotherapy. Besides, RPS15A is also proved by recent research that RPS15A may play a prominent role in heptocarcinogenesis and serve as a potential therapeutic target in hepatocellular carcinoma[24].
The genes in module purple correlated with age and sclera. The GO analysis of module purple was mainly involved in cytoskeleton organization. And this module negatively correlates with and positively correlates with sclera. The hub gene is PTGDS that expressed in the eye and secreted into the aqueous humor (http://www.uniprot.org/uniprot/P41222). Therefore, we inferred that some tissue specific genes might highly express in uveal melanoma.
The genes in module red were mainly involved in inflammatory response and immune response. And the module red correlated with thickness of tumor and tumor location (sclera). The GO analysis and clinic traits indicated that inflammatory and immune response would relate with thickness of tumor and tumor location. The hub gene CD53 might contribute to cell survival in poorly vascularized regions of the tumor mass[25].
Module black is positively associated with LTD and the genes in this module are mainly involved in regulation of transcription. The function of hub gene MSI2 is involved in regulation of the expression of target mRNAs[26]. The LTD is an important indicator for tumor relapse [27]. And the value of LTD is a predictor of survival after treatment of posterior uveal melanoma [4]. After conservative therapy, LTD is associated with increased risk of local tumor recurrence. In this study, we found the relationships between molecular level function and LTD. The regulation of transcription biological process might play a vital role in LTD. MSI2 as a hub gene in black module might be a marker for LTD.
Acknowledgments
Foundations: Supported by the National Natural Science Foundation of China (No. 81271019; No.61463046); Gansu Province Science Foundation for Youths (No.145RJYA282)
Conflicts of Interest: Shi K, None; Bing ZT, None; Cao GQ, None; Guo L, None; Cao YN, None; Jiang HO, None; Zhang MX, None.
REFERENCE
- 1.Singh AD, Bergman L, Seregard S. Uveal melanoma: epidemiologic aspects. Ophthalmol Clin North Am. 2005;1875-84(1):viii. doi: 10.1016/j.ohc.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 2.Damato B. Does ocular treatment of uveal melanoma influence survival? Br J Cancer. 2010;103(3):285–290. doi: 10.1038/sj.bjc.6605765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cree IA. Cell cycle and melanoma-two different tumours from the same cell type. J Pathol. 2000;191(2):112–114. doi: 10.1002/(SICI)1096-9896(200006)191:2<112::AID-PATH592>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 4.Damato B, Coupland SE. A reappraisal of the significance of largest basal diameter of posterior uveal melanoma. Eye (Lond) 2009;23(12):2152–2162. doi: 10.1038/eye.2009.235. [DOI] [PubMed] [Google Scholar]
- 5.Mallikarjuna K, Pushparaj V, Biswas J, Krishnakumar S. Expression of epidermal growth factor receptor, ezrin, hepatocyte growth factor, and c-Met in uveal melanoma: an immunohistochemical study. Curr Eye Res. 2007;32(3):281–290. doi: 10.1080/02713680601161220. [DOI] [PubMed] [Google Scholar]
- 6.Wang F, Bing Z, Zhang Y, Ao B, Zhang S, Ye C, He J, Ding N, Ye W, Xiong J, Sun J, Furusawa Y, Zhou G, Yang L. Quantitative proteomic analysis for radiation-induced cell cycle suspension in 92-1 melanoma cell line. J Radiat Res. 2013;54(4):649–662. doi: 10.1093/jrr/rrt010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan LB, Shi K, Bing ZT, Sun YL, Shen Y. Proteomic analysis of energy metabolism and signal transduction in irradiated melanoma cells. Int J Ophthalmol. 2013;6(3):286–294. doi: 10.3980/j.issn.2222-3959.2013.03.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gangemi R, Mirisola V, Barisione G, Fabbi M, Brizzolara A, Lanza F, Mosci C, Salvi S, Gualco M, Truini M, Angelini G, Boccardo S, Cilli M, Airoldi I, Queirolo P, Jager MJ, Daga A, Pfeffer U, Ferrini S. Mda-9/syntenin is expressed in uveal melanoma and correlates with metastatic progression. PLoS ONE. 2012;7(1):e29989. doi: 10.1371/journal.pone.0029989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barrett T, Troup D, Wilhite S, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Edgar R. NCBI GEO: mining tens of millions of expression profiles-database and tools update. Nucleic Acids Res. 2007;35(Database issue):D760–D765. doi: 10.1093/nar/gkl887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 11.Langfelder P, Horvath S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst Biol. 2007;1:54. doi: 10.1186/1752-0509-1-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keller MP, Choi Y, Wang P, Davis DB, Rabaglia ME, Oler AT, Stapleton DS, Argmann C, Schueler KL, Edwards S, Steinberg HA, Chaibub Neto E, Kleinhanz R, Turner S, Hellerstein MK, Schadt EE, Yandell BS, Kendziorski C, Attie AD. A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res. 2008;18(5):706–716. doi: 10.1101/gr.074914.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2008;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 15.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fuller TF, Ghazalpour A, Aten JE, Drake TA, Lusis AJ, Horvath S. Weighted gene co-expression network analysis strategies applied to mouse weight. Mamm Genome. 2007;18(6–7):463–472. doi: 10.1007/s00335-007-9043-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mason MJ, Fan G, Plath K, Zhou Q, Horvath S. Signed weighted gene co-expression network analysis of transcriptional regulation in murine embryonic stem cells. BMC Genomics. 2009;10:327. doi: 10.1186/1471-2164-10-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plaisier CL, Horvath S, Huertas-Vazquez A, Cruz-Bautista I, Herrera MF, Tusie-Luna T, Aguilar-Salinas C, Pajukanta P. A systems genetics approach implicates USF1, FADS3, and other causal candidate genes for familial combined hyperlipidemia. PLoS Genet. 2009;5(9):e1000642. doi: 10.1371/journal.pgen.1000642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Presson AP, Sobel EM, Papp JC, Suarez CJ, Whistler T, Rajeevan MS, Vernon SD, Horvath S. Integrated weighted gene co-expression network analysis with an application to chronic fatigue syndrome. BMC Syst Biol. 2009;2:95. doi: 10.1186/1752-0509-2-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saris CG, Horvath S, van Vught PW, van Es MA, Blauw HM, Fuller TF, Langfelder P, DeYoung J, Wokke JH, Veldink JH, van den Berg LH, Ophoff RA. Weighted gene co-expression network analysis of the peripheral blood from Amyotrophic Lateral Sclerosis patients. BMC Genomics. 2009;10:405. doi: 10.1186/1471-2164-10-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bing Z, Yang G, Zhang Y, Wang F, Ye C, Sun J, Zhou G, Yang L. Proteomic analysis of effects by x-rays and heavy ion in HeLa cells. Radiol Oncol. 2014;48(2):142–154. doi: 10.2478/raon-2013-0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lavoie C, Tam R, Clark M, Lee H, Sonenberg N, Lasko P. Suppression of a temperature-sensitive cdc33 mutation of yeast by a multicopy plasmid expressing a Deosophila ribosomal protein. J Biol Chem. 1994;269(20):14625–14630. [PubMed] [Google Scholar]
- 23.Lian ZR, Liu J, Li L, Li XX, Tufan NLS, Wu MC, Wang HY, Arbuthnot P, Kew M, Feitelson MA. Human S15a expression is upregulated by hepatitis B virus X protein. Mol Carcinog. 2004;40(1):34–46. doi: 10.1002/mc.20012. [DOI] [PubMed] [Google Scholar]
- 24.Xu M, Wang Y, Chen L, Pan B, Chen F, Fang Y, Yu Z, Chen G. Down-regulation of ribosomal protein S15A mRNA with a short hairpin RNA inhibits human hepatic cancer cell growth in vitro. Gene. 2014;536(1):84–89. doi: 10.1016/j.gene.2013.11.075. [DOI] [PubMed] [Google Scholar]
- 25.Yunta M, Lazo PA. Apoptosis protection and survival signal by the CD53 tetraspanin antigen. Oncogene. 2003;22(8):1219–1224. doi: 10.1038/sj.onc.1206183. [DOI] [PubMed] [Google Scholar]
- 26.Barbouti A, Höglund M, Johansson B, Lassen C, Nilsson PG, Hagemeijer A, Mitelman F, Fioretos T. A novel gene, MSI2, encoding a putative RNA-binding protein is recurrently rearranged at disease progression of chronic myeloid leukemia and forms a fusion gene with HOXA9 as a result of the cryptic t(7;17)(p15;q23) Cancer Res. 2003;63(6):1202–1206. [PubMed] [Google Scholar]
- 27.Kujala E, Kivelä T. Tumor, Node, metastasis classification of malignant ciliary body and choroidal melanoma: evaluation of the 6th edition and future directions. Ophthalmology. 2005;112(6):1135–1144. doi: 10.1016/j.ophtha.2004.11.063. [DOI] [PubMed] [Google Scholar]