

Abstract
Novel target discovery is warranted to improve treatment in adult T-cell acute lymphoblastic leukemia (T-ALL) patients. We provide a comprehensive study on mutations to enhance the understanding of therapeutic targets and studied 81 adult T-ALL patients. NOTCH1 exhibitedthe highest mutation rate (53%). Mutation frequencies of FBXW7 (10%), WT1 (10%), JAK3 (12%), PHF6 (11%), and BCL11B (10%) were in line with previous reports. We identified recurrent alterations in transcription factors DNM2, and RELN, the WNT pathway associated cadherin FAT1, and in epigenetic regulators (MLL2, EZH2). Interestingly, we discovered novel recurrent mutations in the DNA repair complex member HERC1, in NOTCH2, and in the splicing factor ZRSR2. A frequently affected pathway was the JAK/STAT pathway (18%) and a significant proportion of T-ALL patients harboured mutations in epigenetic regulators (33%), both predominantly found in the unfavourable subgroup of early T-ALL. Importantly, adult T-ALL patients not only showed a highly heterogeneous mutational spectrum, but also variable subclonal allele frequencies implicated in therapy resistance and evolution of relapse. In conclusion, we provide novel insights in genetic alterations of signalling pathways (e.g. druggable by γ-secretase inhibitors, JAK inhibitors or EZH2 inhibitors), present in over 80% of all adult T-ALL patients, that could guide novel therapeutic approaches.
Keywords: Acute lymphoblastic leukemia, targeted therapy, T-ALL next generation sequencing, pathways, gene panel
INTRODUCTION
T-cell acute lymphoblastic leukemia (T-ALL) in adults represents a disease with an unfavorable outcome[1]. While the cure rate in pediatric T-ALL patients exceeds 70%, a similar rate in adults is only observed for patients in the favorable risk group of thymic T-ALL as defined by the expression of CD1a[2,3]. Patients within the immature immunophenotypic group of early T-ALL as well as patients of the mature T-ALL subtype show a significantly inferior outcome[1]. Although allogeneic stem cell transplantation (alloSCT) in first complete remission has led to an improved outcome e.g. in the context of the German Multicenter Acute Lymphoblastic Leukemia (GMALL) trials, further therapeutic improvements are urgently warranted, in particular for high risk patients. In B-cell precursor (BCP-) ALL molecularly directed therapies like Rituxumab[4], the bi-specific antibody Blinatumomab[5], or tyrosine kinase inhibitors in Ph+-ALL[6] are well established. In contrast, targeted therapies are not available for T-ALL, with the exception of Nelarabine[7]. In order to identify targets for specific treatment strategies, a better understanding of the molecular background of T-ALL is necessary[8].
Previous to next generation sequencing (NGS), genetic alterations of leukemic blasts were mainly examined by cytogenetics to detect chromosomal rearrangements. In addition, immunophenotypic characterization, gene expression arrays, and copy number alterations were also used to categorize T-ALL[9]. Variations on a single nucleotide level had only been described for very few genes. One of the most relevant and frequent alterations are mutations of NOTCH1 gene occurring in about 60% of all T-ALL cases[10-12]. In addition, mutations of FBXW7, another player in the NOTCH pathway, as well as mutations of WT1 and PTEN were previously described[13-18].
Through NGS, molecular classification of T-ALL has dramatically expanded. Recurrent mutations in T-ALL affect genes involved in transcriptional processes (BCL11B[19], RUNX1[20], GATA3[21]), epigenetic regulation (DNMT3A[20,22], members of polycomb repressor complex (PRC2)[21]), JAK/STAT signalling[21,23-25] (JAK1/2/3, IL7R), ribosomal processes (RPL10, RPL5)[25], and various other functions (e.g. WT1[26], CNOT3[25], PH[[27], MEF2C[28,29], LEF1[30]). These data, predominantly derived from pediatric T-ALL, suggest a highly heterogeneous and complex molecular background of T-ALL. While some of these alterations imply prognostic significance, comprehensive studies with focus on therapeutic targets in larger series of adult T-ALL patients are missing. This is of particular importance as most cancer genes occur at intermediate frequencies of 2-20% or even lower[31].
Thus far, molecular subgroups in T-ALL were defined mainly based on gene expression profile (GEP)[28,32,33] or immunophenotype[2,3]. Both classifications, based on GEP or immunophenotype reflect the physiological T-cell stage, in which growth arrest and malignant transformation occured[34]. The gene signature of the early T-cell precursor (ETP)-ALL reflects the expression profile of early thymocyte progenitors in the double negative (DN)1 stage and ETP-ALL also shows a distinct immunophenotype[35,36]. Importantly, ETP-ALL, which recently gained interest as it represents a subgroup of T-ALL with stem cell and myeloid characteristics[21,36-38], may serve as model for the design of novel molecular therapies. Although the classification of ETP-ALL based on gene expression and immunophenotype were only partly overlapping[29], the subgroup of ETP-ALL is already an ideal model for therapy approaches adapted to its distinct molecular characteristics. Specifically mutated genes, mainly affected in ETP-ALL include members of the (PRC2) or genes reflecting the stem cell and myeloid character of ETP-ALL like FLT3, DNMT3A or KRAS. This mutation pattern of ETP-ALL opens up potential options for targeted therapies[21,22]. This might be of special interest in a minimal residual disease (MRD) setting as a bridging therapy to alloSCT.
Whereas a number of putative driver mutations have been characterized, the spectrum of recurring alterations in larger cohorts and their relevance in different T-ALL subgroups remains unexplored. To unravel this spectrum and to explore potential targets for novel therapeutic interventions, we performed targeted high throughput sequencing of 88 candidate genes in 81 T-ALL samples of adult patients.
RESULTS
Single nucleotide variations and short indels in adult T-ALL
We obtained an average of 1.2 million reads for each sample resulting in an average coverage of 120 reads for the target region. Eighty percent of the targeted region was covered with a minimum of 20 reads (Supplementary Table S1). After exclusion of polymorphisms annotated in dbSNP135, 473 single nucleotide variations (SNVs) and short indels were identified with a minimum call of 20 reads, 313 of those resulted in changes in the coding sequence of the target region. On average, three (3.2) genes per patient were mutated, and 64 (73%) of the 88 genes were mutated in at least one patient (Supplementary Table S2). We identified three patients without any SNVs in the selected genes. One patient showed an aberrantly high rate of SNVs with 21/88 genes being mutated (Supplementary Table S3). The number of mutations in the selected genes did not correlate with the patients' age.
Mutational spectrum of candidate genes in T-ALL
In total, fifteen of the 88 investigated genes were mutated in more than 5% of patients with nine genes showing a mutation frequency of ≥10%. As expected, the highest mutation rate with 53% was found for NOTCH1. Mutation frequencies of FBXW7 (10%), WT1 (10%), JAK3 (12%), and BCL11B (10%) were in the range of previously reported frequencies[21,26,39,40]. Recently identified recurrent alterations in DNM2 (17%), PHF6 (11%), DNMT3A (5%) or RELN (5%) were confirmed in this larger cohort of adult T-ALL patients[20,21,27] (Table 2). Interestingly, genes involved in epigenetic functions such as TET2 (5%), SUZ12 (5%), EP300 (5%) as well as genes that possess transcriptional activity like RUNX1 (9%), PTEN (8%), CBL (5%), or BCOR (4%) were mutated in the range of 5-10% of our T-ALL patients.
Table 2. Characteristics of the investigated adult T-ALL patients.
Abbreviations: WBC, white blood cell count; TCR, T cell receptor
| Number of patients | 81 | |
|---|---|---|
| Sex | male | 67 |
| female | 14 | |
| Age | Median | 35 |
| Range | 17-73 | |
| WBC (/nl) | Median | 41.9 |
| Range | 0.8-332 | |
| Mediastinal mass (n=62) | yes | 42 |
| no | 20 | |
| TCR rearrangement (n=75) | yes | 53 |
| no | 22 | |
| Immunophenotype | thymic | 40 |
| mature | 15 | |
| early | 26 |
Genes previously linked to ETP-ALL were also found to be mutated in the remaining non-ETP T-ALL subgroups including recurring mutations in the histone methyl-transferase MLL2 (11%), frequently mutated in B-cell lymphomas[41-43]. Like in B-cell lymphoma, MLL2 mutations were distributed over the entire gene locus without pointing towards a hot-spot region (Supplementary Figure S1). Similarly, the protocadherins FAT1 (15%) and FAT3 (12%) were altered not only in early T-ALL (FAT1 23%, FAT3 15%), but were also recurrently mutated - though in a lower frequency - in thymic T-ALL (FAT1 15%, FAT3 13%; Table 1).
Table 1. Mutational spectrum and comparison of T-ALL subgroups.
Genes with mutations detected in at least 3% of the examined samples are shown. In parentheses are the percentages for each subgroup
| Mutational spectrum | ||||
|---|---|---|---|---|
| T-ALL subgroups | ||||
| total | thymic | mature | early | |
| n | 81 | 40 | 15 | 26 |
| NOTCH1 | 43 (53.1%) | 27 (67.5%) | 6 (40.0%) | 10 (38.4%) |
| DNM2 | 14 (17.3%) | 7 (17.5%) | 2 (13.3%) | 5 (19.2%) |
| FAT1 | 13 (16.0%) | 6 (15.0%) | 1 (6.7%) | 6 (23.0%) |
| FAT3 | 11 (13.6%) | 5 (12.5%) | 2 (13.3%) | 4 (15.3%) |
| JAK3 | 11 (13.6%) | 3 (7.5%) | 3 (20.0%) | 5 (19.2%) |
| PHF6 | 11 (13.6%) | 5 (12.5%) | 1 (6.7%) | 5 (19.2%) |
| MLL2 | 10 (12.3%) | 5 (12.5%) | 1 (6.7%) | 4 (15.3%) |
| FBXW7 | 9 (11.1%) | 9 (22.5%) | 0 (0.0%) | 0 (0.0%) |
| WT1 | 8 (9.9%) | 4 (10.0%) | 0 (0.0%) | 4 (15.3%) |
| BCL11B | 7 (8.6%) | 5 (12.5%) | 2 (13.3%) | 0 (0.0%) |
| HERC1 | 7 (8.6%) | 4 (10.0%) | 2 (13.3%) | 1 (3.8%) |
| RELN | 7 (8.6%) | 5 (12.5%) | 1 (6.7%) | 1 (3.8%) |
| RUNX1 | 7 (8.6%) | 1 (2.5%) | 2 (13.3%) | 4 (15.3%) |
| PTEN | 6 (7.4%) | 4 (10.0%) | 1 (6.7%) | 1 (3.8%) |
| DNMT3A | 5 (6.2%) | 1 (2.5%) | 1 (6.7%) | 3 (11.5%) |
| CBL | 4 (4.9%) | 1 (2.5%) | 2 (13.3%) | 1 (3.8%) |
| EP300 | 4 (4.9%) | 2 (5.0%) | 1 (6.7%) | 1 (3.8%) |
| JAK1 | 4 (4.9%) | 1 (2.5%) | 1 (6.7%) | 2 (7.6%) |
| MTOR | 4 (4.9%) | 3 (7.5%) | 1 (6.7%) | 0 (0.0%) |
| SUZ12 | 4 (4.9%) | 0 (0.0%) | 1 (6.7%) | 3 (11.5%) |
| TET2 | 4 (4.9%) | 4 (10.0%) | 0 (0.0%) | 0 (0.0%) |
| WHSC1 | 4 (4.9%) | 2 (5.0%) | 0 (0.0%) | 2 (7.6%) |
| BCOR | 3 (3.7%) | 3 (7.5%) | 0 (0.0%) | 0 (0.0%) |
| ETV6 | 3 (3.7%) | 0 (0.0%) | 0 (0.0%) | 3 (11.5%) |
| MTMR3 | 3 (3.7%) | 0 (0.0%) | 2 (13.3%) | 1 (3.8%) |
| PRKCZ | 3 (3.7%) | 3 (7.5%) | 0 (0.0%) | 0 (0.0%) |
| ZRSR2 | 3 (3.7%) | 3 (7.5%) | 0 (0.0%) | 0 (0.0%) |
Mutation frequencies for distinct genes appeared to significantly different across T-ALL subgroups (Figure 1a). This was most remarkable for members of the NOTCH pathway: NOTCH1 showed a higher frequency in thymic (67.5%) compared to early T-ALL (38.4%, P=0.02). Consistent with this mature immunophenotype, NOTCH1 mutation status was significantly linked to a clonal TCR rearrangement (64% clonal TCR rearrangement in NOTCH1mut vs. 36% in NOTCH1wt, P=0.01). FBXW7 mutations, similar to NOTCH1 mutations, occurred exclusively in the subgroup of thymic T-ALL (Table 1). Additional mutations exclusively found in the subgroup of thymic T-ALL included BCL11B, TET2, MTOR, BCOR, and ZSRS2. In contrast, genes of the JAK/STAT pathway (JAK1, JAK3) and the PRC2 complex (EZH2, SUZ12) as well as the transcription factors ETV6 and RUNX1 were predominantly mutated in the immature T-ALL subgroup (Figure 1a).
Figure 1. Comparison of mutation frequencies between the different T-ALL subgroups.
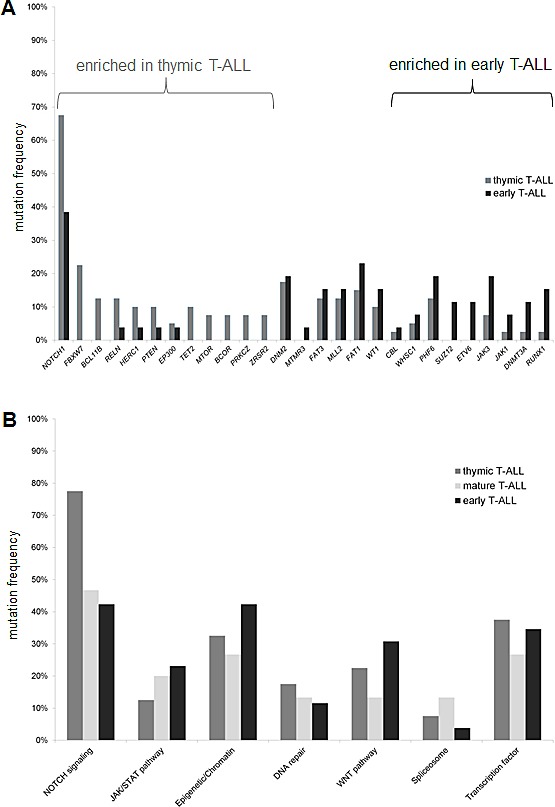
(A) Distribution for single genes and (B) according to the related pathways. Only genes with a mutation rate higher than 3% are shown.
In addition, we found novel mutations in genes which, to our knowledge, have not yet been reported in T-ALL. Among these HERC1, functionally involved in DNA repair, was among the most frequently mutated genes. Other recurrently affected genes included the splicing gene ZRSR2, or PRKCZ, a gene also involved in DNA repair (Supplementary Table S3).
Overall, there was no obvious association between the mutation status of different genes. Some of the genes with low mutation rates occurred exclusively, including genes with redundant functions like e.g. the histone methyltransferases, WHSC1 and MLL2. WHSC1 (also known as NSD2 or MMSET2) is associated with the prognostic unfavourable t(4;14) subgroup in multiple myeloma[44] and only very recently described in T-ALL[45,46]. We found WHSC1 to be mutated in 6% of the patients in our cohort. When combining WHSC1 and MLL2 mutated cases, 17% of all patients revealed alterations of histone methyltransferase genes.
Affected pathways and association with T-ALL subgroups
To address the complexity of this heterogeneous mutational spectrum, we focused on pathways with potential targets. In this study, the NOTCH pathway was affected in about 60% of all T-ALL patients (Figure 1B), including mutations in NOTCH1 and FBXW7 as well as in NOTCH2, NOTCH3, HES1, JAG1, and JAG2 (Supplementary Table S3). Mutations involving the NOTCH pathway were predominant in the thymic subgroup (75%) as compared to the early T-ALL (33%, P=0.004) subgroup. The spectrum of additional mutations between NOTCH1 mutated and NOTCH1 wildtype patients was not significantly different.
Interestingly, over 35% of our T-ALL patients carried lesions in epigenetic modulators. Whereas DNA methylation modifiers (like DNMT3A, TET2, IDH1, IDH2) were affected in 9% of all cases, histone modifiers were even more frequently altered, including members of the PRC such as SUZ12, EZH2, or EP300 and the histone methyltransferases MLL2 and WHSC1 (28%, Figure 2). Interestingly, chromatin modifying genes were slightly more frequently mutated in early compared to thymic T-ALL (42% vs. 32%, n.s.; Figure 1B).
Figure 2. Mutational landscape of adult T-ALL.
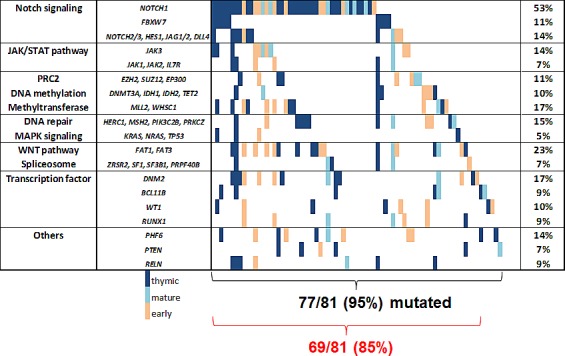
In the right column mutations rates are shown for groups with functional similarity. The red brackets summarize pathways representing potential therapeutic targets and their frequency. Genes with a mutation rate below 5% are grouped with functional similar genes or are not shown.
The JAK/STAT pathway is of particular interest for the design of targeted therapies with the emergence of JAK inhibitors. Mutations in JAK1, JAK2, JAK3, IL7R occurred in 19% of all T-ALL patients, but these preferentially occurred in immature, high risk T-ALL cases. Among those, JAK3 mutations were frequent (14%) and preferentially found in the early (19%) and mature (20%) subgroups compared to thymic T-ALL (8%, n.s., Table 1, Figure 1 and 2).
Another pathway of interest is the WNT pathway with a high rate of mutations in FAT1 and FAT3, which is frequently altered in the immature T-ALL subgroups (Figure 2). The mutation frequency of LEF1, a main player in the WNT pathway, was unexpectedly low (1%), which may be due to the fact that larger deletions could be missed with our NGS approach.
Spliceosome mutations, described for myeloid and mature lymphoid malignancies, were present only in a minority (7.4%) of T-ALL (Figure 1B). Overall, pathways with a potential targeted treatment option were affected in 85% of all T-ALL patients. These included the NOTCH pathway, JAK/STAT pathway, WNT pathway, DNA methylation, chromatin modifying enzymes, spliceosome, and MAPK pathway (Figure 2).
Variable allele frequencies suggest subclonal mutations
To identify mutations that may originate from the founding clone, we analysed the variant allele frequencies (VAFs) of all SNVs. In our cohort, T-ALL samples showed a wide spectrum of VAFs. For a founding clone, VAFs would be expected to be 44% (+/−7%)[47]. Within this T-ALL cohort, samples differed not only in the number of mutated genes, but also in range of VAFs for targeted genes.
The number of mutated genes varied widely across different patients (0-21/patient). The group of patients (n=13) with more than five mutated genes included cases with the majority of mutated genes linked to the founding clone. However, three cases had only one gene with VAFs of greater than 40% and the VAFs of the remaining genes were below 30%, pointing towards a subclonal structure of the leukemia. In patients with three or more mutated genes (n=36), we found at least one gene with VAFs in the range of a founding clone. On the other hand, we found 16 (18%) samples without any alteration with a VAF in the range of a founding clone (Figure 3A). These were all patients with two or less mutated genes and it is likely that the driver mutation was missed due to the gene selection.
Figure 3. Variant allele frequencies (VAFs) of each individual patient (A) and each gene (B) are shown.
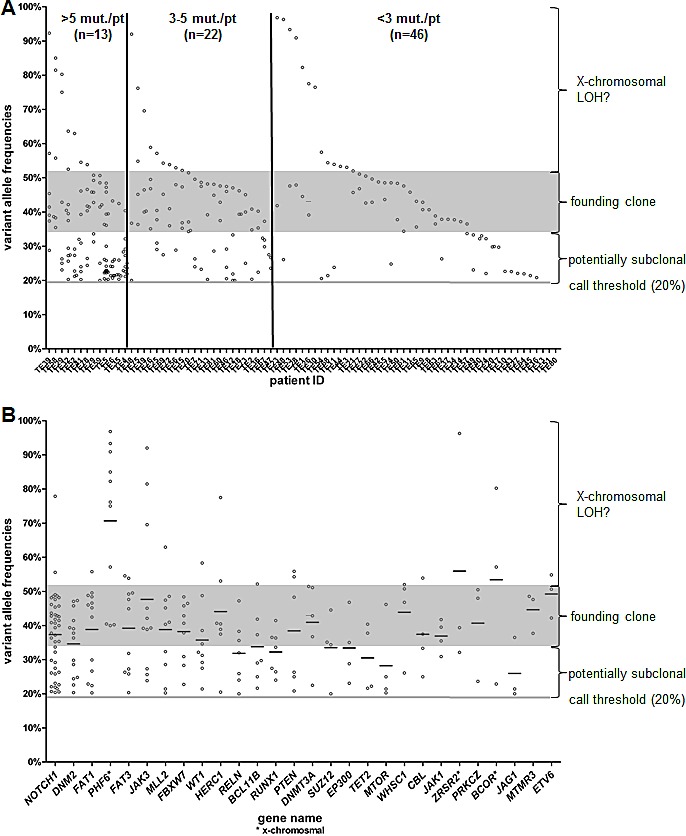
The grey shaded zone displays the expected range for a VAF in the founding clone.
The ideal target for an individualized treatment approach would be a driver mutation with a VAF in a founding clone. Although some of the genes with a mutation rate over 5% had predominantly VAFs over 40%, e.g. NOTCH1 or FAT1, none of those could be exclusively assigned as founding clone. Interestingly, NOTCH1 regarded as a prominent driver in T-ALL was found mutated on a subclonal level in 16 patients (37% of all NOTCH1 mutated patients). Furthermore, in three patients NOTCH1 showed at least two different alterations (Supplementary Figure S1). In all these cases one mutation had a VAF in the range of a founding clone, but the second mutation appeared to be present only within a subclone with a VAF below 30%.
Most of the recurrently mutated genes could be assigned to a founding clone in at least one patient. Some genes only showed low VAFs like e.g. ABL1, FLT3, NRAS or SUZ12 and thus are presumably later events in leukemogenesis. All of these genes were mutated in only three or less T-ALL patients in our cohort. Among the genes with a VAF higher than 50%, PHF6, BCOR, and ZRSR2 are located on the X chromosome (Figure 3B). Taken together, the spectrum of VAFs in T-ALL shows a highly heterogeneous pattern with none of the frequent (>10%) lesions being exclusively present in the founding clone.
DISCUSSION
Although risk stratification and subsequent therapy intensification have led to an improved outcome in adult T-ALL, the cure rate of approximately 50% remains unsatisfactory. Unlike in BCP-ALL with established targeted therapies (Rituximab, TKI, potentially Blinatumomab), no targeted therapy is yet available in T-ALL. Therefore, molecular targets and implementation of individualized treatment options are sorely needed.
In our study, we investigated the mutational spectrum of a large adult T-ALL cohort to identify potential molecular targets. As previously reported, T-ALL shows, despite of common features regarding immunophenotype or gene expression, a highly heterogeneous mutational background[21,25]. However, most of the previously published data are generated in pediatric T-ALL patients. Here, we have investigated in an original and comprehensive study, a large set of candidate genes in a large cohort of adult T-ALL patients. This approach would allow us to identify also recurrent candidate genes altered in lower frequencies[31]. We were able to confirm a broad spectrum of mutations and this provides confidence that the detection of genetic lesions is accurately displayed in sequencing libraries and targeted NGS.
Among the most obvious mutations, we found genetic alterations in over 50% of the patients for NOTCH1, one of the best described events in T-ALL. All other reported recurrent mutations (among others PTEN, PHF6, BCL11B, or WT1) occurred in less than 20% of adult T-ALL patients[33]. The frequency of NOTCH1 mutations as well as mutation rates for other well established genes like WT1, FBXW7, or BCL11B were in the range of previously reported incidences[33]. Another frequent alteration, genomic deletion of CDKN2A, was, however, not covered by our approach.
We also confirmed recurrent mutations in DNM2, PHF6, PTEN, JAK3, and RUNX1, which were only very recently discovered. The cadherins FAT1 and FAT3, mutated in ETP-ALL[22], have not yet been described in non-ETP T-ALL of adults and were identified by our approach to be recurrently mutated across all subgroups of adult T-ALL. FAT1 and its mutational inactivation have been linked to activation of the WNT pathway in solid tumors and to chemoresistance in chronic lymphocytic leukemia[48,49] and could serve as an attractive therapeutic target.
Furthermore, we found a high rate of mutations in MLL2, a histone methyltransferase, frequently mutated in various types of B-cell lymphomas[41-43]. Like in B-cell lymphomas, MLL2 mutations were distributed over the entire gene without any obvious hot-spot region[41,50]. Interestingly, another histone methyltransferase, WHSC1 (also known as MMSET/NSD2), was recurrently mutated in T-ALL and, although in a small number of patients, mutually exclusive within MLL2. WHSC1, associated with the so called Wolf-Hirschhorn syndrome[51], was only very recently found to be mutated in pediatric ALL, particularly in t(12;21) ETV6-RUNX1 ALL[45,46], as well as in mantle cell lymphoma[42]. These results together with mutations in the PRC2 complex and in genes involved in DNA methylation unravel a yet unreported high frequency (of over 25%) of alterations in epigenetic regulators in adult T-ALL. This is in line with other hematologic malignancies like acute myeloid leukemia (AML),myelodysplastic syndrome (MDS) or diffuse large cell lymphoma[41,52,53]. These findings suggest that a very tight regulation of chromatin remodelling, especially for methylation of lysine 27 on histone H3, is required in physiological cell development and correct hematopoietic differentiation.
Interestingly, patients with an immature T-ALL immunophenotype showed a particular high frequency for mutations in epigenetic regulators and thus emphasize the similarity with myeloid malignancies. This is especially striking in the subgroup of ETP-ALL as already described by Zhang and colleagues[21]. We were unable to confirm the high mutation rate in the PRC2 members described for pediatric patients, but we frequently found mutations in regulators of DNA methylation, possibly related to preexisting lesions in hematopoietic progenitors in the elderly[22,54]. Taken together, the high frequency of mutations in epigenetic regulators offers new insights and potential therapeutic applications e.g. of EZH2 inhibitors, histone deacetylase (HDAC) inhibitors or demethylating agents, which should be explored in clinical studies.
Another promising pathway for targeted therapies is the JAK/STAT pathway with frequent JAK3 mutations (13%). This rate is higher than the reported frequency in pediatric ALL patients[21]. For JAK1, varying mutation rates (4-18%) have been published[24,55]; in our cohort we found 4% of JAK1 mutations. In total, 18% of all our adult T-ALL patients carried alterations in the JAK/STAT pathway, predominantly in high-risk patients with an immature immunophenotype that might benefit from the application of molecular directed therapies, including JAK inhibitors[56].
Interestingly, mutations in the spliceosome, which are frequently found in MDS and in subgroups of AML[52,53], were virtually absent in T-ALL patients throughout all subgroups. Therefore, other elementary cellular processes might play a role in T-ALL. In a recent study, alterations in posttranslational mechanisms were suggested[25]. Unfortunately, these findings were reported after the design of our study and genes of interest, like RPL5, RPL10, or CNOT3, were not included in our gene panel.
In contrast to the work of de Keersmaecker and colleagues, we did not observe an age dependent distribution of mutation frequencies[25]. However this is likely due to our study design focused on candidate genes potentially enriched for driver mutations. The difference in the mutation frequencies in unbiased whole exome approaches and the frequencies in selected gene panels raises the question, whether the higher rate of mutations in prima vista not T-ALL associated genes simply reflects the altered hematopoiesis in elderly or possesses itself a leukemogenic potential[57-59].
NGS techniques are becoming widely available and are about to guide treatment decisions. This offers the opportunity not only to identify targets but also to unravel the spectrum of subclonal architecture that likely affects the response to targeted therapies. In addition, the mutational spectrum of leukemic cell changes during the progression of the disease and relapses are frequently harbored in preexisting subclones[47,57]. It has been shown that specific mutations, which are only present in a minor subclone at diagnosis, could lead to relapse due to chemotherapy resistance[60,61]. Therefore, the sole assessment of mutated genes might insufficient to select an optimal targeted therapy and determination of mutation frequency might be necessary to predict responses and the risk of relapse. This additional level of complexity in describing mutational landscapes for each individual patient is explored in our study and emphasizes that reported drivers not only occurred in the founding clone, but also in subclones. Thus, the sole restriction to gene panel assays for diagnostic purposes will likely not be sufficient to capture the wide clonal diversity and thus will likely miss mutations in the founding clone and even more in the subclones[47].
Adult T-ALL reveals a highly heterogeneous and individual spectrum of candidate gene mutations. Here, we provide an original and comprehensive overview of recurring mutations that unravel altered pathways enriched in specific leukemic subgroups. In addition, we identified novel candidate genes with potential therapeutic implications (FAT1, MLL2, HERC1). These mutations have to be further validated in larger patient cohorts accompanied by functional assays regarding their value as potential therapeutic targets. The identification of individual lesions in daily clinical routine, their clonal evolution, and the incorporation of highly individualized therapies in study trials will be a future challenge.
PATIENTS AND METHODS
Patients and treatment
We investigated bone marrow samples from 81 adult T-ALL patients with material sent to the reference laboratory of the GMALL study group and with sufficient genomic DNA quality, quantity and blast count (>80%) to perform NGS (Supplementary Table S4). Immunophenotyping of fresh samples was centrally performed in the GMALL reference laboratory at the Charité, University Hospital Berlin, Germany. Immunophenotyping was carried out as previously described[62,63]. T-lineage leukemia was subclassified into pre-T-ALL or early T-ALL (cyCD3+, CD7+, CD5+/−, CD2−, sCD3−, CD4−/+, CD8−/+, CD1a− or cyCD3+, CD7+, CD5−, CD2+, sCD3−, CD4−, CD8−, CD1a−), thymic T-ALL (cyCD3+, CD7+, CD5+/−, CD2+/−, sCD3+/−, CD4+, CD8+, CD1a+), and mature T-ALL (cyCD3+, CD7+, CD5+, CD2+, sCD3+/−, CD4+/−, CD8+/−, CD1a−). The immature subtypes of pre-T-ALL and early T-ALL are merged to a combined early T-ALL group. In addition, patients were classified to have an ETP-ALL within early T-ALL according to the criteria originally used by Couston-Smith and colleagues[36]. In detail, all samples were positive for cyCD3 and CD7, with absence of CD1a and CD8 (less than 5% of all lymphoblasts were positive), and weak expression of CD5 (i.e. less than 75% of all lymphoblasts were positive). Furthermore, the immunophenotype was characterized by the expression (i.e. more than 25% of the lymphoblasts positive) of at least one myeloid or stem cell marker (CD13, CD33, CD65, CD117, CD34, HLA-DR).
Patients' characteristics
Of the 81 adult T-ALL patients examined in this study, 40 patients showed an immunophenotype of thymic T-ALL, 15 of mature T-ALL and 26 of early T-ALL, and amongst the latter group, 20 had an ETP-ALL immunophenotype. The median age was 35 years (range 17-73) and 83% of the patients were male. The median white blood cell count (WBC) at diagnosis was 41.9/nL (range 0.8-332; Table 2). For all 81 samples the TCR rearrangement status was available[64].
Selection of candidate genes
The 88 genes selected for targeting sequencing are listed in the supplement (Supplementary Table S2). We selected genes known to be recurrently mutated in T-ALL, but also other genes, frequently mutated in BCP-ALL, (AML), and (MDS) were incorporated into the analysis[21,25,52,65]. Furthermore, genes with functions in epigenetic regulations, like members of the (PRC2) or in the splicing machinery, were included. Also, we selected candidate genes, based on our analysis of five exomes of adult ETP-ALL[22]. The targeted region of the 88 genes covered 1427 coding exons and 311 Kb of sequence (Supplementary Table S2).
Targeted sequencing of candidate genes
We constructed libraries from 3 μg of genomic DNA, which were labeled by barcode indices (length: 6 bp). Customized biotinylated RNA oligo pools (SureSelect, Agilent) were used to hybridize the target regions comprising the 88 selected genes. We performed 76-bp paired-end sequencing on an Illumina Genome Analyzer IIx platform. Reads were mapped to NCBI hg19 RefSeq. For a variant call, we required at least a read depth of 30, an allele frequency of 20% and an average base calling quality of Q13. Polymorphisms annotated in dbSNP 135 were excluded (Supplementary Figure S2).
Statistical analyses
Differences in the clinical characteristics were tested by the Pearson χ2 test resp. Fisher test. Differences in the mutation rate were analyzed by the Pearson χ2 test. For all tests a P-value < 0.05 (two-sided) was considered to indicate a significant difference. All calculations were performed using the SPSS software version 17 (SPSS Inc., Chicago, IL, USA) and GraphPad Prism® software version 5 (GraphPad Software Inc., La Jolla, CA, USA).
SUPPLEMENTARY INFORMATION TABLES AND FIGURES
Acknowledgments
We want to thank Liliana H. Mochmann for critical reading the manuscript. This work was supported by research grants from the Deutsche Krebshilfe (Mildred Scheel Professur), Wilhem-Sander-Stiftung, Novartis and Gutermuth-Stiftung to CD Baldus, Deutsche Krebshilfe grant 70-2657-Ho2 to Dieter Hoelzer and partly BMBF 01GI 9971 to Dieter Hoelzer and Nicola Gökbuget and Deutsche Krebshilfe grant 109031 to PA Greif and SK Bohlander.
REFERENCES
- 1.Gokbuget N, Hoelzer D. Treatment of adult acute lymphoblastic leukemia. Semin. Hematol. 2009;46:64–75. doi: 10.1053/j.seminhematol.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 2.Gokbuget N, Raff R, Brugge-Mann M, Flohr T, Scheuring U, Pfeifer H, Bartram CR, Kneba M, Hoelzer D. Risk/MRD adapted GMALL trials in adult ALL. Ann. Hematol. 2004;83(Suppl 1):S129–S131. doi: 10.1007/s00277-004-0850-2. [DOI] [PubMed] [Google Scholar]
- 3.Marks DI, Paietta EM, Moorman AV, Richards SM, Buck G, DeWald G, Ferrando A, Fielding AK, Goldstone AH, Ketterling RP, Litzow MR, Luger SM, McMillan AK, Mansour MR, Rowe JM, Tallman MS, et al. T-cell acute lymphoblastic leukemia in adults: clinical features, immunophenotype, cytogenetics, and outcome from the large randomized prospective trial (UKALL XII/ECOG 2993) Blood. 2009;114:5136–5145. doi: 10.1182/blood-2009-08-231217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomas DA, O'Brien S, Faderl S, Garcia-Manero G, Ferrajoli A, Wierda W, Ravandi F, Verstovsek S, Jorgensen JL, Bueso-Ramos C, Andreeff M, Pierce S, Garris R, Keating MJ, Cortes J, Kantarjian HM. Chemoimmunotherapy with a modified hyper-CVAD and rituximab regimen improves outcome in de novo Philadelphia chromosome-negative precursor B-lineage acute lymphoblastic leukemia. J. Clin. Oncol. 2010;28:3880–3889. doi: 10.1200/JCO.2009.26.9456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Topp MS, Gokbuget N, Zugmaier G, Degenhard E, Goebeler ME, Klinger M, Neumann SA, Horst HA, Raff T, Viardot A, Stelljes M, Schaich M, Kohne-Volland R, Bruggemann M, Ottmann OG, Burmeister T, et al. Long-term follow-up of hematologic relapse-free survival in a phase 2 study of blinatumomab in patients with MRD in B-lineage ALL. Blood. 2012;120:5185–5187. doi: 10.1182/blood-2012-07-441030. [DOI] [PubMed] [Google Scholar]
- 6.Ottmann OG, Wassmann B, Pfeifer H, Giagounidis A, Stelljes M, Duhrsen U, Schmalzing M, Wunderle L, Binckebanck A, Hoelzer D. Imatinib compared with chemotherapy as front-line treatment of elderly patients with Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ALL) Cancer. 2007;109:2068–2076. doi: 10.1002/cncr.22631. [DOI] [PubMed] [Google Scholar]
- 7.Gokbuget N, Basara N, Baurmann H, Beck J, Bruggemann M, Diedrich H, Guldenzoph B, Hartung G, Horst HA, Huttmann A, Kobbe G, Naumann R, Ratei R, Reichle A, Serve H, Stelljes M, et al. High single-drug activity of nelarabine in relapsed T-lymphoblastic leukemia/lymphoma offers curative option with subsequent stem cell transplantation. Blood. 2011;118:3504–3511. doi: 10.1182/blood-2011-01-329441. [DOI] [PubMed] [Google Scholar]
- 8.de Keersmaecker K, de Bock CE, Cools J. The biology of T-cell acute lymphoblastic leukemia. Hematology Education:the Education Program for Theannual Congress of the EuropeanHematology Association. 2013;2013:1–7. [Google Scholar]
- 9.Mullighan CG, Downing JR. Genome-wide profiling of genetic alterations in acute lymphoblastic leukemia: recent insights and future directions. Leukemia. 2009;23:1209–1218. doi: 10.1038/leu.2009.18. [DOI] [PubMed] [Google Scholar]
- 10.Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 11.Breit S, Stanulla M, Flohr T, Schrappe M, Ludwig WD, Tolle G, Happich M, Muckenthaler MU, Kulozik AE. Activating NOTCH1 mutations predict favorable early treatment response and long-term outcome in childhood precursor T-cell lymphoblastic leukemia. Blood. 2006;108:1151–1157. doi: 10.1182/blood-2005-12-4956. [DOI] [PubMed] [Google Scholar]
- 12.Ferrando A. NOTCH mutations as prognostic markers in T-ALL. Leukemia. 2010;24:2003–2004. doi: 10.1038/leu.2010.237. [DOI] [PubMed] [Google Scholar]
- 13.Baldus CD, Thibaut J, Goekbuget N, Stroux A, Schlee C, Mossner M, Burmeister T, Schwartz S, Bloomfield CD, Hoelzer D, Thiel E, Hofmann WK. Prognostic implications of NOTCH1 and FBXW7 mutations in adult acute T-lymphoblastic leukemia. Haematologica. 2009;94:1383–1390. doi: 10.3324/haematol.2008.005272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Asnafi V, Buzyn A, Le Noir S, Baleydier F, Simon A, Beldjord K, Reman O, Witz F, Fagot T, Tavernier E, Turlure P, Leguay T, Huguet F, Vernant JP, Daniel F, Bene MC, et al. NOTCH1/FBXW7 mutation identifies a large subgroup with favorable outcome in adult T-cell acute lymphoblastic leukemia (T-ALL): a Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL) study. Blood. 2009;113:3918–3924. doi: 10.1182/blood-2008-10-184069. [DOI] [PubMed] [Google Scholar]
- 15.Palomero T, Dominguez M, Ferrando AA. The role of the PTEN/AKT Pathway in NOTCH1-induced leukemia. Cell Cycle. 2008;7:965–970. doi: 10.4161/cc.7.8.5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zuurbier L, Homminga I, Calvert V, te Winkel ML, Buijs-Gladdines JG, Kooi C, Smits WK, Sonneveld E, Veerman AJ, Kamps WA, Horstmann M, Petricoin EF, III, Pieters R, Meijerink JP. NOTCH1 and/or FBXW7 mutations predict for initial good prednisone response but not for improved outcome in pediatric T-cell acute lymphoblastic leukemia patients treated on DCOG or COALL protocols. Leukemia. 2010;24:2014–2022. doi: 10.1038/leu.2010.204. [DOI] [PubMed] [Google Scholar]
- 17.Heesch S, Goekbuget N, Stroux A, Tanchez JO, Schlee C, Burmeister T, Schwartz S, Blau O, Keilholz U, Busse A, Hoelzer D, Thiel E, Hofmann WK, Baldus CD. Prognostic implications of mutations and expression of the Wilms tumor 1 (WT1) gene in adult acute T-lymphoblastic leukemia. Haematologica. 2010;95:942–949. doi: 10.3324/haematol.2009.016386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jotta PY, Ganazza MA, Silva A, Viana MB, da Silva MJ, Zambaldi LJ, Barata JT, Brandalise SR, Yunes JA. Negative prognostic impact of PTEN mutation in pediatric T-cell acute lymphoblastic leukemia. Leukemia. 2010;24:239–242. doi: 10.1038/leu.2009.209. [DOI] [PubMed] [Google Scholar]
- 19.Gutierrez A, Kentsis A, Sanda T, Holmfeldt L, Chen SC, Zhang J, Protopopov A, Chin L, Dahlberg SE, Neuberg DS, Silverman LB, Winter SS, Hunger SP, Sallan SE, Zha S, Alt FW, et al. The BCL11B tumor suppressor is mutated across the major molecular subtypes of T-cell acute lymphoblastic leukemia. Blood. 2011;118:4169–4173. doi: 10.1182/blood-2010-11-318873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grossmann V, Haferlach C, Weissmann S, Roller A, Schindela S, Poetzinger F, Stadler K, Bellos F, Kern W, Haferlach T, Schnittger S, Kohlmann A. The molecular profile of adult T-cell acute lymphoblastic leukemia: mutations in RUNX1 and DNMT3A are associated with poor prognosis in T-ALL. Genes Chromosomes. Cancer. 2013;52:410–422. doi: 10.1002/gcc.22039. [DOI] [PubMed] [Google Scholar]
- 21.Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, Lu C, Chen SC, Wei L, Collins-Underwood JR, Ma J, Roberts KG, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neumann M, Heesch S, Schlee C, Schwartz S, Gokbuget N, Hoelzer D, Konstandin NP, Ksienzyk B, Vosberg S, Graf A, Krebs S, Blum H, Raff T, Bruggemann M, Hofmann WK, Hecht J, et al. Whole-exome sequencing in adult ETP-ALL reveals a high rate of DNMT3A mutations. Blood. 2013;121:4749–4752. doi: 10.1182/blood-2012-11-465138. [DOI] [PubMed] [Google Scholar]
- 23.Shochat C, Tal N, Bandapalli OR, Palmi C, Ganmore I, te KG, Cario G, Cazzaniga G, Kulozik AE, Stanulla M, Schrappe M, Biondi A, Basso G, Bercovich D, Muckenthaler MU, Izraeli S. Gain-of-function mutations in interleukin-7 receptor-alpha (IL7R) in childhood acute lymphoblastic leukemias. J. Exp. Med. 2011;208:901–908. doi: 10.1084/jem.20110580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flex E, Petrangeli V, Stella L, Chiaretti S, Hornakova T, Knoops L, Ariola C, Fodale V, Clappier E, Paoloni F, Martinelli S, Fragale A, Sanchez M, Tavolaro S, Messina M, Cazzaniga G, et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J. Exp. Med. 2008;205:751–758. doi: 10.1084/jem.20072182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De KK, Atak ZK, Li N, Vicente C, Patchett S, Girardi T, Gianfelici V, Geerdens E, Clappier E, Porcu M, Lahortiga I, Luca R, Yan J, Hulselmans G, Vranckx H, Vandepoel R, et al. Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic leukemia. Nat. Genet. 2013;45:186–190. doi: 10.1038/ng.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tosello V, Mansour MR, Barnes K, Paganin M, Sulis ML, Jenkinson S, Allen CG, Gale RE, Linch DC, Palomero T, Real P, Murty V, Yao X, Richards SM, Goldstone A, Rowe J, et al. WT1 mutations in T-ALL. Blood. 2009;114:1038–1045. doi: 10.1182/blood-2008-12-192039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van VP, Palomero T, Khiabanian H, Van der Meulen J, Castillo M, Van RN, De MB, Philippe J, Gonzalez-Garcia S, Toribio ML, Taghon T, Zuurbier L, Cauwelier B, Harrison CJ, Schwab C, Pisecker M, et al. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat. Genet. 2010;42:338–342. doi: 10.1038/ng.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Homminga I, Pieters R, Langerak AW, de Rooi JJ, Stubbs A, Verstegen M, Vuerhard M, Buijs-Gladdines J, Kooi C, Klous P, van Vlierberghe P, Ferrando AA, Cayuela JM, Verhaaf B, Beverloo HB, Horstmann M, et al. Integrated transcript and genome analyses reveal NKX2-1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer Cell. 2011;19:484–497. doi: 10.1016/j.ccr.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 29.Zuurbier L, Gutierrez A, Mullighan CG, Cante-Barrett K, Gevaert AO, de RJ, Li Y, Smits WK, Buijs-Gladdines JG, Sonneveld E, Look AT, Horstmann M, Pieters R, Meijerink JP. Immature MEF2C-dysregulated T-cell leukemia patients have an early T-cell precursor acute lymphoblastic leukemia gene signature and typically have non-rearranged T-cell receptors. Haematologica. 2014;99:94–102. doi: 10.3324/haematol.2013.090233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gutierrez A, Sanda T, Ma W, Zhang J, Grebliunaite R, Dahlberg S, Neuberg D, Protopopov A, Winter SS, Larson RS, Borowitz MJ, Silverman LB, Chin L, Hunger SP, Jamieson C, Sallan SE, et al. Inactivation of LEF1 in T-cell acute lymphoblastic leukemia. Blood. 2010;115:2845–2851. doi: 10.1182/blood-2009-07-234377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC, Behm FG, Pui CH, Downing JR, Gilliland DG, Lander ES, Golub TR, Look AT. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. 2002;1:75–87. doi: 10.1016/s1535-6108(02)00018-1. [DOI] [PubMed] [Google Scholar]
- 33.Van VP, Ferrando A. The molecular basis of T cell acute lymphoblastic leukemia. J. Clin. Invest. 2012;122:3398–3406. doi: 10.1172/JCI61269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meijerink JP. Genetic rearrangements in relation to immunophenotype and outcome in T-cell acute lymphoblastic leukaemia. Best. Pract. Res. Clin. Haematol. 2010;23:307–318. doi: 10.1016/j.beha.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 35.Rothenberg EV. Transcriptional drivers of the T-cell lineage program. Curr. Opin. Immunol. 2012;24:132–138. doi: 10.1016/j.coi.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D, Cheng C, Su X, Rubnitz JE, Basso G, Biondi A, Pui CH, Downing JR, Campana D. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009;10:147–156. doi: 10.1016/S1470-2045(08)70314-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neumann M, Heesch S, Gokbuget N, Schwartz S, Schlee C, Benlasfer O, Farhadi-Sartangi N, Thibaut J, Burmeister T, Hoelzer D, Hofmann WK, Thiel E, Baldus CD. Clinical and molecular characterization of early T-cell precursor leukemia: a high-risk subgroup in adult T-ALL with a high frequency of FLT3 mutations. Blood Cancer J. 2012;2:e55. doi: 10.1038/bcj.2011.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haydu JE, Ferrando AA. Early T-cell precursor acute lymphoblastic leukaemia. Curr. Opin. Hematol. 2013;20:369–373. doi: 10.1097/MOH.0b013e3283623c61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O'Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters R, Draetta G, Sears R, Clurman BE, Look AT. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J. Exp. Med. 2007;204:1813–1824. doi: 10.1084/jem.20070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thompson BJ, Buonamici S, Sulis ML, Palomero T, Vilimas T, Basso G, Ferrando A, Aifantis I. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J. Exp. Med. 2007;204:1825–1835. doi: 10.1084/jem.20070872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, Jackman S, Krzywinski M, Scott DW, Trinh DL, Tamura-Wells J, Li S, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bea S, Valdes-Mas R, Navarro A, Salaverria I, Martin-Garcia D, Jares P, Gine E, Pinyol M, Royo C, Nadeu F, Conde L, Juan M, Clot G, Vizan P, Di CL, Puente DA, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc. Natl. Acad. Sci. U. S. A. 2013 doi: 10.1073/pnas.1314608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, Cruz-Gordillo P, Knoechel B, Asmann YW, Slager SL, Novak AJ, Dogan A, Ansell SM, Link BK, Zou L, Gould J, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl. Acad. Sci. U. S. A. 2012;109:3879–3884. doi: 10.1073/pnas.1121343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martinez-Garcia E, Popovic R, Min DJ, Sweet SM, Thomas PM, Zamdborg L, Heffner A, Will C, Lamy L, Staudt LM, Levens DL, Kelleher NL, Licht JD. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood. 2011;117:211–220. doi: 10.1182/blood-2010-07-298349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jaffe JD, Wang Y, Chan HM, Zhang J, Huether R, Kryukov GV, Bhang HE, Taylor JE, Hu M, Englund NP, Yan F, Wang Z, Robert ME, III, Wei L, Ma J, Easton J, et al. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat. Genet. 2013;45:1386–1391. doi: 10.1038/ng.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oyer JA, Huang X, Zheng Y, Shim J, Ezponda T, Carpenter Z, Allegretta M, Okot-Kotber CI, Patel JP, Melnick A, Levine RL, Ferrando A, Mackerell AD, Jr., Kelleher NL, Licht JD, Popovic R. Point mutation E1099K in MMSET/NSD2 enhances its methyltranferase activity and leads to altered global chromatin methylation in lymphoid malignancies. Leukemia. 2014;28:198–201. doi: 10.1038/leu.2013.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walter MJ, Shen D, Shao J, Ding L, White BS, Kandoth C, Miller CA, Niu B, McLellan MD, Dees ND, Fulton R, Elliot K, Heath S, Grillot M, Westervelt P, Link DC, et al. Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia. 2013;27:1275–1282. doi: 10.1038/leu.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morris LG, Kaufman AM, Gong Y, Ramaswami D, Walsh LA, Turcan S, Eng S, Kannan K, Zou Y, Peng L, Banuchi VE, Paty P, Zeng Z, Vakiani E, Solit D, Singh B, et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat. Genet. 2013;45:253–261. doi: 10.1038/ng.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Messina M, Del G I, Khiabanian H, Rossi D, Chiaretti S, Rasi S, Spina V, Holmes AB, Marinelli M, Fabbri G, Piciocchi A, Mauro FR, Guarini A, Gaidano G, Dalla-Favera R, Pasqualucci L, et al. Genetic lesions associated with chronic lymphocytic leukemia chemo-refractoriness. Blood. 2014 Feb. doi: 10.1182/blood-2013-10-534271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miyake N, Koshimizu E, Okamoto N, Mizuno S, Ogata T, Nagai T, Kosho T, Ohashi H, Kato M, Sasaki G, Mabe H, Watanabe Y, Yoshino M, Matsuishi T, Takanashi J, Shotelersuk V, et al. MLL2 and KDM6A mutations in patients with Kabuki syndrome. Am. J. Med. Genet. A. 2013;161:2234–2243. doi: 10.1002/ajmg.a.36072. [DOI] [PubMed] [Google Scholar]
- 51.Bergemann AD, Cole F, Hirschhorn K. The etiology of Wolf-Hirschhorn syndrome. Trends Genet. 2005;21:188–195. doi: 10.1016/j.tig.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 52.Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, Chalkidis G, Suzuki Y, Shiosaka M, Kawahata R, Yamaguchi T, Otsu M, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 54.Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, Mollica L, Li J, Viale A, Heguy A, Hassimi M, Socci N, Bhatt PK, Gonen M, Mason CE, Melnick A, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 2012;44:1179–1181. doi: 10.1038/ng.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Asnafi V, Le NS, Lhermitte L, Gardin C, Legrand F, Vallantin X, Malfuson JV, Ifrah N, Dombret H, MacIntyre E. JAK1 mutations are not frequent events in adult T-ALL: a GRAALL study. Br. J. Haematol. 2010;148:178–179. doi: 10.1111/j.1365-2141.2009.07912.x. [DOI] [PubMed] [Google Scholar]
- 56.Vainchenker W, Constantinescu SN. JAK/STAT signaling in hematological malignancies. Oncogene. 2013;32:2601–2613. doi: 10.1038/onc.2012.347. [DOI] [PubMed] [Google Scholar]
- 57.Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, Kandoth C, Fulton RS, McLellan MD, Dooling DJ, Wallis JW, Chen K, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McFarland CD, Korolev KS, Kryukov GV, Sunyaev SR, Mirny LA. Impact of deleterious passenger mutations on cancer progression. Proc. Natl. Acad. Sci. U. S. A. 2013;110:2910–2915. doi: 10.1073/pnas.1213968110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, Drier Y, Zou L, Ramos AH, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meyer JA, Wang J, Hogan LE, Yang JJ, Dandekar S, Patel JP, Tang Z, Zumbo P, Li S, Zavadil J, Levine RL, Cardozo T, Hunger SP, Raetz EA, Evans WE, Morrison DJ, et al. Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat. Genet. 2013;45:290–294. doi: 10.1038/ng.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tzoneva G, Perez-Garcia A, Carpenter Z, Khiabanian H, Tosello V, Allegretta M, Paietta E, Racevskis J, Rowe JM, Tallman MS, Paganin M, Basso G, Hof J, Kirschner-Schwabe R, Palomero T, Rabadan R, et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat. Med. 2013;19:368–371. doi: 10.1038/nm.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schwartz S, Rieder H, Schlager B, Burmeister T, Fischer L, Thiel E. Expression of the human homologue of rat NG2 in adult acute lymphoblastic leukemia: close association with MLL rearrangement and a CD10(−)/CD24(−)/CD65s(+)/CD15(+) B-cell phenotype. Leukemia. 2003;17:1589–1595. doi: 10.1038/sj.leu.2402989. [DOI] [PubMed] [Google Scholar]
- 63.Bene MC, Castoldi G, Knapp W, Ludwig WD, Matutes E, Orfao A, Van't Veer MB. Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL) Leukemia. 1995;9:1783–1786. [PubMed] [Google Scholar]
- 64.Neumann M, Coskun E, Fransecky L, Mochmann LH, Bartram I, Sartangi NF, Heesch S, Gokbuget N, Schwartz S, Brandts C, Schlee C, Haas R, Duhrsen U, Griesshammer M, Dohner H, Ehninger G, et al. FLT3 mutations in early T-cell precursor ALL characterize a stem cell like leukemia and imply the clinical use of tyrosine kinase inhibitors. PLoS. One. 2013;8:e53190. doi: 10.1371/journal.pone.0053190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kon A, Shih LY, Minamino M, Sanada M, Shiraishi Y, Nagata Y, Yoshida K, Okuno Y, Bando M, Nakato R, Ishikawa S, Sato-Otsubo A, Nagae G, Nishimoto A, Haferlach C, Nowak D, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat. Genet. 2013;45:1232–1237. doi: 10.1038/ng.2731. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.