

Summary
Introduction
Hemostasis is a rapid response by the body to stop bleeding at sites of vessel injury. Both platelets and fibrin are important for the formation of a hemostatic plug. Mice have been used to uncover the molecular mechanisms that regulate the activation of platelets and coagulation under physiologic conditions. However, measurements of hemostasis in mice are quite variable, and current methods do not quantify platelet adhesion or fibrin formation at the site of injury.
Methods
We describe a novel hemostasis model that uses intravital fluorescence microscopy to quantify platelet adhesion, fibrin formation and time to hemostatic plug formation in real time. Repeated vessel injuries of ~ 50–100 μm in diameter were induced with laser ablation technology in the saphenous vein of mice.
Results
Hemostasis in this model was strongly impaired in mice deficient in glycoprotein Ibα or talin-1, which are important regulators of platelet adhesiveness. In contrast, the time to hemostatic plug formation was only minimally affected in mice deficient in the extrinsic tissue factor (TFlow) or the intrinsic factor IX coagulation pathways, even though platelet adhesion was significantly reduced. A partial reduction in platelet adhesiveness obtained with clopidogrel led to instability within the hemostatic plug, especially when combined with impaired coagulation in TFlow mice.
Conclusions
In summary, we present a novel, highly sensitive method to quantify hemostatic plug formation in mice. On the basis of its sensitivity to platelet adhesion defects and its real-time imaging capability, we propose this model as an ideal tool with which to study the efficacy and safety of antiplatelet agents.
Keywords: blood coagulation, blood platelets, fluorescence imaging, hemostasis, mice
Introduction
Platelets are critical in the first phase of hemostatic plug formation, as they quickly form aggregates on the exposed subendothelium. Although platelet aggregates may be sufficient to transiently stop bleeding, the formation of a stable hemostatic plug requires blood coagulation, i.e. the generation of thrombin and the conversion of fibrinogen to crosslinked fibrin. Once formed, fibrin serves as a glue that reinforces the platelet plug.
Platelet integrins play an essential role in hemostasis, as they mediate platelet adhesion to the subendothelium and platelet–platelet cohesion [1], both in arterioles and in venules. The major platelet integrin is αIIbβ3, which is a receptor for fibrinogen and von Willebrand factor (VWF) deposited at sites of vascular injury. Binding of αIIbβ3 to these ligands depends on its ability to change from a low-affinity to a high-affinity state, a process controlled by intracellular signaling events [2]. Critical to this affinity modulation is talin-1, an adapter protein that connects the integrin to the cytoskeleton. Platelet-specific deletion of talin-1 leads to markedly impaired hemostasis, spontaneous hemorrhage, and protection from thrombosis [3,4]. Glycoprotein (GP)Ib–V–IX, another receptor for VWF, is expressed in a high-affinity state on resting platelets. The GPIb–VWF interaction mediates transient platelet adhesion (tethering), and is particularly important for thrombus formation under conditions of high shear stress [5,6]. Hemostasis is less impaired in mice with defects in the expression or function of GPIb–V–IX than in mice lacking αIIbβ3 or talin-1 [7].
At sites of vascular injury, the extrinsic, tissue factor (TF)-dependent pathway triggers coagulation, whereas the intrinsic pathway is critical for amplification of the clotting response. TF-dependent activation of coagulation is required for hemostasis [8]. Low-level expression of human TF (~ 1% relative to mouse TF) rescued the lethality in TF null embryos [9]. TF-mediated activation of factor X leads to the generation of a small amount of thrombin and the deposition of some fibrin at the margins of wounds [10]. This extrinsic pathway fibrin is not sufficient for hemostasis, as shown by the bleeding in thrombocytopathies that are associated with thrombin generation defects [11]. Thrombin generation through the intrinsic pathway is required for normal hemostasis, and deficiency in FXI, FIX or FVIII leads to bleeding [12-14].
Mice are the most widely used animals for studying the molecular mechanisms underlying hemostasis [15]. It is important to remember, however, that conclusions drawn from studies in mice have to be interpreted with caution, as there are significant differences between the hemostatic systems of mice and humans. These differences include the absence of protease-activated receptor 1 [16] and Fcγ receptor RIIA from mouse platelets [17], the absence of protein C inhibitor in mouse plasma [18], and differences in the TF pathway inhibitor isoforms expressed in mouse and human tissue [19]. The mouse model, however, has several important advantages, including the availability of genetically modified mice, the relatively low costs of mice, and the strong similarities in the vascular system between mice and humans. Measuring hemostasis, however, is challenging in these small animals. In the tail clip assay, the most commonly used method to study hemostasis in mice [20], the tail tip is amputated, and the bleeding time and the amount of blood loss are recorded. Given the severity of the lesion and the many reported modifications of the model, however, it is not surprising that there is substantial variability in results, both within the same group of study mice and between different laboratories. A less severe and more standardized hemostasis model is based on repeated puncture injury to the saphenous vein [21]. Bleeding times in this model are generally < 100 s, reflecting the smaller injuries than with tail transection. Another important advantage of this model is the ability to repeatedly disrupt the hemostatic plug, i.e. the ability to obtain multiple bleeding times from the same animal. Just like the tail bleeding time assay, however, this model does not quantify platelet adhesion or thrombin-dependent fibrin formation at the site of injury.
We describe a novel method for the quantitative analysis of platelet adhesion, thrombin/fibrin formation and bleeding time in mice. In this model, a standardized vascular injury to the saphenous vein is induced with laser ablation technology and hemostatic plug formation and bleeding are assessed by intravital microscopy.
Materials and methods
Materials
Low molecular weight heparin (enoxaparin sodium) and clopidogrel bisulfate (Plavix) were from Sanofi-Aventis (Bridgewater, NJ, USA). Heparin-coated capillaries were from VWR (West Chester, PA, USA). Bovine serum albumin (fraction V), prostacyclin (prostaglandin I2), and ADP (Sigma-Aldrich, St. Louis, MO, USA). An antibody against GPIX was from Emfret Analytics (Wuerzburg, Germany). An antibody against fibrin was a kind gift from R. Camire (Children’s Hospital of Philadelphia).
Animals
Talin1f/fPF4-Cre [22], IL4R/GPIb-tg [7], FIX−/− [13], TFlow [9] and wild-type (WT) control mice were bred on a C57BL6/J strain background, and housed in the mouse facility of the University of North Carolina at Chapel Hill. Genotypes were verified by PCR analysis. All experimental procedures were approved by the Animal Care and Use Committee of the University of North Carolina at Chapel Hill. Where indicated, mice were treated (by oral gavage) with clopidogrel bisulfate (75 mg kg−1 body weight) 24 h and 3 h before the experiment, to inhibit P2Y12 signaling in circulating platelets [23].
Laser-induced injury to the saphenous vein
Mice (12–14 weeks of age) were anesthetized by intraperitoneal injection of ketamine/xylazine (100 and 10 mg kg−1, respectively) (Med-Vet International, Mettawa, IL, USA). An Alexa488-labeled antibody against GPIX (2.5 μg per mouse) and an Alexa647-labeled antibody against fibrin (4 μg per mouse) were administered by retro-orbital injection. The saphenous vein was exposed as described recently [17], and the vessel was exposed to two maximum-strength 532-nm laser pulses (70 μJ; 100 Hz; duration, 7 ns; interval between pulses. 10 ms) (Ablate! photoablation system; Intelligent Imaging Innovations, Denver, CO, USA), focused through a × 20 water immersion objective lens (Zeiss, Jena, Germany) with a numerical aperture of 1 and a working distance of 1.8 mm. Fluorescence signals were recorded for 4.5 min before the same site was reinjured as described above. After 5 min of recording, another injury was inflicted at the same site, and fluorescence signals were recorded for another 5 min. A schematic representation of the main components of the microscopy setup and the time-line of the repeated injury model are provided in Fig. S1A, B. Similar injuries were performed at up to five distinct locations along the saphenous vein. The bleeding time was recorded when occlusion of the lesion lasted for > 15 s. To avoid effects from earlier injuries, new injuries were performed upstream of the previous injury. No trends in platelet accumulation (Fig. S2) or bleeding times (not shown) were observed between different injury locations along the saphenous vein. Throughout the experiment, constant perfusion drip was maintained on the exposed saphenous vein with a physiologic salt solution containing 32 mm NaCl, 4.7 mm KCl, 1.2 mm MgSO4, 2 mm CaCl2, and 18 mm NaHCO3 (pH 7.4), which was bubbled with 5% CO2/95% N2 for 15 min prior to the start of the experiment. The physiologic salt solution was maintained at 37 °C by perfusion through a Sloflo In-line solution heater (SF-28) (Warner Instruments, Hamden, CT, USA). Fluorescence signals within the boundaries of the saphenous vein were recorded with a Zeiss Axio Examiner Z1 microscope (Intelligent Imaging Innovations) equipped with a multi-color LED light source (Lumencor) and a × 20/1 numerical aperture water immersion objective lens (Zeiss). All data were analyzed with SLIDEBOOK 5.0 (Intelligent Imaging Innovations).
Statistics
Statistical analyses were performed with prism (GraphPad Software, Inc., La Jolla, CA, USA) or slidebook 5.0 (Intelligent Imaging Innovations). A P-value of ≤ 0.05 was considered to be significant. All data are presented as mean ± standard error of the mean.
Results
Novel method to visualize hemostatic plug formation
Fluorescently labeled antibodies against GPIX, a subunit of the VWF receptor complex, and against fibrin were infused into anesthetized mice, and the saphenous vein was exposed as described recently [21]. A top-down injury to the vessel wall was induced by laser ablation, resulting in vascular denudation (lesion area, ~ 1800 μm2; diameter, ~ 48 μm) and blood loss from the site of injury (Fig. 1; Video S1). Platelet (Fig. 1A,B) and fibrin (Fig. 1A,C) accumulation occurred rapidly at the site of injury, and hemostasis was achieved ~ 10 s after laser injury (Fig. 1D). Platelet accumulation (anti-GPIX fluorescence) reached its maximum ~ 1 min after laser-induced damage, before diminishing over time (Fig. 1B). Fibrin accumulation was slightly delayed relative to platelet accumulation, and was predominantly observed in a ring around the platelet plug (Fig. 1C). To determine whether a repeated injury would lead to increased fibrin generation and more robust fibrin binding within the platelet plug, we reinjured the initial site, and again monitored platelet plug and fibrin formation for an additional 5 min. Upon the second injury (lesion area, ~ 2600 μm2; diameter, ~ 57 μm), significant increases in the accumulation of platelets and the accumulation of fibrin were observed. However, fibrin was still mostly seen in a ring around the platelet plug. A third injury (lesion area, ~ 3300 μm2; diameter, ~ 65 μm) was performed at the initial site, which again led to increased platelet and fibrin accumulation (Fig. 1A). Repeated laser damage to the same site led to a small but significant prolongation of the bleeding time (Fig. 1D).
Fig. 1.
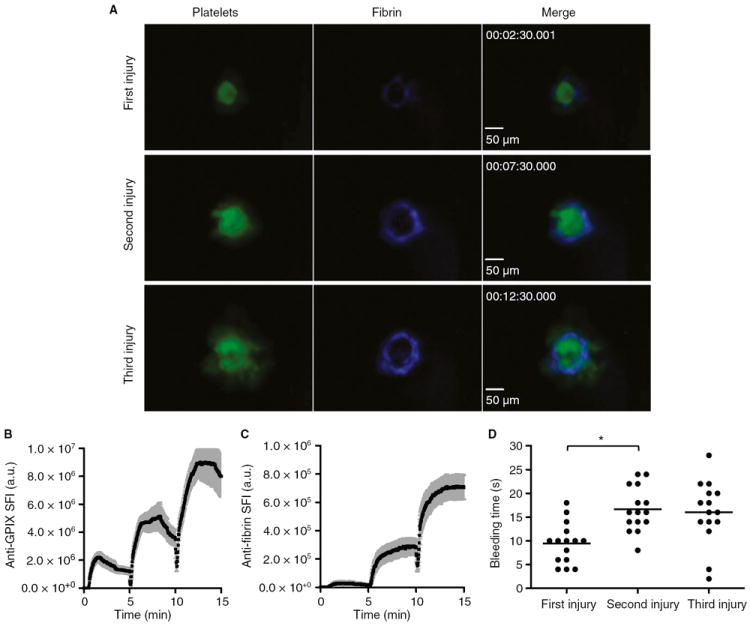
Quantitative analysis of platelet accumulation, fibrin generation and bleeding time after laser injury to the saphenous vein of wild-type mice. Mice were injected with Alexa647-labeled antibodies against fibrin and Alexa488-labeled antibodies against glycoprotein (GP)IX to monitor fibrin generation and platelet accumulation, respectively. Repeated vascular injury was induced by laser ablation. (A) Representative images at the indicated times after the start of the recording. (B, C) Accumulation of platelets (B) and fibrin (C). The sum fluorescence intensity (SFI) ± standard error of the mean is shown. Total number of injuries at distinct locations: n = 15 (obtained in four experimental mice). (D) Bleeding time. *P < 0.05.
Platelets play a critical role in small injury hemostasis
To assess the contribution of platelets to hemostasis after laser injury to the saphenous vein, we first examined platelet plug and fibrin formation in mice lacking talin-1 in megakaryocytes/platelets only (talin1f/fPF4-Cre). As shown previously, platelets lacking talin-1 fail to aggregate in response to various agonists, including thrombin, thromboxane A2, and collagen [22]. As expected, platelet adhesion was markedly impaired and hemostasis was not achieved at sites of laser injury in talin1f/fPF4-Cre mice (Fig. 2A,B,D; Video S2). The continued blood loss into the surrounding tissue led to a dramatic increase in fibrin formation (Fig. 2A,C). Hemostasis was also impaired in mice lacking the extracellular domain of GPIbα (IL4R/GPIb-tg mice) (Fig. 2D) [7]. However, platelet accumulation at the site of injury was greater than in talin1f/fPF4-Cre mice, leading to a narrowing of the opening in the vessel wall (Fig. 2A,B; Video S3). Consistent with the fact that IL4R/GPIb-tg platelets aggregate normally in response to agonist stimulation [6], large thrombi were observed in the extravascular space. As in talin1f/fPF4-Cre mice, fibrin accumulation was markedly increased at sites of injury in IL4R/GPIb-tg mice (Fig. 2A,C). Together, these studies show that: (i) platelet adhesion/aggregation plays a critical role in small injury hemostasis as tested in this novel model; and (ii) shear stress in the saphenous vein must be high enough for platelet adhesion to be (partially) dependent on GPIb–V–IX.
Fig. 2.
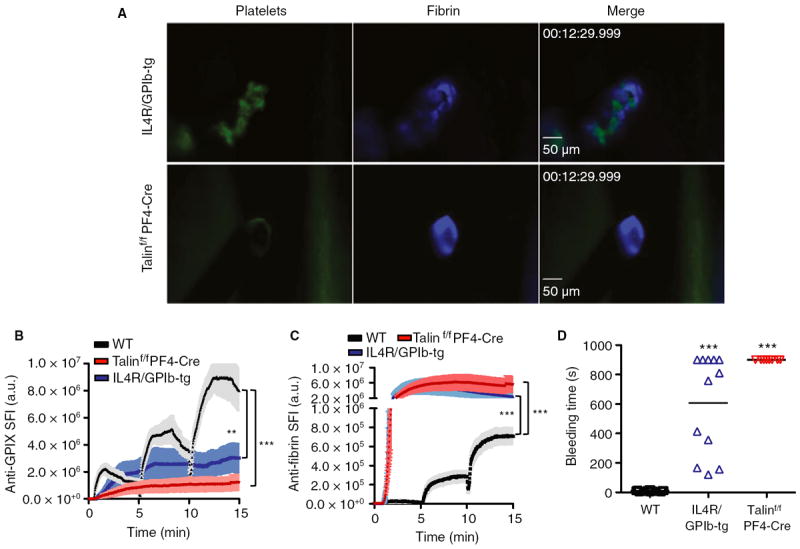
Contribution of platelets to hemostasis after laser injury. Vascular lesions were induced in wild-type (WT) (black line/symbols), talin1f/fPF4-Cre (red) and IL4R/GPIb-tg (blue) mice. (A) Representative images. (B, C) Accumulation of platelets (B) and fibrin (C). Sum fluorescence intensity (SFI) ± standard error of the mean is shown. Talin1f/fPF4-Cre: n = 9 (four mice). IL4R/GPIb-tg: n = 10 (four mice). (D) Bleeding time. **P < 0.001, ***P < 0.0001.
Coagulation augments small injury hemostasis
TF is a critical component of the extrinsic coagulation pathway and the initiation of coagulation at sites of vascular injury [8]. To evaluate the contribution of TF to hemostasis after laser injury, we examined mice deficient in mouse TF but expressing low levels (~ 1% of normal) of human TF [9] in our model. Surprisingly, hemostasis at sites of laser injury in these TFlow mice was comparable to that in WT mice (Fig. 3A–D; Video S4), although a small but significant prolongation of the bleeding time was observed after the second and third injuries (Fig. 3D). Consistent with the predicted role of TF in thrombin generation at sites of vascular injury, the accumulation of both platelets and fibrin was significantly reduced in these mice (Fig. 3B,C). As in the TFlow mice, the bleeding time in mice deficient in the intrinsic coagulation pathway (FIX−/−) was only minimally prolonged (Fig. 3D). Whereas platelet accumulation was significantly lower at sites of laser injury in FIX−/− mice (Fig. 3B), fibrin formation was increased as compared with controls (Fig. 3C; Video S5). These findings are consistent with a model in which the intrinsic pathway is important for sustained thrombin generation and full platelet activation at sites of injury. The increased fibrin levels may reflect plasma leakage through the porous plug, leading to TF-dependent thrombin generation in the extravascular space.
Fig. 3.
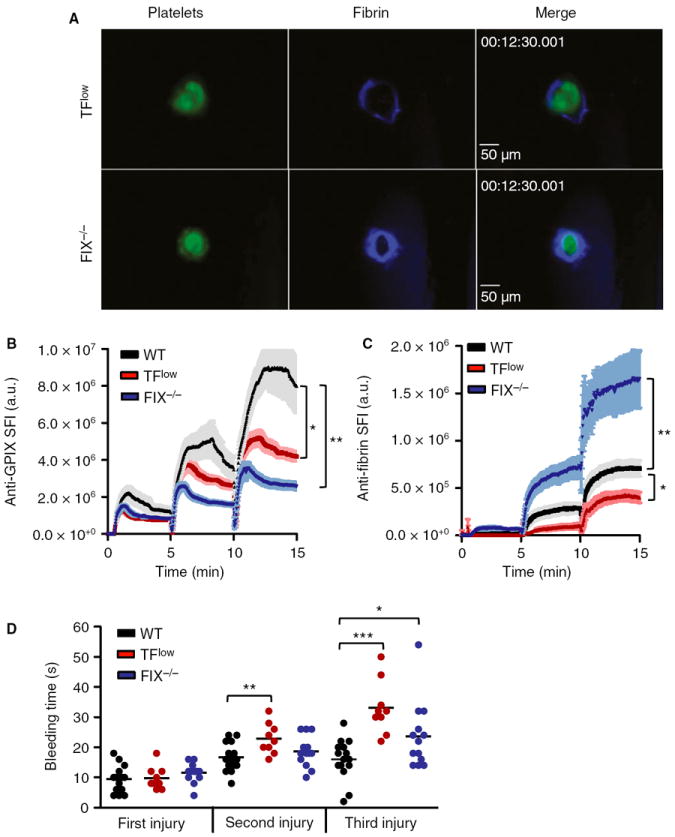
Contribution of the extrinsic and intrinsic coagulation pathways to hemostasis after laser injury. Vascular lesions were induced in wild-type (WT) (black line/symbols), TFlow (red) and FIX−/− (blue) mice. (A) Representative images. (B, C) Accumulation of platelets (B) and fibrin (C). Sum fluorescence intensity (SFI) ± standard error of the mean is shown. TFlow: n = 9 (three mice). FIX−/−: n = 13 (three mice). (D) Bleeding time. *P < 0.05, **P < 0.001, ***P < 0.0001. GP, glycoprotein; TF, tissue factor.
Analysis of combined defects in platelet function and coagulation
Intrigued by the findings in the TFlow and FIX−/− mice, we investigated whether a proper coagulation response would be critical for small injury hemostasis in mice with partially defective platelet function. To address this question, we studied hemostatic plug formation in WT and TFlow mice pretreated with the P2Y12 inhibitor clopidogrel bisulfate. P2Y12 inhibition significantly reduced platelet accumulation (Fig. 4A,B) but not fibrin accumulation (Fig. 4C) in WT mice. The bleeding time in WT/clopidogrel mice was minimally prolonged (Figs. 4D) and thrombus stability was decreased, especially following the first insult to the vessel (Video S6; numbers in the bar graph in Fig. 4D indicate the percentage of injuries that reopened after hemostatic plug formation). Interestingly, clopidogrel treatment led to a significantly increased bleeding time resulting from hemostatic plug instability in TFlow mice as compared with WT controls (Fig. 4G; Video S7), demonstrating the importance of both thrombin generation and feedback signaling by ADP in this model of small injury hemostasis. Consistently, platelet accumulation at the site of injury tended to be lower in clopidogrel-treated TFlow mice than in untreated TFlow mice (Fig. 4E), whereas fibrin formation was slightly increased at sites of injury in clopidogrel-treated TFlow mice (Fig. 4F).
Fig. 4.
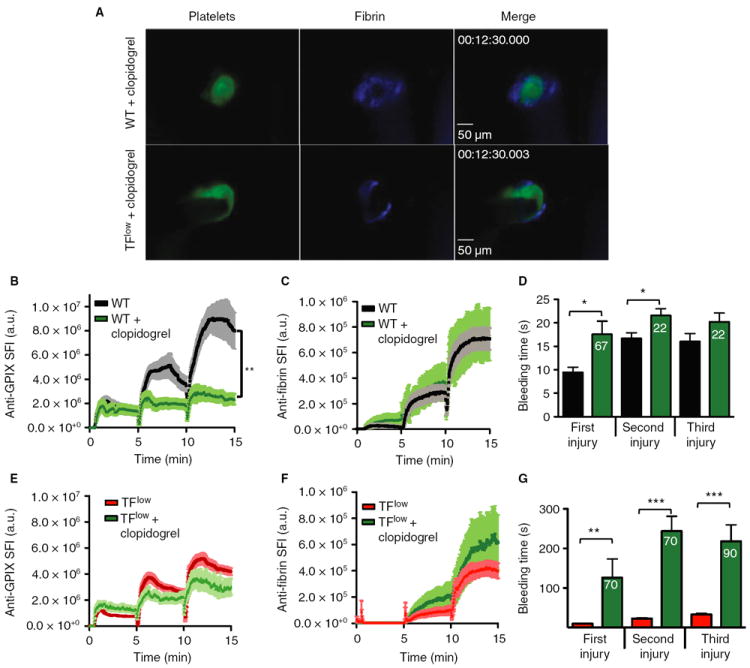
Effect of clopidogrel on hemostasis in wild-type (WT) and TFlow mice. WT and TFlow mice were treated with the P2Y12 inhibitor clopidogrel bisulfate prior to laser injury. (A) Representative images. (B–D) Platelet accumulation (B), fibrin accumulation (C) and bleeding time (D) in WT (black line/bars) and WT/clopidogrel (green) mice. (E–G) Platelet accumulation (E), fibrin accumulation (F) and bleeding time (G) in TFlow (red line/bars) or TFlow/clopidogrel (green) mice. Sum fluorescence intensity (SFI) ± standard error of the mean is shown. *P < 0.05, **P < 0.001, ***P < 0.0001. WT/clopidogrel: n = 10 (three mice). TFlow/clopidogrel: n = 10 (three mice). Numbers in bars indicate the incidence of repeated bleeding during the 5-min observation period following laser injury. The bleeding time was recorded when occlusion of the lesion lasted for > 15 s. GP, glycoprotein; TF, tissue factor.
Discussion
We describe a new model for the quantitative analysis of platelet accumulation, thrombin/fibrin generation and bleeding at a site of vascular injury. This novel method has several defining characteristics, including the use of intravital fluorescence microscopy to monitor the hemostatic process, the novel use of laser ablation technology to generate vascular lesions with a defined diameter, the ability to repeatedly disrupt the hemostatic process at the same site of injury, and the possibility of performing multiple injuries along the exposed saphenous vein. Thus, whereas classic murine hemostasis models, such as the tail clip assay, provide only one data point per mouse, our model can yield up to 15 bleeding time measurements per animal, and at the same time provide real-time analysis of platelet accumulation and fibrin formation at the site of injury.
The tail clip assay is the most widely used method to measure hemostasis in mice. However, there are many variables, including where and how the injury is inflicted, the method of blood collection, and vascular spasms associated with the transection of the various tail vessels, that make standardization of this assay a big challenge [20,24]. In contrast, laser ablation generates a small lesion area, does not transect the vessel, and does not cause visible vascular spasms. As a result, we obtained very reproducible bleeding time measurements for most genotypes tested in this study. Mice lacking the extracellular domain of GPIbα constituted an exception, as they were characterized by large variability in the bleeding times. As shown by intravital microscopy, this variability in hemostatic function is a reflection of the delayed adhesion of IL4R/GPIb-tg platelets and the resulting gradual occlusion of the vascular lesion. However, large thrombi accumulated in the surrounding tissue, confirming previous findings that the hemostatic defect in these mice is not the result of impaired aggregation of these platelets [6].
In addition to classic bleeding time models, investigators have used various intravital microscopy-based methods to monitor thrombus formation at sites of vascular injury [25,26]. The FeCl3-induced and the laser-induced cremaster models are both considered to be thrombosis models, not hemostasis models. Neither injury leads to significant blood loss at the site of injury, unlike in our model, where we observe bleeding from the injured saphenous vein. Recent work by Motto et al. further suggests that FeCl3 injury does not lead to significant exposure of subendothelial matrix, but that it causes the formation of complexes between red blood cells and ferric ions along the exposed vasculature. These complexes ultimately recruit platelets from the flowing blood [27]. Unlike laser injury, FeCl3 leads to the formation of occlusive thrombi, in both small and large vessels. A vascular injury model, induced by puncture with a micropipette, has been described by van Gestel et al. [28,29]. In this model, 6–8-μm injuries are made in small mesenteric arterioles, 20–40 μm in diameter. In contrast, we induce larger injuries (~ 50 μm in diameter) in the much larger saphenous vein. Consistently, bleeding times in the micropipette model are very short (1–2 s), and P2Y12 inhibition had no significant effect on the bleeding time or the rate of rebleeding. In contrast, our studies showed a significant prolongation of the bleeding time and an increased rate of rebleeding in mice treated with clopidogrel. Both studies demonstrate that P2Y12 inhibition reduces the size of the platelet plug after vessel wall injury. We acknowledge that our model is dependent on the availability of rather expensive laser ablation technology. As compared with the micropipette puncture model, however, it has the advantage that the same optics are used to injure the endothelium and to monitor platelet and fibrin accumulation (Fig. S1). This ability to injure while we perform real-time imaging allows us to ‘surgically’ remove an existing plug by repeated laser exposure.
Our studies in mice with severe defects in platelet adhesion or coagulation suggest that the laser injury model mimics hemostatic defects induced by small-vessel lesions in humans. Specifically, we observed a critical role for platelets, but not coagulation, in the formation of a hemostatic plug after laser injury to the saphenous vein. Similarly, impaired platelet function frequently leads to prolonged bleeding times at sites of small-vessel lesions in humans (skin cuts and mucocutaneous lesions) [30]. In contrast, patients with coagulation disorders frequently show delayed occurrence of large hematomas, which are thought to reflect the critical role of fibrin in the stabilization of larger thrombi. Delayed bleeding was also described for mice with impaired clotting function after challenge in the tail clip assay, i.e. a more severe injury model [31]. When the bleeding time is used as a read-out, hemostasis after laser injury is largely independent of coagulation. The ability to visualize hemostatic plug formation in real time, however, provided additional information on the hemostatic process in these mice. Platelet accumulation was significantly reduced in both TFlow and FIX−/− mice, even though the bleeding time was not prolonged. In contrast, fibrin accumulation was markedly increased in lesions of talin1f/fPF4-Cre, IL4R/GPIb-tg and FIX−/− mice, whereas it was decreased in TFlow mice. These findings suggest that prolonged plasma exposure to TF leads to increased thrombin and fibrin generation in the surrounding tissue. As the bleeding time in FIX−/− mice was only minimally increased, we propose that hemostatic plugs in these animals are more porous than those in control mice, a conclusion that would be consistent with the increased plasma leakage into the joints of hemophilic patients. Further studies will be required to test this hypothesis.
The laser injury model also allows for repeated injuries to the same site, leading to a progressive increase in the lesion area and thus increased platelet/fibrin signals and prolonged bleeding times. This graded injury response provides additional sensitivity in situations where a single injury did not reveal marked defects in hemostatic plug formation. For example, a single injury to the saphenous vein of TFlow mice did not lead to an increase in the bleeding time as compared with control mice. However, a small but significant increase in the bleeding time was observed after the second and third injuries, consistent with the minor hemostatic defect observed in these mice after tail transection [32]. A further increase in the sensitivity of our assay to clotting defects was observed in mice treated with the P2Y12 inhibitor clopidogrel bisulfate. Whereas clopidogrel administration had only a small effect on clot stability and the bleeding time after laser injury in WT mice, it markedly impaired hemostasis in TFlow mice. This finding is in good agreement with the markedly increased bleeding observed in hemophilic patients who are given platelet inhibitors [33]. Thus, partial inhibition of platelet function provides a powerful tool with which to sensitize the laser injury hemostasis model for coagulation defects.
In summary, we describe here a novel method to monitor and quantify the hemostatic process in mice. Small injuries are generated along the exposed saphenous vein with the use of a laser ablation system, and platelet adhesion and fibrin accumulation are monitored by intravital microscopy. Studies in mutant mice defective in platelet adhesion or coagulation further suggest this assay as a model for hemostasis after small-vessel lesions in humans, i.e. a situation where hemostasis is largely dependent on the ability of platelets to form a hemostatic plug. On the basis of this sensitivity to platelet adhesion defects and its real-time imaging capability, we propose this model as an ideal tool with which to study the efficacy and safety of antiplatelet agents.
Supplementary Material
Fig. S1. Laser injury model.
Fig. S2. Platelet accumulation at three different injury locations along the saphenous vein.
Video S1. Formation of a hemostatic plug in an injured venule of a wild-type mouse.
Video S2. Formation of a hemostatic plug in an injured venule of a talin1f/fPF4-Cre mouse.
Video S3. Formation of a hemostatic plug in an injured venule of an IL4R/GPIb-tg mouse.
Video S4. Formation of a hemostatic plug in an injured venule of a TFlow mouse.
Video S5. Formation of a hemostatic plug in an injured venule of an FIX−/− mouse.
Video S6. Formation of a hemostatic plug in an injured venule of a wild-type mouse treated with clopidogrel bisulfate.
Video S7. Formation of a hemostatic plug in an injured venule of a TFlow mouse treated with clopidogrel bisulfate.
Acknowledgments
We thank A. Cholka for providing excellent animal husbandry services, and Y. Boulaftali and D. Paul for help with clopidogrel studies and antibody labeling, and helpful discussions. We thank J. Ware for providing IL4R/GPIb-tg mice.
This work was supported by the American Heart Association (14EIA18910004 to W. Bergmeier) and the National Heart, Lung, and Blood Institute, NIH, grants HL106009 (W. Bergmeier), HL117659 (D. Monroe), and HL006350 (N. Mackman).
Footnotes
Addendum
T. Getz and W. Bergmeier designed the study, performed experiments, analyzed the data, and wrote the manuscript. R. Piatt performed experiments and analyzed data. B. G. Petrich provided transgenic mice and reviewed the manuscript. D. Monroe and N. Mackman provided transgenic mice and contributed to data interpretation and the writing of the manuscript.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Disclosure of Conflict of Interests
The authors state that they have no conflict of interest.
References
- 1.Broos K, Feys HB, De Meyer SF, Vanhoorelbeke K, Deckmyn H. Platelets at work in primary hemostasis. Blood Rev. 2011;25:155–67. doi: 10.1016/j.blre.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 2.Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol. 2010;11:288–300. doi: 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petrich BG, Marchese P, Ruggeri ZM, Spiess S, Weichert RAM, Ye F, Tiedt R, Skoda RC, Monkley SJ, Critchley DR, Ginsberg MH. Talin is required for integrin-mediated platelet function in hemostasis and thrombosis. J Exp Med. 2007;204:3103–11. doi: 10.1084/jem.20071800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nieswandt B, Moser M, Pleines I, Varga-Szabo D, Monkley S, Critchley D, Fassler R. Loss of talin1 in platelets abrogates integrin activation, platelet aggregation, and thrombus formation in vitro and in vivo. J Exp Med. 2007;204:3113–18. doi: 10.1084/jem.20071827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Savage B, Almus-Jacobs F, Ruggeri ZM. Specific synergy of multiple substrate–receptor interactions in platelet thrombus formation under flow. Cell. 1998;94:657–66. doi: 10.1016/s0092-8674(00)81607-4. [DOI] [PubMed] [Google Scholar]
- 6.Bergmeier W, Piffath CL, Goerge T, Cifuni SM, Ruggeri ZM, Ware J, Wagner DD. The role of platelet adhesion receptor GPIbalpha far exceeds that of its main ligand, von Willebrand factor, in arterial thrombosis. Proc Natl Acad Sci USA. 2006;103:16900–5. doi: 10.1073/pnas.0608207103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanaji T, Russell S, Ware J. Amelioration of the macrothrombocytopenia associated with the murine Bernard–Soulier syndrome. Blood. 2002;100:2102–7. doi: 10.1182/blood-2002-03-0997. [DOI] [PubMed] [Google Scholar]
- 8.Mackman N. The role of tissue factor and factor VIIa in hemostasis. Anesth Analg. 2009;108:1447–52. doi: 10.1213/ane.0b013e31819bceb1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parry GC, Ehrlich JH, Carmeliet P, Luther T, Mackman N. Low levels of tissue factor are compatible with development and hemostasis in mice. J Clin Invest. 1998;101:560–9. doi: 10.1172/JCI814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sixma JJ, van den Berg A. The haemostatic plug in haemophilia A: a morphological study of haemostatic plug formation in bleeding time skin wounds of patients with severe haemophilia A. Br J Haematol. 1984;58:741–53. doi: 10.1111/j.1365-2141.1984.tb06121.x. [DOI] [PubMed] [Google Scholar]
- 11.Satta N, Toti F, Fressinaud E, Meyer D, Freyssinet JM. Scott syndrome: an inherited defect of the procoagulant activity of platelets. Platelets. 1997;8:117–24. doi: 10.1080/09537109709169326. [DOI] [PubMed] [Google Scholar]
- 12.Bi L, Lawler AM, Antonarakis SE, High KA, Gearhart JD, Kazazian HH. Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat Genet. 1995;10:119–21. doi: 10.1038/ng0595-119. [DOI] [PubMed] [Google Scholar]
- 13.Lin HF, Maeda N, Smithies O, Straight DL, Stafford DW. A coagulation factor IX-deficient mouse model for human hemophilia B. Blood. 1997;90:3962–6. [PubMed] [Google Scholar]
- 14.Gailani D, Lasky NM, Broze GJ. A murine model of factor XI deficiency. Blood Coagul Fibrinolysis. 1997;8:134–44. doi: 10.1097/00001721-199703000-00008. [DOI] [PubMed] [Google Scholar]
- 15.Thijs T, Deckmyn H, Broos K. Model systems of genetically modified platelets. Blood. 2012;119:1634–42. doi: 10.1182/blood-2011-10-381715. [DOI] [PubMed] [Google Scholar]
- 16.Nakanishi-Matsui M, Zheng YW, Sulciner DJ, Weiss EJ, Ludeman MJ, Coughlin SR. PAR3 is a cofactor for PAR4 activation by thrombin. Nature. 2000;404:609–13. doi: 10.1038/35007085. [DOI] [PubMed] [Google Scholar]
- 17.McKenzie SE. Humanized mouse models of FcR clearance in immune platelet disorders. Blood Rev. 2002;16:3–5. doi: 10.1054/blre.2001.0170. [DOI] [PubMed] [Google Scholar]
- 18.Hayashi T, Nishioka J, Kamada H, Asanuma K, Kondo H, Gabazza EC, Ido M, Suzuki K. Characterization of a novel human protein C inhibitor (PCI) gene transgenic mouse useful for studying the role of PCI in physiological and pathological conditions. J Thromb Haemost. 2004;2:949–61. doi: 10.1111/j.1538-7836.2004.00733.x. [DOI] [PubMed] [Google Scholar]
- 19.Wood JP, Ellery PER, Maroney SA, Mast AE. Biology of tissue factor pathway inhibitor. Blood. 2014;123:2934–43. doi: 10.1182/blood-2013-11-512764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greene TK, Schiviz A, Hoellriegl W, Poncz M, Muchitsch EM. Animal models subcommittee of the Scientific and Standardization Committee of the ISTH. Towards a standardization of the murine tail bleeding model. J Thromb Haemost. 2010;8:2820–2. doi: 10.1111/j.1538-7836.2010.04084.x. [DOI] [PubMed] [Google Scholar]
- 21.Pastoft AE, Lykkesfeldt J, Ezban M, Tranholm M, Whinna HC, Lauritzen B. A sensitive venous bleeding model in haemophilia A mice: effects of two recombinant FVIII products (N8 and Advate(®)) Haemophilia. 2012;18:782–8. doi: 10.1111/j.1365-2516.2012.02780.x. [DOI] [PubMed] [Google Scholar]
- 22.Stefanini L, Ye F, Snider AK, Sarabakhsh K, Piatt R, Paul DS, Bergmeier W, Petrich BG. A talin mutant that impairs talin–integrin binding in platelets decelerates αIIbβ3 activation without pathological bleeding. Blood. 2014;123:2722–31. doi: 10.1182/blood-2013-12-543363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stolla M, Stefanini L, Roden RC, Chavez M, Hirsch J, Greene T, Ouellette TD, Maloney SF, Diamond SL, Poncz M, Woulfe DS, Bergmeier W. The kinetics of alphaIIbbeta3 activation determines the size and stability of thrombi in mice: implications for antiplatelet therapy. Blood. 2011;117:1005–13. doi: 10.1182/blood-2010-07-297713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baumgartner B, Jaki T, Wolfsegger MJ, Eder B, Schiviz A, Schwarz HP, Muchitsch EM. Optimization, refinement and reduction of murine in vivo experiments to assess therapeutic approaches for haemophilia A. Lab Anim. 2010;44:211–17. doi: 10.1258/la.2010.009113. [DOI] [PubMed] [Google Scholar]
- 25.Westrick RJ, Winn ME, Eitzman DT. Murine models of vascular thrombosis. Arterioscler Thromb Vasc Biol. 2007;27:2079–93. doi: 10.1161/ATVBAHA.107.142810. [DOI] [PubMed] [Google Scholar]
- 26.Denis CV, Wagner DD. Platelet adhesion receptors and their ligands in mouse models of thrombosis. Arterioscler Thromb Vasc Biol. 2007;27:728–39. doi: 10.1161/01.ATV.0000259359.52265.62. [DOI] [PubMed] [Google Scholar]
- 27.Barr JD, Chauhan AK, Schaeffer GV, Hansen JK, Motto DG. Red blood cells mediate the onset of thrombosis in the ferric chloride murine model. Blood. 2013;121:3733–41. doi: 10.1182/blood-2012-11-468983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Gestel MA, Heemskerk JWM, Slaaf DW, Heijnen VVT, Sage SO, Reneman RS, oude Egbrink MGA. Real-time detection of activation patterns in individual platelets during thromboembolism in vivo: differences between thrombus growth and embolus formation. J Vasc Res. 2002;39:534–43. doi: 10.1159/000067208. [DOI] [PubMed] [Google Scholar]
- 29.van Gestel MA, Heemskerk JWM, Slaaf DW, Heijnen VVT, Reneman RS, oude Egbrink MGA. In vivo blockade of platelet ADP receptor P2Y12 reduces embolus and thrombus formation but not thrombus stability. Arterioscler Thromb Vasc Biol. 2003;23:518–23. doi: 10.1161/01.ATV.0000057809.32354.22. [DOI] [PubMed] [Google Scholar]
- 30.George JN. Platelets. Lancet. 2000;355:1531–9. doi: 10.1016/S0140-6736(00)02175-9. [DOI] [PubMed] [Google Scholar]
- 31.Broze GJ, Yin ZF, Lasky N. A tail vein bleeding time model and delayed bleeding in hemophiliac mice. Thromb Haemost. 2001;85:747–8. [PubMed] [Google Scholar]
- 32.Pawlinski R, Pedersen B, Erlich J, Mackman N. Role of tissue factor in haemostasis, thrombosis, angiogenesis and inflammation: lessons from low tissue factor mice. Thromb Haemost. 2004;92:444–50. doi: 10.1160/TH04-05-0309. [DOI] [PubMed] [Google Scholar]
- 33.Kaneshiro MM, Mielke CH, Kasper CK, Rapaport SI. Bleeding time after aspirin in disorders of intrinsic clotting. N Engl J Med. 1969;281:1039–42. doi: 10.1056/NEJM196911062811904. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Laser injury model.
Fig. S2. Platelet accumulation at three different injury locations along the saphenous vein.
Video S1. Formation of a hemostatic plug in an injured venule of a wild-type mouse.
Video S2. Formation of a hemostatic plug in an injured venule of a talin1f/fPF4-Cre mouse.
Video S3. Formation of a hemostatic plug in an injured venule of an IL4R/GPIb-tg mouse.
Video S4. Formation of a hemostatic plug in an injured venule of a TFlow mouse.
Video S5. Formation of a hemostatic plug in an injured venule of an FIX−/− mouse.
Video S6. Formation of a hemostatic plug in an injured venule of a wild-type mouse treated with clopidogrel bisulfate.
Video S7. Formation of a hemostatic plug in an injured venule of a TFlow mouse treated with clopidogrel bisulfate.