

Abstract
At the dawn of a new era in label-free quantitation on high-resolution MS instruments, classical methods such as iTRAQ continue to provide very useful insights in comparative proteomics. The potential to multiplex samples makes this reporter-based labeling technique highly suited for method optimization as demonstrated here by a set of standard series. Instead of studying ratios of annotated proteins, we propose an alternative method, based on the analysis of the average reporter ratios of all the spectra from a sample or a large distinct subset herein. This strategy circumvents the bias, associated with the annotation and iTRAQ quantitation, leading to increased adequacy in measuring yield differences between workflows. As gel electrophoresis prior to MS analysis is highly beneficial, for example, as a fractionation step, the approach was applied to evaluate the influence of several parameters of the established in-gel digestion protocol. We quantified the negative effect of SYPRO Ruby staining and the positive effect of gel fixation prior to digestion on peptide yield. Finally, we emphasize the benefits of adding CaCl2 and ACN to a tryptic in-gel digest, resulting in an up to tenfold enhanced peptide recovery and fewer trypsin missed cleavages.
Keywords: Gel fractionation, In-gel digestion, iTRAQ, Method optimization, Quantification, Technology
Over the years, isobaric labeling techniques have been applied on a large scale in different proteome studies where relative quantitation is required. Although label-free methods, such as SWATH (AB SCIEX) and HDMSE (Waters Corporation) are gaining popularity, label-based strategies remain important. The reporter-based labeling methods still have the ability to give complementary insights, especially in terms of minimizing technical variation by parallel quantitation of multiple samples 1,2.
One of the main challenges in interpreting iTRAQ data is the underestimation of the fold change, partially caused by interfering masses in the silent region of the reporters and mixed MS/MS 3,4. Here, we present a new workflow for method optimization by means of isobaric tags (such as iTRAQ) to overcome these challenges without the need of complex data analysis tools. In-depth protocol knowledge can be achieved by dividing a (standard) peptide mixture into equal parts and differentially labeling them for each experimental condition under investigation. In the event of a digestion optimization, a protein sample can be split into equal parts, which are labeled and pooled after digestion 5. The relative yield of each condition is then defined by all the associated reporter ions in a run instead of only focusing on the identified proteins. Additionally, the effect of different experimental conditions on a specific peptide set with similar physicochemical classes can be evaluated after annotation.
We first analyzed a standard series of known ratios to test the preciseness of this approach and more specifically validate the influence of contaminating peaks in the reporter region on quantitation accuracy on our lower resolution ESI-Q-TOF MS platform. A well-defined protein mixture digest (Dionex no. 161088) was equally split into four, differentially labeled according to the manufacturer's protocol and mixed in different ratios (Table1). For LC–MS/MS analysis, peptides dissolved in 0.1% formic acid were analyzed on a Dionex U3000, coupled to a Q-TOF Premier (Waters Corporation). To create high-quality spectra and abundant reporter peaks, an enhanced TIC threshold of the peptide precursors was set for MS/MS selection during the data-dependent acquisition.
Table 1.
Validation of the iTRAQ method
| Sample | Label distribution | Theoretical ratio | Experimental ratio | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 114 | 115 | 116 | 117 | log 114/115 116/117 114/117 116/115 | log 114/115 | SD | log 116/117 | SD | log 114/117 | SD | log 116/115 | SD | |
| S1 | 1 | 9 | 1 | 9 | −0.954 | −0.930 | 0.065 | −0.855 | 0.115 | −0.867 | 0.377 | −0.867 | 0.106 |
| S2 | 2 | 8 | 2 | 8 | −0.602 | −0.616 | 0.076 | −0.532 | 0.087 | −0.585 | 0.076 | −0.562 | 0.087 |
| S3 | 3 | 7 | 3 | 7 | −0.368 | −0.382 | 0.067 | −0.319 | 0.079 | −0.363 | 0.063 | −0.337 | 0.085 |
| S4 | 4 | 6 | 4 | 6 | −0.176 | −0.192 | 0.071 | −0.114 | 0.071 | −0.173 | 0.058 | −0.132 | 0.086 |
| S5 | 5 | 5 | 5 | 5 | 0.000 | −0.025 | 0.079 | 0.067 | 0.070 | 0.011 | 0.060 | 0.031 | 0.091 |
| S6 | 6 | 4 | 6 | 4 | 0.176 | 0.158 | 0.096 | 0.253 | 0.067 | 0.188 | 0.059 | 0.224 | 0.110 |
| S7 | 7 | 3 | 7 | 3 | 0.368 | 0.333 | 0.077 | 0.449 | 0.064 | 0.387 | 0.063 | 0.395 | 0.084 |
| S8 | 8 | 2 | 8 | 2 | 0.602 | 0.576 | 0.102 | 0.656 | 0.077 | 0.600 | 0.083 | 0.631 | 0.092 |
| S9 | 9 | 1 | 9 | 1 | 0.954 | 0.888 | 0.126 | 1.096 | 0.093 | 1.045 | 0.080 | 0.935 | 0.138 |
| Linear regression | |||||||||||||
| r2 | 0.9997 | 0.9981 | 0.9965 | 0.9993 | |||||||||
| Slope | 0.9643 | 1.0160 | 0.9995 | 0.9621 | |||||||||
Identical parts of a peptide mixture were labeled with a different reporter label of the iTRAQ 4plex Kit (AB SCIEX, Framingham, MA, USA). Each sample (S1–S9) consists of a different combination of the four labels resulting in known (theoretical) ratios. After MS analysis, the average 114/115, 116/117, 114/117, and 116/115 log ratios and the associated SD are calculated from the spectra with reporter areas above the 0.3 threshold for each sample. For the linear regression analysis, both the slope and r2 values are presented for each ratio.
Using Mascot Distiller (Matrix Science), the raw data were processed into a .mgf peak list according to parameters that were optimized to increase the quality of the iTRAQ peaks (Supporting Information Methods 1 for experimental details). Subsequently, the areas from the 114.1, 115.1, 116.1, and 117.1 iTRAQ reporter peaks were extracted from each MS/MS spectrum by a freely available CompOmics script 6. To compensate for the isotope carry-over, the correction factors of the manufacturer's certificate of analysis were applied. MS/MS spectra containing reporter masses with an area value below 0.3 were not taken into account for quantitation, as these are more susceptible to variation 3,4. For data analysis, each data point represents the average of all the log ratios in one run since iTRAQ in method development requires comparison of all the spectra in a sample instead of defining quantitative changes of individual proteins. Apart from yielding accurate quantitative data on the optimization, iTRAQ coordinately circumvents the variation in precursor mass selection found in repeated runs of conventional analyses: an absent reporter indicates an absent peptide, a conclusion that cannot be drawn when two sequential runs are compared.
When plotting the theoretical against the experimental ratios of all the MS/MS spectra over a 1–10 range, a strong correlation was demonstrated. The high r2 values (e.g. 0.9997 for 114/115) displaying the goodness of fit of the linear regression, indicate that the given iTRAQ ratios predict the actual relationship between two labeled samples on our system. Low SD displays the high precision of this approach, and the slopes of the regression lines around 1 (results in Table1) demonstrate the capability to quantify small differences between two ratios, leading to accurate quantification. Most importantly, peptide identification is not required, as reporter intensities can be extracted separately. To target a specific set of proteins or peptides with, for example, similar physicochemical properties, protein annotation is possible. Note that the iTRAQ multiplex provides the opportunity to have duplicates in each run wherein one can coordinately discriminate the labels that are most impacted by near-isobaric masses and mixed MS/MS 2. This is, for example, reflected in the slight difference in the slope and the 95% confidence interval between the 114/115 and 116/117 regression line as shown in Fig.1, probably caused by label-specific effects 7. However, all the labels are suited for optimization studies as the results of the regression analysis of all the calculated ratios indicated.
Figure 1.
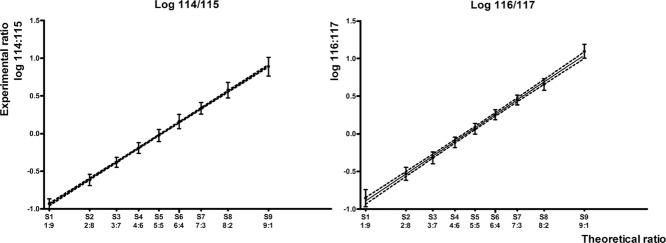
The plotted relationship of the theoretical and experimental ratios (data in Table1) of two of the four ratios. Each data point represents the average of the log ratios of all MS/MS spectra. Error bars indicate the SD. The linear regression of the means results in an r2 value of 0.9997 (114/115) and 0.9981 (116/117). The dashed lines define the 95% confidence band of the regression line.
Next, we used this iTRAQ approach to optimize our in-gel digestion protocol, the technique that bridges the gap between two keystone methodologies in proteomics: gel electrophoresis and MS. Despite the gain in popularity of the gel-free methods, particularly due to highly sensitive mass spectrometers, gel electrophoresis still has an added value as a molecular weight fractionator, a purification step and in the study of more hydrophobic (membrane) proteins 8. One very interesting application lies in PTM research where isobaric labels are used to measure stoichiometry: iTRAQ after gel fractionation is capable of reducing interfering precursors and thus enhancing quantitation accuracy 7,9. Most protocols separate the proteins by SDS-PAGE and visualize the fixed proteins by CBB, silver stain, or popular SYPRO Ruby (SR) stain 10. After imaging, proteins in the gel bands are classically destained, reduced, and alkylated before digestion 11. Numerous in-gel digestion strategies and optimized protocols are available: some focus on the reduction of the digestion time, while others implement alternative reagents and multiple proteases to augment peptide recovery 12. However, the consequence of several well-established steps in many protocols on the robustness of the technique is not well documented. Yet, a large (interrun) variation in the yield of peptide extraction is a well-known downside of many in-gel digestion protocols and challenges optimization 11.
Here, we examine the impact of protein fixation and SR staining and the possible benefits of ACN and CaCl2 on digest efficiency when gel electrophoresis is implemented as a sample preparation step prior to MS analysis. A HepG2 cell lysate was equally divided over 16 wells of two gels for the electrophoresis. For each condition, the extracted peptides were iTRAQ labeled and pooled immediately after the digest (Fig.2A). Every experiment was carried out four times in parallel to allow swapping iTRAQ labels to compensate for possible variation in label efficiency or label accuracy due to label-specific contaminating peaks.
Figure 2.

(A) Left panel: Equal amounts of a HepG2 cell lysate are divided over 16 lanes of two gels, whereby a loaded lane is alternated with an empty one to allow easy cutting of the gel (schematic of one gel is shown). After electrophoresis, the gels are cut around the 50 kDa marker to create a high (A) and low (B) molecular weight fraction. The different lanes were excised and in-gel digestion was performed on the different gel bands according to different conditions and pooled for each replicate after labeling. Right panel: Condition 1—digestion of a fixed and SR-stained gel, marked as the standard (ST) protocol. Condition 2—digestion of a fixed, nonstained gel. Condition 3—digestion of a nonfixed, nonstained gel. Condition 4—ST supplemented with 1 mM CaCl2 and 5% ACN. Extracted peptides from bands 1 to 4 were labeled and pooled according to the presented schedule, together forming four high and four low molecular weight samples. S1–S4: different replicates. (B) Each bar represents the average and SD of all the reporters of one of the four replicas. The different conditions are compared to the standard situation where the gel is fixed and SR stained. Despite a large variation, ratios indicate that SR has a negative effect on peptide recovery and fixation a positive influence. Addition of CaCl2 and ACN during trypsin digestion increases the peptide yield over sevenfold in average. Asterisk: The positive effect of gel fixation is verified by three of four replicates from the “fixation no SR/no fixation no SR” ratios.
The gels were cut around the 50 kDa marker to create two fractions and test the possible impact of the protein molecular weight on the digest conditions. Next, the individual lanes were excised to be digested under the different conditions. For the standard condition 1, proteins were fixed within the gel with a 7% acetic acid, 10% MeOH solution twice for 10 min. After a short wash with Milli-Q, the gel bands were incubated overnight with the SR gel stain in the dark at room temperature. For alternative conditions 2 and 3, the staining (2) or fixation and staining step (3) were skipped. The next day, the gel bands were washed, reduced, alkylated, and dehydrated before modified trypsin (Promega) was added for the overnight digestion at 37°C. The digest was performed with reagents analogous to the standard iTRAQ protocol from AB SCIEX (see Supporting Information Methods 1 for experimental details). Alternatively, for condition 4, 1 mM CaCl2 and 5% ACN were added to the trypsin buffer on a fixed and stained gel (Fig.2A, right). After digestion, peptides were extracted with ACN in three steps, labeled, and pooled according to Fig.2A. Data analysis was performed as described above.
With no obvious differences in the calculated average ratios between high and low molecular weight fractions, these data files were merged for further analysis. In Fig.2B, the conditions 2–4 are compared against condition 1, the standard protocol where proteins are extracted from a fixed and SR-stained gel. iTRAQ quantified large variations in peptide recovery between different gel digests. This coordinately emphasizes the importance of replicate analysis, especially during technical optimization. However, the main advantage of applying this strategy for method development is that the entire peptide ion yield is taken into account. Unlike most studies, we do not rely on the amount of identified proteins directly affected by unexpected modifications and inherent to different steps under investigation such as gel staining and fixation. SR staining clearly has a negative influence on peptide recovery in each replicate as shown by the increased “fixation no SR/standard” ratios where no staining was applied. Peptide loss is a known downside of fluorescent methods, yet here we show that this is not due to unexpected modifications, but rather to a loss of on average ±40% of the extracted peptides. When implementing gel electrophoresis as a fractionation or purification step, staining should thus be avoided. Surprisingly, however, fixation of a gel with acetic acid and MeOH hinted toward a positive effect on peptide recovery as suggested by three of the four significantly positive “fixation no SR/no fixation no SR” ratios (one-sample t-test, p < 0.0001; Fig.2B, asterisk). As mentioned earlier, when looking at annotated peptides one can select certain populations of peptides within the ratio distribution (e.g., left and right of the mean; Supporting Information Methods 2A for details) 6. When these subsets of labeled peptides are compared based on certain properties such as the number of missed cleavages and grand average of hydrophobicity (GRAVY) scores, one can coordinately define the gain of a certain methodology for specific characteristic sequences. Since fixation yielded a higher amount of peptides, we hypothesize that skipping fixation possibly results in spontaneous migration of proteins and peptides out of the gel. On the other hand, gel fixation resulted in an increased number of missed cleavages, which suggests that fixation could restrict the number of accessible clipping sites. Finally, despite the use of a modified trypsin, addition of CaCl2 and ACN at the time of digestion was found to be very beneficial to the yield of the digestion resulting in an up to tenfold increase in peptide recovery. To our knowledge, this is the first time that the advantages of those additives are quantified specifically for in-gel digestion. The advantage of including Ca2+ was previously explained by the beneficial effect on trypsin autocleavage during in-solution digestion protocols but is often overlooked since modified trypsin which is less susceptible to autocleavage, became a golden standard 11. This significant difference is probably induced by the stabilizing effect of Ca2+ ions on trypsin and improvement of the protein accessibility in the presence of ACN 13. CaCl2 and ACN supplementation does not result in recovery of more hydrophobic peptides as shown by the similar GRAVY scores but does diminish the amount of trypsin-missed cleavages from 26 to 17% (Supporting Information Methods 2B).
Isobaric tags are mostly applied to study the proteome expression between different biological samples. The label-based bias or variation induced by the required annotation of the proteins is often overlooked or corrected for by using complex postacquisition tools. Our standard series emphasize the accuracy of the iTRAQ technique in quantifying small loading differences between samples when analyzing the average ratios of all the data in a run. iTRAQ allows for multiplexing and internal replicates, which makes the technique highly suitable for method optimization. Using all the data excludes the variation associated with protein identification and quantitation, caused by unexpected modifications or fragmentation. For gel electrophoresis, particularly when used as a fractionation and purification tool for subsequent MS analysis, we recommend fixation of the gel and skipping SR staining before digestion. Finally, we corroborate the positive influence of CaCl2 and ACN addition in tryptic in-gel digestion protocols to increase reproducibility, above all in automated workflows 7.
The authors have declared no conflict of interest.
Glossary
- Q
quadrupole
- SR
SYPRO Ruby
Additional supporting information may be found in the online version of this article at the publisher's web-site
References
- [1].DeSouza LV, Siu KWM. Mass spectrometry-based quantification. Clin. Biochem. 2013;46:421–431. doi: 10.1016/j.clinbiochem.2012.10.025. [DOI] [PubMed] [Google Scholar]
- [2].Evans C, Noirel J, Ow SY, Salim M. An insight into iTRAQ: where do we stand now. Anal. Bioanal. Chem. 2012;404:1011–1027. doi: 10.1007/s00216-012-5918-6. [DOI] [PubMed] [Google Scholar]
- [3].Hultin-Rosenberg L, Forshed J, Branca RM, Lehtio J, Johansson HJ. Defining, comparing, and improving iTRAQ quantification in mass spectrometry proteomics data. Mol. Cell. Proteomics: MCP. 2013;12:2021–2031. doi: 10.1074/mcp.M112.021592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ow SY, Salim M, Noirel J, Evans C. iTRAQ underestimation in simple and complex mixtures: “the good, the bad and the ugly”. J. Proteome Res. 2009;8:5347–5355. doi: 10.1021/pr900634c. [DOI] [PubMed] [Google Scholar]
- [5].Burkhart JM, Vaudel M, Zahedi RP, Martens L, Sickmann A. iTRAQ protein quantification: a quality-controlled workflow. Proteomics. 2011;11:1125–1134. doi: 10.1002/pmic.201000711. [DOI] [PubMed] [Google Scholar]
- [6].Barsnes H, Vaudel M, Colaert N, Helsens K. compomics-utilities: an open-source Java library for computational proteomics. BMC Bioinformatics. 2011;12:1–6. doi: 10.1186/1471-2105-12-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Schmidt C, Hesse D, Raabe M, Urlaub H, Jahn O. An automated in-gel digestion/iTRAQ-labeling workflow for robust quantification of gel-separated proteins. Proteomics. 2013;13:1417–1422. doi: 10.1002/pmic.201200366. [DOI] [PubMed] [Google Scholar]
- [8].Shevchenko A, Loboda A, Ens W, Schraven B. Archived polyacrylamide gels as a resource for proteome characterization by mass spectrometry. Electrophoresis. 2001;22:1194–1203. doi: 10.1002/1522-2683()22:6<1194::AID-ELPS1194>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- [9].Przybylski C, Junger MA, Aubertin J, Radvanyi F. Quantitative analysis of protein complex constituents and their phosphorylation states on a LTQ-Orbitrap instrument. J. Proteome Res. 2010;9:5118–5132. doi: 10.1021/pr1003888. [DOI] [PubMed] [Google Scholar]
- [10].White IR, Pickford R, Wood J, Skehel JM. A statistical comparison of silver and SYPRO Ruby staining for proteomic analysis. Electrophoresis. 2004;25:3048–3054. doi: 10.1002/elps.200405947. [DOI] [PubMed] [Google Scholar]
- [11].Granvogl B, Ploscher M, Eichacker LA. Sample preparation by in-gel digestion for mass spectrometry-based proteomics. Anal. Bioanal. Chem. 2007;389:991–1002. doi: 10.1007/s00216-007-1451-4. [DOI] [PubMed] [Google Scholar]
- [12].Switzar L, Giera M, Niessen WM. Protein digestion: an overview of the available techniques and recent developments. J. Proteome Res. 2013;12:1067–1077. doi: 10.1021/pr301201x. [DOI] [PubMed] [Google Scholar]
- [13].Sipos T, Merkel JR. An effect of calcium ions on the activity, heat stability, and structure of trypsin. Biochemistry. 1970;9:2766–2775. doi: 10.1021/bi00816a003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.