

Abstract
The immunosuppressive agent cyclosporin A (CsA), a calcineurin inhibitor which blocks T cell activation has provided the pharmacologic foundation for organ transplantation. CsA exerts additional effects on non-immune cell populations and may adversely effect microvascular endothelial cells, contributing to chronic rejection, a long-term clinical complication and significant cause of mortality in solid-organ transplants, including patients with small bowel allografts. Growth of new blood vessels, or angiogenesis, is a critical homeostatic mechanism in organs and tissues, and regulates vascular populations in response to physiologic requirements. We hypothesized that CsA would inhibit the angiogenic capacity of human gut microvessels. Primary cultures of human intestinal microvascular endothelial cells (HIMEC) were used to evaluate CsA's effect on four in vitro measures of angiogenesis, including endothelial stress fiber assembly, migration, proliferation and tube formation, in response to the endothelial growth factor VEGF. We characterized the effect of CsA on intracellular signaling mechanisms following VEGF stimulation. CsA affected all VEGF induced angiogenic events assessed in HIMEC. CsA differentially inhibited signaling pathways which mediated distinct steps of the angiogenic process. CsA blocked VEGF induced nuclear translocation of the transcription factor NFAT, activation of p44/42 MAPK, and partially inhibited JNK and p38 MAPK. CsA differentially affected signaling cascades in a dose dependent fashion and completely blocked expression of COX-2, which was integrally linked to HIMEC angiogenesis. These data suggest that CsA inhibits the ability of microvascular endothelial cells to undergo angiogenesis, impairing vascular homeostatic mechanisms and contributing to the vasculopathy associated with chronic rejection.
Background
The calcineurin inhibitor cyclosporin A (CsA) is a potent immunosuppressive agent that has formed the pharmacologic cornerstone of solid organ transplantation. CsA prevents the activation of lymphokine genes essential for T cell proliferation by disrupting calcium-dependent signal transduction pathways in leukocytes [1]. Although pharmacologic studies of CsA have focused primarily on T cell responses, there is emerging evidence that this agent may exert potent effects on blood vessels, promoting arterial hypertension, inducing long-term vascular dysfunction, and contributing to obliterative vasculopathy in chronic transplant rejection [2-5]. At the present time, chronic rejection with its associated vasculopathy, is the major cause of late allograft dysfunction, including patients with intestinal transplants [6,7].
In solid-organ transplantation, the vascular endothelium has received attention because of its unique role as the interface between the donor graft and the host's circulating immune cells, and as a focus of acute rejection [8,9]. More recent investigation has demonstrated that the endothelium plays a central role in chronic rejection, where inappropriate activation of endothelial cells results in obliterative vasculopathy and accelerated post-transplant atherosclerosis [10], a major cause of morbidity and mortality in solid organ transplant recipients. Activation of graft endothelium in chronic rejection may result from host/graft immunologic attack, as well as dysfunction associated with transplant immunosuppression [5]. In transplantation of the small bowel, microvascular dysfunction may contribute to more significant problems with both acute and chronic rejection in these patients. Indeed, small bowel transplantation has been one of the more problematic clinical areas in the realm of solid organ grafts, where patients require increased immunosuppressive regimens and have had overall, less successful clinical outcomes [11-13].
The growth of new microvessels, or angiogenesis, is now appreciated to be a critical biologic process involved in tissue homeostasis. Angiogenesis is initiated by local activation of genes encoding diffusable angiogenic factors, or by the release of vascular growth factors which subsequently act on local microvasular cell populations, as well as by a decrease in local angiostatic factors, including interferon beta [14]. Angiogenesis involves an orchestrated sequence of steps which include endothelial activation, stress fiber assembly, fibrinolysis, proteolytic degradation of the basement membrane and the extracellular matrix, migration, proliferation and neovascularization [15]. One of the major angiogenic growth factors is the vascular endothelial growth factor (VEGF), which selectively induces activation, migration, proliferation and tube formation in endothelial cells in vitro. VEGF is a 34–42 kDa glycoprotein which exerts its biological effects on endothelial cells through its two major tyrosine kinase receptors, VEGFR1/Flt-1 (fetal liver kinase-1) and VEGFR2/Flk-1/KDR (kinase insert domain containing receptor). By binding to these receptors, VEGF activates various signaling cascades, including the mitogen activated protein kinase family (ERK1/2, p38 MAPK and SAPK/JNK) and phosphoinositol3-kinase (PI3 kinase) [16,17]. A downstream result of these signaling events is the expression of COX-2, which plays an integral role in VEGF induced angiogenesis [18-20]. Finally, investigation has demonstrated a pivotal role for the transcription factor nuclear factor activated in T-cells (NFAT) in the angiogenic signaling of VEGF in human umbilical vein endothelial cells (HUVEC) [21], which is inhibited by CsA. Thus, there is a potential role for CsA in blocking angiogenesis in microvascular cell populations, potentially through its effect on NFAT.
An integrated analysis of the effect of CsA on the multiple stages of the angiogenic process in human organ specific microvascular endothelial cells has not been performed to date. Studies evaluating the effect of CsA on human umbilical vein endothelial cells [22] may not accurately reflect microvascular events, and are further complicated by their wide functional variability in tissue culture, causing discrepant results reported by different laboratories [23]. In addition, it is also now appreciated that human microvascular endothelial cells derived from differentiated, organ specific vascular beds have been shown to differ significantly from HUVEC in their responsiveness to cytokines, the expression profile of antigens, and the elaboration of secretory products [24-27]. We investigated the mechanisms of VEGF-induced endothelial cell signaling and angiogenesis in HIMEC, an organ specific microvascular cell population, and the effect of CsA on four in vitro components of angiogenesis, including stress fiber assembly, migration, proliferation/growth and tube formation.
Results
VEGF, but not TNF-α/LPS, enhances growth of HIMEC
We have previously shown that VEGF is a potent stimulus for in vitro growth and proliferation of HIMEC [28,29]. Additional studies have demonstrated that in vitro activation of HIMEC with TNF-α/LPS will result in inflammatory activation, expression of cell adhesion molecules and increased leukocyte binding which is mediated by activation of NF-κB and mitogen activated protein kinases [30]. Because MAPK activation has been linked to proliferation of various cell types, including endothelial cells, we performed in vitro growth studies to assess the effect of VEGF and TNF-α/LPS on HIMEC. HIMEC monolayers stimulated with VEGF for 24 hr resulted in a significant increase in cell number, while TNF-α/LPS had essentially no effect and was similar to unstimulated cell growth (Figure 1). As expected, VEGF stimulation of HIMEC resulted in activation of all three members of MAPK family members (p44/42 MAPK, JNK, p38 MAPK; Figure 2), which was similar to that achieved by stimulation with TNF-α/LPS [30]. This data demonstrates that MAPK activation is a common pathway involved in both the inflammatory and angiogenic activation of HIMEC, but also suggested that growth and proliferation would involve additional intracellular signaling mechanisms.
Figure 1.

VEGF, but not TNF-α/LPS stimulation of HIMEC results in enhanced cell growth. HIMEC monolayers stimlulated with VEGF (50 ng/ml) for 24 hr resulted in a significant increase in cell number, while TNF-α/LPS had essentially no effect and was similar to unstimulated cell growth.
Figure 2.

VEGF enhances phosphorylation of MAPK family members of signaling proteins in HIMEC. Activation of p44/42 MAPK, JNK and p38 MAPK in HIMEC by VEGF was assessed using Western blot analysis. Total cell lysates from cultured HIMEC stimulated with VEGF (10–50 ng/ml), were subjected to SDS-PAGE and immunoblotted with phospho-specific anti-ERK 1/2, JNK and p38 MAPK antibodies. VEGF enhances phosphorylation of all three MAPK members, as compared with total ERK. The time of induction ranged from 1 to 120 min as detailed in the materials and methods section. Maximum detection of p42/44 MAPK was seen at 30 min (shown); Maximum activation of p38 MAPK was detected at 10 min in response to VEGF; JNK maximum activation was seen at 5 min following VEGF activation. Coomassie staining of the polyacrylamide gel confirms equal amounts of total protein loaded. The representative figure shown is from one of three independent experiments.
Effect of CsA on VEGF induced stress fiber assembly in HIMEC
CsA exerts potent inhibitory effects on TNF-α/LPS induced activation of HIMEC, by inhibiting p38 MAPK activation [30]. We began experiments investigating the effect of CsA on VEGF induced angiogenic activation in HIMEC by assessing endothelial stress fiber assembly and cytoskeletal architectural rearrangement, an early step in angiogenesis. HIMEC rapidly demonstrated stress fiber assembly following 1–10 ng/ml VEGF stimulation (Figure 3). However, higher dosages of VEGF (i.e. 50 ng/ml) resulted in a reduction in stress fiber assembly, suggesting that the effects of this growth factor follow a bimodal distribution, where higher concentrations result in diminished biologic effects. CsA decreased actin stress fiber formation following VEGF which was similar to the effect of the p38 MAPK inhibitor SB203580. The p44/42 MAPK inhibitor PD098059 failed to affect VEGF induced stress fiber assembly. VEGF (10 ng/ml) induced stress fiber assembly in HIMEC was abolished by receptor blockade using neutralizing monoclonal antibodies, suggesting that this phenomenon was mediated through VEGFR2.
Figure 3.

Stress fiber assembly in HIMEC following VEGF stimulation. VEGF (10 ng/ml) induced stress fiber assembly was attenuated by anti-VEGFR2, CsA and SB203580 but not PD098059. The effect of VEGF on stress fiber polymerization in HIMEC was assessed by fluorescence staining with fluorescein phalloidin, a substance which specifically detects F-actin. Confluent HIMEC monolayers were grown on fibronectin-coated glass chamber slides, and stimulated with VEGF (1–10 ng/ml, 15 min) prior to staining. Control, minimal stress fibers are demonstrated in unstimulated HIMEC (negative control). VEGF, at 10 ng/ml but not at 50 ng/ml strongly increased stress fiber assembly. At some intercellular junctions, a marked retraction of endothelial cells is notable, leading to scattered interruptions in continuity of the endothelial cell monolayer. Pre-incubation of HIMEC with SB203580 (p38 MAPK inhibitor, 5 μM), CsA (0.1 μM) and neutralizing anti-VEGFR2 antibody (10 μg/ml, 15 min, 37°C) markedly attenuated stress fiber assembly by VEGF (note diminished fluorescence intensity and less pronounced stress fiber induction). Fluorescence microscopic images were obtained with an original magnification of 400×, at a fixed shutter speed. Figure shows representative images from one of three independent experiments.
Immunosuppressive agents inhibit VEGF induced migration, proliferation and growth of HIMEC
Angiogenesis involves multiple events in endothelial cells, including migration and proliferation. HIMEC migration demonstrated a bimodal distribution in response to increasing dosages of VEGF, where 1–10 ng/ml resulted in a higher response compared to 50 ng/ml (Figure 4 left panel). CsA significantly inhibited VEGF induced migration of HIMEC, which was similar to the p38 MAPK inhibitor SB203580 (Figure 4 right panel). FK506, an additional calcineurin inhibitor and potent immunosuppressive agent, exhibited a similar effect compared to CsA, which exhibited a low level increase in baseline migration, which was similar to that observed in VEGF stimulated cells. Rapamycin, an immunosuppressive agent which functions through a distinct mechanism, interacting with m-TOR (mammalian target of rapamycin), demonstrated a stimulatory effect on resting cells and a diminished inhibition of VEGF induced HIMEC migration compared to CsA and FK506. Taken together with the previous experiments, these data suggest that CsA is a potent inhibitor of both stress fiber formation and cell migration, which is potentially mediated through effects on p38 MAPK activation.
Figure 4.

VEGF at low concentration (1–10 ng/ml) is a potent chemoattractant for HIMEC migration in vitro. Effect of VEGF on HIMEC migration assessed by a Transwell chemotaxis assay. HIMEC migrating across fibronectin-coated polycarbonate filters (pore size, 8 μm) were quantified using modified Wright's stain (Diff-Quik) and bright field microscopy. Left panel; VEGF (1–10 ng/ml) elicits strong chemotactic properties in HIMEC. Note the biphasic dose response, as higher concentrations of VEGF (50 ng/ml and higher) do not result in marked chemotaxis in HIMEC. Right panel; VEGF (10 ng/ml) induced chemotaxis in HIMEC is decreased by specific inhibitors of p38 MAPK (SB203580), CsA and FK506 but not Rapamycin. As shown in the right panel, VEGF acts as a potent chemoattractant for HIMEC. This chemotactic response was potently diminished by SB203580 (5 μM), CsA (0.1 μM) and FK506 (50 nM). * = p < 0.05 versus positive control; n.s. = not significant. No VEGF, denotes the negative control while VEGF (10 ng/ml) served as the positive control. All conditions were assessed in triplicate, and data are expressed as mean number of migrated cells per high-power field (200×) ± S.E.
Cell cycle re-entry and DNA replication in endothelial cells is a requisite step in angiogenesis. Experiments were performed on HIMEC proliferation assessing 3H thymidine uptake in response to low (10 ng/ml) and high (50 ng/ml) dose VEGF. Proliferation in response to VEGF (10 ng/ml) revealed a complete inhibition of increased thymidine uptake in HIMEC treated with CsA and the COX-2 inhibitor NS398 (Figure 5 right panel). In contrast to previous studies in stress fiber assembly and migration, the p44/42 MAPK inhibitor PD098059 was more effective than the p38 MAPK inhibitor SB203580 in blocking thymidine uptake in HIMEC stimulated with 10 ng/ml VEGF. At high dosages of VEGF (i.e. 50 ng/ml), CsA continued to block proliferation, which was partially inhibited by SB203580 and PD098059 (Figure 5 left panel).
Figure 5.

VEGF enhances the proliferation rate of HIMEC in vitro. VEGF-induced HIMEC proliferation was assessed by 3H thymidine uptake. 4 × 104 HIMEC/well were stimulated for 12 hr with increasing concentrations of VEGF (0, 1, 10, and 100 ng/ml) and pulsed for 6 hr with 1 μCi/ml 3H thymidine. 3H thymidine uptake in total cell lysates was measured in a β-counter. left; VEGF enhanced HIMEC proliferation at 50 ng/ml while low concentrations (i.e. 10 ng/ml VEGF) did not. right; The inhibitory effects of PD098059, CsA and NS398 but not SB203580 on VEGF (50 ng/ml) induced proliferation response in HIMEC. HIMEC pre-incubation with PD098059, CsA and NS398 significantly inhibited VEGF induced proliferation response in HIMEC. All conditions were assessed in triplicate, and data are expressed as percent of control 3H thymidine uptake (0 ng/ml VEGF) ± S.E. *, p < 0.05 versus control.
Experiments evaluating endothelial growth in a wounded monolayer, with cell expansion across a leading edge, again demonstrated a potent angiogenic effect of VEGF compared to cells which were unstimulated (Figure 6). CsA treated HIMEC monolayers were completely unresponsive to VEGF, and grew at rates similar to untreated cells. In these experiments, the most potent inhibitor of monolayer expansion was the p44/42 MAPK inhibitor PD098059, while SB203580 failed to block this phenomenon.
Figure 6.

HIMEC growth across a leading edge demonstrated that VEGF (10 ng/ml) (■) resulted in increased monolayer expansion compared to cells which were unstimluated (◆). CsA (0.1 μM) treated monolayers completely blocked the effect of VEGF, and grew at rates similar to untreated cells (●). The most potent inhibitor of monolayer expansion was the p44/42 MAPK inhibitor PD098059 (▲), whereas p38 MAPK inhibitor SB203580 had no effect on HIMEC monolayer expansion (□).
Matrigel™ In vitro-Tube Formation in HIMEC
In vitro-angiogenesis of HIMEC was assessed by tube formation assays. HIMEC seeded onto Matrigel™ in complete growth medium display formation of robust tube-like structures within 8 hr (data not shown), with further maturation after 16 hr (Fig 7 upper panel). CsA (0.1 μM), PD098059 (10 μM) and NS398 (10 μM) exhibited a marked inhibitory effect on the formation of tube-like structures by HIMEC, visible by the disruption of these capillary-like microvessels and cells remaining coherent in spherical clusters (Fig 7 lower panel). These findings indicate that activation of p44/42 MAPK and COX-2 is required for endogenous in vitro-tube formation in HIMEC.
Figure 7.
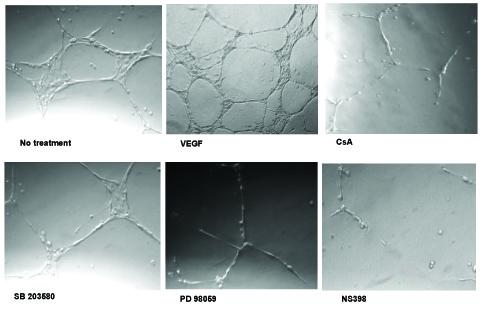
In vitro tube formation of HIMEC requires activation of ERK 1/2 (p44/42 MAPK). In vitro endothelial tube formation assays employed Matrigel™ as a three-dimensional extracellular matrix. HIMEC in complete growth medium (5 × 104) were seeded onto 24-well plates containing Matrigel™ (5 mg/ml). Where indicated, the medium was supplemented with CsA (0.1 μM), p38 MAPK inhibitor SB203580 (5 μM), specific inhibitors of MAPK kinase (PD098059, 10 μM), and COX-2 inhibitor NS398 (10 μM), respectively. Cells receiving DMSO served as a vehicle control, and were equivalent to no treatment (data not shown). Naive HIMEC seeded in complete growth medium display robust tube formation. Addition of VEGF to growth medium resulted in enhanced tube formation. All three inhibitors markedly abrogated spontaneous tube formation of HIMEC seeded in complete growth medium. Note the cells remaining adherent in spherical clusters, lacking mature tube-like structures.
VEGF enhances phosphorylation of MAPK
The ability of VEGF to induce MAPK phosphorylation was assessed by immunoblot analysis using phospho-specific antibodies. VEGF stimulation of HIMEC leads to a marked phosphorylation and activation of extracellular signal regulated kinase (ERK 1/2, p44/42 MAPK), SAPK/JNK and p38 MAPK. ERK 1/2 phosphorylation by VEGF in HIMEC was time and dose dependent, reaching its maximum at 30 min of stimulation by 50 ng/ml of VEGF (Figure 8). PD098059 and CsA abolished p44/42 MAPK phoshorylation in HIMEC whereas SB203580 did not. Equal loading of protein was assured by western blotting after stripping and reprobing membranes with non-phospho-specific-MAPK antibodies along with Coomassie staining of the polyacrylamide gel after separation of proteins. Phosphorylation of p38 MAPK by VEGF in HIMEC was maximal at 5 min, and SB203580, a specific inhibitor of p38 MAPK resulted in inhibition of the p38 MAPK activity but not its phosphorylation. CsA partially inhibited p38 MAPK activation in HIMEC in response to VEGF (Figure 9). Control experiments were performed which demonstrated no effect of SB203580, PD098059 on the non-VEGF stimulated HIMEC (data not shown).
Figure 8.

VEGF enhanced phosphorylation of ERK 1/2 (p44/42 MAPK) and p38 MAPK in HIMEC is diminished by specific inhibitors of ERK 1/2, p38 MAPK and CsA. Activation of ERK 1/2, a member of the MAPK superfamily, in HIMEC by VEGF was assessed using Western blot analysis. Total cell lysates from VEGF stimulated HIMEC with or without inhibitors were subjected to SDS-PAGE and immunoblotted with phosphorylated and non-phosphorylated specific anti-ERK 1/2 antibodies. VEGF enhanced phosphorylation of ERK 1/2 was diminished by CsA and PD098059, as compared with total ERK 1/2. TNF-α/LPS stimulation of HIMEC served as a positive control for ERK 1/2 activation.
Figure 9.

Activation of p38 MAPK, a member of the MAPK superfamily, in HIMEC by VEGF was assessed using Western blot analysis, as described above using phosphorylated and non-phosphorylated specific anti-p38 MAPK antibodies. VEGF enhanced phosphorylation of p38 MAPK is diminished by CsA and decreased by SB203580 (inhibits the p38 MAPK activity but not its phosphorylation), as compared with total p38 MAPK. Again, TNF-α/LPS activation served as a positive control. Coomassie blue staining of the polyacrylamide gel confirms equal amounts of total protein loaded. The representative figure shown is from one of three independent experiments.
Effect of VEGF on Nuclear Factor of Activated T cells (NFAT) in HIMEC
VEGF stimulation of HIMEC leads to dephosphorylation of NFATp and its nuclear translocation from the cytosol as detected by western blot analysis using specific anti-NFAT antibodies. Stimulation of HIMEC with 50 ng/ml VEGF resulted in a complete NFATp translocation to the nucleus within 15 min which was sustained for 2 hr. Nuclear NFATp declined to undetectable levels after 5 hr (Figure 10). Pre-treatment of HIMEC with 0.1 μM of CsA prevented NFATp translocation to the nucleus in VEGF stimulated cells, as shown by western blot analysis of nuclear extracts (Figure 11). Proinflammatory activation of HIMEC using a combination of TNF-α/LPS did not result in a nuclear translocation of NFATp, demonstrating the specificity of this signaling pathway in angiogenesis.
Figure 10.

Effect of VEGF on Nuclear Factor of Activated Tcells (NFAT) activation in HIMEC. VEGF stimulation of HIMEC leads to dephosphorylation of NFATp and its nuclear translocation from the cytosol as detected by western blot analysis using specific anti-NFAT antibodies. Stimulation of HIMEC with 50 ng/ml VEGF resulted in a complete NFATp translocation to the nucleus within 15 min which was sustained for 2 hr. Nuclear NFATp declined to undetectable levels after 5 hr.
Figure 11.

Pre-treatment of HIMEC with 0.1 μM of CsA prevented NFATp translocation to the nucleus in VEGF stimulated cells, as shown by Western blot analysis of nuclear extracts. Proinflammatory activation of HIMEC using combination TNF-α/LPS did not result in a nuclear translocation of NFATp.
DNA-binding activity of NFATp, NF-κB and AP-1 in VEGF activated HIMEC
Having shown that VEGF activates NFATp in HIMEC, we next determined the binding of the transcription factor to an NFAT site of the human COX-2 promoter (nucleotides -117 to -91), NF-κB (nucleotides -277 to-211 containing the NFκB site of the human COX-2 promoter) and AP-1 (nucleotides -82 to -58 containing the NFAT-AP1 site of the human COX-2 promoter) [22]. Figure 12 shows the electrophoretic mobility shift assay analysis of the nuclear extracts from VEGF activated HIMEC which demonstrate a marked increase in NFAT sequences of the COX-2 promoter bound to nuclear proteins of VEGF activated but not TNF-α/LPS stimulated HIMEC. The NFAT DNA-binding activity in HIMEC was inhibited by 0.1 μM of CsA pre-treatment (Figure 12 left panel). Next we studied the effect of VEGF on NF-κB activation in HIMEC. Figure 12 (middle panel) demonstrates that VEGF fails to activate NF-κB in HIMEC. This was in contrast to TNF-α/LPS activation of HIMEC which resulted in marked NF-κB activation. CsA had no effect on TNF-α/LPS induced binding of NF-κB (Figure 12 middle panel). VEGF stimulation of HIMEC also resulted in increased AP-1 activity, as shown in Figure 12 (right panel), AP-1 probe efficiently and specifically bound to nuclear proteins and this binding was not sensitive to CsA pre-treatment
Figure 12.

Electrophoretic mobility shift assay of NFATp, NFκB and AP-1 in VEGF and TNF-α/LPS stimulated HIMEC. Nuclear extracts from VEGF (50 ng/ml) or TNF-α/LPS stimulated HIMEC with or without CsA (0.1 μM) were subjected to EMSA using probes to left panel; the NFAT site of human COX-2 promoter (nucleotides -117 to -91). Middle panel; the NFκB site of the human COX-2 promoter (nucleotides -135 to -123). Right panel; the NFAT-AP1 site of the human COX-2 promoter probe (nucleotides -82 to -58). CsA inhibited NFAT DNA binding following VEGF activation in HIMEC (A). NFAT was not activated by TNF-α/LPS. VEGF did not activate NFκB in HIMEC, which was activated following TNF-α/LPS (B). VEGF activated AP-1, which was not inhibited by CsA (C). These data demonstrate the selective inhibitory effect of CsA on NFAT acativation following VEGF stimulation in HIMEC.
To visualize the nuclear translocation of NFAT, the p65 subunit of NF-κB and the AP-1 components c-fos and c-Jun, we performed immunofluorescence staining of VEGF stimulated HIMEC using specific antibodies (i.e. anti-NFATp, p65, c-fos and c-Jun). As shown in Figure 13, in unstimulated HIMEC, NFATp was detected in the cytoplasm and following VEGF activation was translocated to the nucleus, whereas TNF-α/LPS activation did not result in nuclear translocation of NFAT. CsA pre-treatment resulted in abrogation of the NFATp nuclear translocation (Figure 13). Thus, the immunofluorescence results confirmed the data obtained from western blot analysis. Interestingly, VEGF did not lead to translocation of the NF-κB-subunit p65, whereas TNF-α/LPS activation of HIMEC resulted in p65 translocation from cytosol to the nucleus and was unaffected by CsA (Figure 14). VEGF stimulation of HIMEC also resulted in nuclear translocation of c-fos and c-Jun, which was also not affected by CsA (Figure 15).
Figure 13.

Effect of VEGF on NFAT, p65 subunit of NFκB, c-fos and c-Jun in HIMEC. Immunofluorescence staining was used to visualize nuclear translocation following VEGF induced activation of NFAT, and the p65 subunit of NFκB and AP-1 component (c-fos and c-Jun) of HIMEC. In unstimulated HIMEC, NFATp was detected in cytoplasm, and following VEGF activation was translocated to the nucleus, whereas TNF-α/LPS activation of HIMEC did not result in nuclear NFAT translocation. CsA pre-treatment resulted in abrogation of the NFATp nuclear translocation following VEGF stimulation.
Figure 14.

VEGF did not lead to translocation of the NFκB-subunit p65, whereas TNF-α/LPS activation of HIMEC resulted in p65 subunit translocation from cytosol to the nucleus in a CsA-insensitive manner.
Figure 15.

VEGF stimulation of HIMEC also resulted in nuclear translocation of c-fos and c-Jun independent of CsA effect.
VEGF increases COX-2 gene expression in HIMEC
Activation of HIMEC with VEGF also resulted in enhanced COX-2 expression at the mRNA level as detected by RT-PCR using COX-2 specific primers. Enhanced COX-2 expression was time dependent, increased from 3–6 hr which was maximal, declining from 12 to 24 hr. Pre-treatment of HIMEC with 0.1 μM of CsA, 10 μM NS398 and 10 μM PD098059 completely abolished VEGF induction of COX-2, whereas SB203580 did not exert an inhibitory effect on expression of this gene product (Figure 16). There was no detectable change in the level of COX-1 mRNA after VEGF stimulation. β-actin gene expression was used as an internal control in these experiments.
Figure 16.

Effect of VEGF on COX-1 and COX-2 mRNA and protein expression in HIMEC. Detection of COX-1 and COX-2 mRNA in HIMEC by semi-quantitative reverse transcriptase-PCR using specific primers. HIMEC constitutively express mRNA for COX-1 but not COX-2. Stimulation (3 h) of HIMEC with VEGF (50 ng/ml) led to marked up-regulation of COX-2 gene expression, whereas COX-1 was unaffected. CsA, NS398 and PD098059 pre-treatment of HIMEC inhibited COX-2 mRNA expression following VEGF. β-actin, was used as loading control. Representative figure for HIMEC isolates from five different patients.
The effect of various cytokines on COX-2 protein expression was determined. As demonstrated in Figure 17 both TNF-α/LPS and VEGF were strong activators of COX-2 expression in HIMEC. Enhanced COX-2 protein expression in HIMEC following VEGF activation was time dependent, which increased at 3 hr and declined after 24 hr, as detected by western blotting using a specific anti-COX-2 antibody. In marked contrast, levels of COX-1 protein expression remained unchanged following VEGF stimulation of HIMEC (Figure 18). Consistent with the gene expression data, pre-treatment of HIMEC with 0.1 μM of CsA completely abolished VEGF induction of COX-2 protein expression (Figure 18).
Figure 17.

VEGF enhances COX-2 protein expression in HIMEC. Western blot analysis of HIMEC stimulated by various cytokines and chemokines revealed that VEGF and TNF-α/LPS activation resulted in equally enhanced COX-2 protein expression. HIMEC were stimulated with TNF-α/LPS, IL-1β, IL-8 and IL-1β/IL-8 for 3 – 12 h (Data shown is 3 h activation time period, which was maximal).
Figure 18.

Time course of enhanced COX-2 protein expression by VEGF in HIMEC. VEGF (50 ng/ml) resulted in a maximal increase of COX-2 protein at 3 hr which declined after 24 hr as detected by western blotting. There were no changes in COX-1 protein expression in VEGF stimulated HIMEC. Pretreatment of HIMEC with 0.1 μM CsA diminished COX-2 protein expression following VEGF. Both NS398 and PD098059 were strong inhibitors of COX-2 protein expression in VEGF stimulated HIMEC (not shown).
Discussion
In this study, we demonstrate that CsA exerts potent effects on the angiogenic capacity of human microvascular endothelial cells, differentially inhibiting multiple stages in the in vitro angiogenic process. This inhibitory effect of CsA on VEGF function in HIMEC involved 1) actin assembly and stress fiber formation, 2) cell migration, 3) proliferation and monolayer expansion (growth), and 4) tube formation. Furthermore, experiments focusing on the effect of CsA on signaling events following VEGF induced HIMEC activation demonstrated blockade of the DNA binding activity of the transcription factor NFAT, complete inhibition of p44/42 MAPK activation, partial inhibition of p38 MAPK, and complete inhibition of COX-2, with no effect on AP-1 activation. Furthermore, our data demonstrate that these signaling cascades play specific roles in the four components of angiogenesis which were modeled in our human organ specific microvascular system, and were differentially affected by CsA. Thus, CsA effects multiple signaling pathways in VEGF induced angiogenesis, which may ultimately impact on vessel growth, an important component of vascular homeostasis and may contribute to the vasculopathy of chronic rejection.
Receptor tyrosine kinases and their ligands play a crucial role in vascular development, and considerable progress has been made toward understanding the cellular and molecular events in angiogenesis [31]. Activation of endothelial signal transduction pathways are essential aspects of neovascularization, as endothelial activation must occur for cells to undergo angiogenesis. Although the biological functions of VEGF and its role in the angiogenic process have been extensively explored, the intracellular signaling pathways that lead to distinct gene expression patterns and altered endothelial physiology in response to VEGF in human organ specific microvascular endothelial cell populations remain incompletely defined, nor has the VEGF induced signaling in endothelial cells been characterized in an integrated fashion. Data obtained in this study demonstrate that VEGF activation of HIMEC results in activation all three MAPK members (ERK1/2, p38 MAPK and SAPK/JNK) and resulted in increased both migration and proliferation. Signaling through MAPKs are heavily dependent on the immediate environment which surrounds the endothelial cell. ERK1/2 is generally involved with cell growth and proliferation, whereas SAPKs are usually known to transduce stress signals [32]. Here we have shown that biological effects of VEGF on endothelial cells are concentration dependent, as 10 ng/ml VEGF resulted in HIMEC migration, p38 MAPK activation and actin/stress fiber formation while higher concentrations (i.e. 50 ng/ml) resulted in activation of p44/42 MAPK, COX-2 expression, NFAT activation and increased cell proliferation. This is in agreement with Rousseau et al. [33] and Seetharam et al. [34], who have shown that exposure of human umbilical vein endothelial cells (HUVEC) to concentrations of VEGF that increase cell migration also promotes actin polymerization, formation of stress fibers and recruitment of vinculin to ventral plaques. Actin fibers disappeared in HIMEC exposed to higher concentrations of VEGF (50–100 ng/ml) and cell migration was also diminished. SB203580, a p38 MAPK specific inhibitor blocked both cell migration and actin polymerization without affecting cell proliferation. Thus, our data suggest that p38 MAPK activation by VEGF plays a critical role in HIMEC migration by regulating actin polymerization dynamics and organization. Interestingly, another study by Tanaka and coworkers has demonstrated the importance of SAPK/p38 MAPK activation as a modulator of endothelial cell migration in response to bFGF [35].
Treatment of HIMEC with neutralizing VEGFR2 antibody abolished the cell migration, which was consistent with the observation that VEGFR2 is a positive regulator of angiogenesis [33]. However, the biochemical events that couple VEGFR2 to p38 MAPK activation and cell migration are still unknown. Studies have shown that VEGFR1 is involved in the down-regulation of the VEGFR2 which is required for angiogenesis [33]. Petrova et al. [36] demonstrated that VEGFR1 has a higher affinity than VEGFR2 for VEGF and can titrate VEGF by competing with VEGFR2, so at low concentration of VEGF, this will establish a threshold for the onset of angiogenesis and allows a down regulation of signal later in the process [33].
VEGF stimulation of endothelial cells results in dephosphorylation and translocation of NFATp from the cytoplasm to the nucleus in a time dependent fashion. The calcineurin inhibitor CsA blocked the translocation of NFATp. It has been shown that in T lymphocytes and fibroblasts, elevations of intracellular calcium levels result in calcineurin activation and subsequent activation and nuclear localization of NFAT proteins [37,38]. These data suggest that VEGF activation of the calcineurin pathway in HIMEC leads to the translocation of NFATp to the nucleus, where it is phosphorylated and then exported to the cytoplasm. Therefore, it appears that NFATp activation in HIMECs is regulated by similar mechanisms like those operating in T cells or fibroblasts.
NFAT proteins are the major targets of the calcineurin inhibitors CsA and FK506, and NFAT activation has been shown to affect the activation of other transcription factors which may in part explain the potent effect of CsA and FK506 as immunosuppressive agents [39,40]. CsA has been shown to play a role in the expression of cytokines produced by endothelial cells including IL-1, IL-6, and IL-8 in different cell types [41,42], thus NFAT may be potentially involved in the transcriptional regulation of these cytokine genes in endothelial cells. Our data corroborate previous studies demonstrating that NFAT activation is a key component of the angiogenic response induced by VEGF in endothelial cells [22], and modulation of this pathway may represent an adverse effect of calcineurin inhibition during transplant immunosuppression. Indeed, an increasing body of evidence suggests that the pharmacologic strategies which effectively inhibit acute rejection, may in fact be playing a central role in the etiogenesis of vascular dysfunction which is central to the pathology of chronic rejection, and is emerging as an important cause of morbidity and mortality in solid organ transplant patients in the years following successful engraftment.
The cyclooxygenase enzymes COX-1 and COX-2 have been shown to play an important role in the regulation of angiogensis [43]. These enzymes catalyze the conversion of arachidonic acid to PGH2, the first step in the biosynthesis of the PGs thromboxane and prostacyclin [44]. In endothelial cells COX-1 is constitutively expressed, whereas COX-2 is inducible in response to various activators such as mitogens, hormones and inflammatory cytokines [45]. The human COX-2 promoter contains binding sites for cAMP response element (CRE), NF-IL6 (C/EBP), and NFκB [46]. An essential role for NFAT in the induction of COX-2 gene by VEGF in HUVEC has been reported [22]. However, these investigators did not evaluate the activation of alternate signaling mechanisms which undergo activation in response to VEGF, specifically the MAPK family members. Our present study confirms that COX-2 and NFAT played an important role in microvascular endothelial angiogenesis induced by VEGF, but in addition demonstrated a differential contribution of p44/42 MAPK and p38 MAPK in various cellular physiologic steps which comprise angiogenesis.
VEGF activation of MAPKs (ERKs, JNKs/SAPKs, and p38 MAPK) signal transduction pathways in endothelial cells [47,48] are consistent with the VEGF activation of AP-1 that we show here. The MAPKs regulate the AP-1 family members at the transcriptional and posttranscriptional levels [49,50]. We have shown the induction of AP-1 by the AP-1 DNA-binding activity and the translocation of c-Fos and c-jun from cytoplasm to the nucleus upon VEGF activation of HIMEC. AP-1 DNA binding was not inhibited by CsA. Given the major role of VEGF in physiological and pathological angiogenesis, the identification of NFAT and AP-1 as transcription factors that couple VEGF signaling to the transcriptional gene response may help to localize therapeutic targets to antagonize or modulate the angiogenic process and to further delineate the upstream signaling pathways and the specific gene expression program triggered by VEGF in endothelial cells.
Perhaps the most important finding in our paper, is the demonstration of the differential effect of CsA on distinct signaling pathways which play integral roles in the complex biologic process of angiogenesis. CsA is known to exert specific effects on the intracellular signaling cascades, as previously published data from our laboratory demonstrated that CsA inhibits p38 MAPK activation in response to TNF-α+ LPS activation in HIMECs [33]. We evaluated a series of key steps in VEGF induced angiogenesis, and found that various members of the MAPK family, NFAT, AP-1 and COX-2 played primary roles in different stages of the cell physiology. p38 MAPK played an early role in stress fiber assembly, and also played a key role in endothelial cell migration. Monolayer expansion and proliferation of endothelial cells was dependent on p44/42 MAPK and COX-2. Maturation of microvascular endothelial cells in the context of in vitro tube formation was dependent on p44/42 MAPK, COX-2, NFAT but not p38 MAPK. Our data also demonstrated that NFκB did not play a role in the VEGF induced angiogenic response, nor did c-Fos or c-Jun, the components of AP-1. Integrating the specific effect of CsA on these various stages of angiogenesis has not been previously described. A major challenge in defining the regulation of intracellular cellular activation remains the process of characterizing the simultaneous activation and interaction of signaling cascades and networks. Our data demonstrates that MAPK signaling cascades will be involved in multiple steps in angiogenesis, playing distinct roles sequentially in the process. Finally, we have also demonstrated that the signaling responses in HIMEC were dependent on VEGF concentration, as higher amounts of this growth factor resulted in a paradoxical decrease in activation, a phenomenon previously described for the angiogenic chemokines [51,14,29].
Our study demonstrates that different signaling pathways will play dominant roles in individual steps which are involved in a complex biologic process, such as angiogenesis. The use of complementary in vitro assays for the assessment of angiogenesis allowed us to characterize the signaling pathways which were interacting during VEGF activation. If only one assay had been emphasized, the overall interplay of signaling cascade activation would not have been appreciated. At the present time, there is intense interest in defining the ability of signaling cascades to interact in a coordinated fashion, and likewise, an attempt to characterize the effect of pharmacologic agents on cellular activation.
There are important clinical ramifications which can be envisioned related to the observations in our study. Pharmacologic approaches using CsA and COX-2 inhibition may exert potent effects on blood vessels, which are underappreciated at the present time. The importance of angiogenesis in neoplastic disease and wound healing are two immediate areas where these pharmacologic agents may exert beneficial, or potentially deleterious effects. Studies focusing on organ specific microvascular populations may help in determining new clinical indications for the use of pharmacologic agents targeting blood vessels.
Conclusion
In summary, CsA exerts potent effects on human microvascular endothelial cells, inhibiting all of the stages of in vitro angiogenesis which were evaluated. More importantly, the effect of CsA was exerted through differential inhibitory effects on an interplay of signaling cascades, which contributed to distinct components of angiogenesis, including stress fiber assembly, migration, endothelial monolayer expansion and proliferation, and tube formation. Our data suggests that CsA exerts potent effects on non-immune vascular cell populations which may contribute to microvascular dysfunction and the vasculopathy which characterizes chronic rejection following solid organ transplantation. Our findings might help explain the limited clinical success of human small intestinal transplantation, as the calcineurin inhibitors required for post-transplant immunosuppression adversely affect angiogenic and vascular homeostatic mechanisms in intestinal microvascular endothelial cells. Finally, defining the effect of the calcineurin inhibitors on human microvascular endothelial cells may ultimately open the route for novel anti-angiogenic strategies using these agents as inhibitors of vessel proliferation for therapeutic benefit, potentially as adjunctive treatments of adenocarcinoma.
Material and Methods
Antibodies and reagents
VEGF and anti-human VEGF antibodies (VEGFR1 and VEGFR2) were from R&D Systems (Minneapolis, MN). Cyclosporine A (CsA) was from Alexis (San Diego, CA). Rapamycin, FK506 and the MAPK inhibitors (PD098059, SB203580) were obtained from Calbiochem (La Jolla, CA). The selective COX-2 inhibitor NS398 was from Cayman Chemical Co. (Ann Arbor, MI). Antibodies against the MAPK superfamily members (p44/42 MAPK, p38 MAPK, and c-jun NH2-terminal MAPK (SAPK)) were from New England Biolabs (Beverly, MA). Antibodies to NFAT, p65 subunit of NF-κB and COX-1/-2 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Fluorescein-conjugated phalloidin was from Molecular Probes, Inc. (Eugene, OR). Fluorescein-conjugated streptavidin was from Pierce (Rockford, IL). Endothelial Cell Growth Supplement (ECGS) was from Upstate Biotech Inc. (Lake Placid, NY). MCDB-131 medium, porcine heparin, PSF (penicillin/streptomycin/fungizone), and soybean trypsin inhibitor were from Sigma (St. Louis, MO). Fetal bovine serum (FBS) was from Bio Whittaker (Walkersville, MD). Collagenase type II was from Worthington Biochemical Corporation (Lakewood, NJ), and bovine serum albumin (BSA, Fraction V) was obtained from Fisher Scientific (Fair Lawn, NJ). Human plasma fibronectin was from Chemicon International (Temecula, CA). Oligonucleotide and primers were from Operon (Alameda, CA).
Primary culture of human intestinal microvascular endothelial cells (HIMEC)
Macroscopically normal small intestinal specimens for HIMEC isolation were obtained from patients undergoing scheduled bowel resection. The use of human tissues was approved by the Institutional Review Board of the Medical College of Wisconsin. HIMEC were isolated from surgical specimens and maintained as described earlier [28]. HIMEC cultures were recognized by microscopical features and modified lipoprotein uptake (Dil-ac-LDL, Biomedical Technology, Inc., Stoughton, MA) and expression of Factor VIII-associated antigen. All experiments were carried out using primary endothelial cell cultures between passages 8–12.
Endothelial cell chemotaxis assay
Chemotaxis assay was carried out as described earlier [29]. Briefly, using polycarbonate filters (8 μm pore size, Becton Dickinson Labware, Franklin Lakes, NJ) 5 × 105 cells were added to the upper chamber, and chemotaxis buffer (1000 μl) containing VEGF (1–50 ng/ml) was filled into the lower compartment of the 12-well plates. After 3 hr of incubation at 37°C, cell culture inserts were removed, wiped and stained with DiffQuik (Baxter Scientific, McGraw, IL). Migrated HIMEC adherent to the lower side of the membrane were counted (ten random high-power-fields (HPF; 40×) per condition in a blinded fashion). In inhibition studies, resuspended cells were incubated with neutralizing anti-VEGFR2 antibody, isotype control antibody, CsA (0.1 μM), FK506 (50 nM), Rapamycin (20 nM), SB203580 (5 μM), PD098059 (10 μM) or the selective COX-2 inhibitor, NS398 (10 μM), for 30 min at 37°C. Cell viability was >95% as assessed by trypan blue exclusion. Each condition was assessed in triplicate.
Cell proliferation assay
3 × 104 HIMEC per well were seeded onto fibronectin-coated 24-well plates using growth medium without ECGS as described earlier [29]. After pre-treatment with the indicated inhibitors (CsA 0.1 μM, PD098059 10 μM, SB230580 5 μM, NS398 10 μM) for 30 min at 37°C, cells were stimulated with VEGF (50 ng/mL) for 24, 48, and 72 hr, or left untreated. Following complete detachment, cells were re-suspended and counted in a Coulter Counter (Coulter, Brea, CA). In parallel experiments, cell viability was assessed by trypan blue exclusion and was greater than 95%. Each condition was assessed in triplicate.
Cellular DNA synthesis was assessed by 3H-thymidine uptake in HIMEC as described earlier [29]. HIMEC were pulsed with 3H-thymidine (1 μCi/ml; Amersham, Arlington Heights, IL), washed twice on ice with 5% (v/v) trichloroacetic acid prior to fixation. DNA was then released from precipitated material by alkaline lysis in 0.5 N NaOH, and supernatants were quantified in a beta-counter. Each condition was assessed in triplicate.
Microscopic wounding assay
To assess HIMEC migration in response to angiogenic stimuli, a microscopic wounding assay was performed as described earlier [52]. In brief, a HIMEC confluent monolayer was scraped along a straight line, and the remaining monolayer was then incubated with growth medium (without ECGS), and cells were pretreated for 30 min at 37°C with or without CsA (0.1 μM), SB203580 (5 μM) or PD098059 (10 μM). Then, cells were stimulated by addition of VEGF (10 ng/ml) or left untreated. The migration of HIMEC across the demarcation line was monitored using an inverted microscope. At each time point (24, 48, and 72 hr), 10 random fields using an ocular grid were counted in a blinded fashion. Data were expressed as cells/mm2, and each condition was assessed in triplicate.
Matrigel™ in vitro-tube formation assay
Endothelial tube formation was assessed using Matrigel™, a solubilized extracellular basement membrane matrix extracted from the Engelbreth-Holm-Swarm mouse sarcoma, as described previously [53]. HIMEC resuspended in complete growth medium which were seeded at a density of 5 × 104 cells per well. Where indicated, the growth medium was supplemented with CsA (0.1 μM), PD098059 (10 μM), SB203580 (5 μM) and NS398 (10 μM). Control wells contained no inhibitors. Endothelial tube formation on Matrigel™ after 16 hr was assessed by inverted phase contrast microscopy and photographed with an inverted tissue culture microscope. Five high power fields per condition were examined and experiments were repeated in two independent HIMEC cultures.
Cellular fractionation and western blot analysis
Confluent HIMEC monolayers in 35-mm culture dishes (one dish per condition) were pre-treated with various inhibitors for 30 min or left untreated before VEGF activation (50 ng/ml) for different time periods (1, 5,10, 15, 20, 30, 60, and 120 min) as described previously [54]. The homogenates were centrifuged and supernatants (cytosolic fraction) were removed and protein concentrations were determined using a Bradford Assay (Bio-Rad, Hercules, CA). Equal amounts of protein were separated by SDS-PAGE and transferred to nitrocellulose membranes [30]. The membranes were blocked for 3 hr at room temperature in 3% (w/v) BSA, 3% (w/v) nonfat dry milk in Tris-buffered saline (50 mM Tris-HCl, 150 mM NaCl, pH 7.4) containing 0.1% (v/v) Tween 20(TBS-T), then were incubated with specific primary antibodies to NFATp, COX-2, phosphorylated and non-phosphorylated MAPK (ERK1/2, p38 MAPK and JNK) at 4°C overnight as specified. Detection was by secondary antibody coupled to horseradish peroxidase (HRP) and ECL™ (Amersham Pharmacia Biotech; Arlington Heights, IL).
Electrophoretic mobility shift assay (EMSA)
Nuclear protein extraction was performed as described previously [30]. In brief, HIMEC monolayers were lysed in hypotonic lysis buffer and cell nuclei were collected and frozen immediately in liquid nitrogen until further usage. Protein concentrations were determined using a Bradford assay (Bio-Rad, Hercules, CA). The samples were then incubated on ice with 32P-labeled double stranded oligonucleotide for 30 min and DNA-protein complexes were separated by SDS-PAGE. Dried gels were exposed to X-ray film to detect DNA binding of NFAT, NF-κB and AP-1. The synthetic oligonucleotides [22] used as probes in electrophoretic mobility shift assays (EMSAs) were as follows: NFAT; 5'-tcgaCAAGGGGAGAGGAGGGAAAAATTTGTGGC-3' (nucleotides -117 to -91 containing the NFAT site of the human COX-2 promoter); NF-κB; 5'-gatcAGTGGGGACTACCCCCT-3' (nucleotides -277 to-211 containing the NF-κB site of the human COX-2 promoter) and for AP-1; 5'-tcgaCAAAAGGCGGAAAGAAACAGTCATTTC-3' (nucleotides -82 to -58 containing the NFAT-AP1 site of the human Cox-2 promoter).
Immunofluorescence staining
F-Actin polymerization was assessed in subconfluent HIMEC seeded on fibronectin-coated glass chamber slides (LabTek; Nalge Nunc, Naperville, IL) as described previously [29]. Cells were cultured in MCDB-131 without FBS for 12 hr and pretreated for 30 min at 37°C with or without CsA (0.1 μM), SB203580 (5 μM) or PD098059 (10 μM). HIMEC were then stimulated with 10–50 ng/ml recombinant human VEGF (1–15 min), and fixed with 3.7% (v/v) formaldehyde in PBS for 20 min at room temperature. Cells were washed and permeabilized with Triton X-100 (0.1% (v/v) in PBS) for 10 min, blocked with 2.5% (w/v) BSA/PBS, and stained with fluorescein-phalloidin (Molecular Probes, Eugene, OR). After washing, slides were air dried and mounted with Fluoromount-G (Southern Biotechnology, Birmingham, AL) and examined with a fluorescence microscope (Olympus BX-40) using a fixed shutter speed to allow for comparison of fluorescence intensity. In some experiments, stress fiber assembly was blocked by pre-incubation of cells with 10 μg/ml VEGFR2 antibody, (30 min at 37°C).
For immunofluoresence staining to demonstrate translocation of NFAT, p65 (NF-κB subunit), c-Jun and c-Fos (AP-1 subunit), cells were grown as above, left untreated or pre-treated with CsA (0.1 μM) before VEGF (50 ng/ml) or TNF-α/LPS (TNF-α 100 units/ml, LPS 1 μg/ml) for 30 min. Cells were then fixed with 3% (w/v) paraformaldehyde in phosphate-buffered saline (PBS) for 15 min at room temperature and washed three times (5 min each) with washing buffer (PBS, 0.01% (v/v) Nonidet P-40 [NP-40]). After blocking for 30 min as above, the cover slips were incubated with the anti-NFATp, p65, c-Jun, phospho-c-Jun and c-Fos antibodies for 60 min at room temperature. Unbound antibody was removed by rinsing three times with washing buffer and the cover slips were incubated for 30 min with a fluorescein-conjugated secondary antibody (Santa Cruz), washed three times, mounted and visualized as above.
RNA preparation and semi-quantitative RT-PCR
For determination of COX-1 and COX-2 mRNA expression, confluent HIMEC cultures were serum starved for 12 hr and stimulated with VEGF (50 ng/ml) with or without inhibitors for 3, 6 and 12 hr. Total RNA was extracted using TRIzol (Life Technologies, Rockville, MD) and treated with Deoxyribonuclease I, Amplification Grade (Life Technologies) according to the manufacturer's instruction. The complementary DNA (cDNA) was generated from 1 μg of total RNA with oligo (dT) primer using Superscript First-Strand Synthesis System for RT-PCR (Life Technologies) according to the protocol and in a total volume of 80 μl. Four μl of cDNA solution were used for polymerase chain reaction (PCR) in a total volume of 40 μl containing 1.5 mM MgCl2, 0.2 mM dNTP mix, 0.5 μM of sense and anti-sense primers, respectively, and 1 IU of Taq polymerase (Life Technologies). PCR amplifications were performed 30 cycles for COX-1 (94°C for 1 min, 56°C for 1 min, 72°C for 1 min), 35 cycles for COX-2 (94°C for 1 min, 54°C for 1 min, 72°C for 1 min) and 25 cycles for β-actin (94°C for 1 min, 60°C for 1 min, 72°C for 1 min), using COX-1, COX-2 and β-actin specific primers (below) and followed by final extension for 7 min at 72°C. Twenty μl of PCR product were visualized on 1.2% agarose gels stained with ethidium bromide. RNA solution without reverse transcription was used as negative control (no RT), and β-actin served as an internal control. Primer sequences used are shown in table 1.
Table 1.
Human COX-1, COX-2 and Bata-actin primers
| COX-1 forward (5'-TGCCCAGCTCCTGGCCCGCCGCT-3') |
| COX-1 reverse (5'-TTCAAATGAGATTGTGGGAAAATTGTC-3') |
| COX-2 forward (5'-TCAAATGAGATTGTGGGAAAATTG-3') |
| COX-2 reverse 5'-TCTAGTAGAGACGGACTCATAGAA-3') |
| β-actin forward (5'-CCAGAGCAAGAGAGGCATCC-3') |
| β-actin reverse (5'-CTGTGGTGGTGAAGCTGTAG-3') |
Analysis of data
Statistical Analysis was performed using Statview 4.5 and superANOVA software for the Macintosh. When single comparisons were made, t-tests were used, applying paired or unpaired analysis as appropriate. When multiple comparisons between groups were performed one way or two-way analysis of variance was used as appropriate followed by the Student-Newman Keuls test. P ≤ 0.05 was considered significant.
Abbreviations
CsA; Cyclosporin A, HIMEC; human intestinal microvascular endothelial cells; EC: endothelial cell; MAPK: mitogen-activated protein kinases; SAPK/JNK: stress activated protein kinase/c-Jun kinase; ERK: extracellular signal-regulated kinase, TNF-α; tumor necrosis factor alpha, LPS; lipopolysaccharide, NFκB; nuclear factor kappa B, NFAT; nuclear factor of activated T cells, COX; cyclooxygenase, PMSF; phenylmethylsulfonylfluoride.
Competing interests
None declared.
Authors' contributions
PR and DGB were responsible for overall experimental design, analysis, and writing the manuscript. JH, HO, NAJ, PJF, MSL, MFO, and CPJ contributed to performance of experiments.
Acknowledgments
Acknowledgments
We thank H. Brandenburg for expert assistance in preparation of the manuscript. This work was supported by the Medical College of Wisconsin Cancer Center (P.R., D.G.B., M.F.O.), the National Institute of Health grants DK 057139, the Crohn's and Colitis Foundation of America and the Digestive Disease Center and Cancer Center of the Medical College of Wisconsin (D.G.B., P.R.).
Contributor Information
Parvaneh Rafiee, Email: prafiee@mcw.edu.
Jan Heidemann, Email: jan_heidemann@hotmail.com.
Hitoshi Ogawa, Email: hogawa6820@yahoo.com.jp.
Nathan A Johnson, Email: najohnson@mcw.edu.
Pamela J Fisher, Email: pjkexel@yahoo.com.
Mona S Li, Email: mli@mail.mcw.edu.
Mary F Otterson, Email: otterson@mcw.edu.
Christopher P Johnson, Email: cjohnson@mcw.edu.
David G Binion, Email: dbinion@mcw.edu.
References
- Goldfeld AE, Tsai E, Kincaid R, Belshaw PJ, Schrieber SL, Strominger JL, Rao A. Calcineurin mediates human tumor necrosis factor alpha gene induction in stimulated T and B cells. J Exp Med. 1994;180:763–768. doi: 10.1084/jem.180.2.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao SZ, Schroeder JS, Alderman EL, Hunt SA, Valantine HA, Wiederhold V, Stinson EB. Prevalence of accelerated coronary artery disease in heart transplant survivors. Comparison of cyclosporine and azathioprine regimens. Circulation. 1989;80:100–105. [PubMed] [Google Scholar]
- Textor SC, Canzanello VJ, Taler SJ, Wilson DJ, Schwartz LL, Augustine JE, Raymer JM, Romero JC, Wiesner RH, Krom RA, Burnett J. Cyclosporine-induced hypertension after transplantation. Mayo Clin Proc. 1994;69:1182–1193. doi: 10.1016/s0025-6196(12)65772-3. [DOI] [PubMed] [Google Scholar]
- Ventura HO, Malik FS, Mehra MR, Stapleton DD, Smart FW. Mechanisms of hypertension in cardiac transplantation and the role of cyclosporine. Curr Opin Cardiol. 1997;12:375–381. [PubMed] [Google Scholar]
- Shears LL, Kawaharada N, Tzeng E, Billiar TR, Watkins SC, Kovesdi I, Lizonova A, Pham SM. Inducible nitric oxide synthase suppresses the development of allograft arteriosclerosis. J Clin Invest. 1997;100:2035–2042. doi: 10.1172/JCI119736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaus A, Margreiter R, Pernthaler H, Klima G, Offner FA. Diffuse mesenterial sclerosis: a characteristic feature of chronic small-bowel allograft rejection. Virchows Arch. 2003;442:48–55. doi: 10.1007/s00428-002-0715-9. [DOI] [PubMed] [Google Scholar]
- Parizhskaya M, Redondo C, Demetris A, Jaffe R, Reyes J, Ruppert K, Martin L, Abu-Elmagd K. Chronic rejection of small bowel grafts: pediatric and adult study of risk factors and morphologic progression. Pediatr Dev Pathol. 2003;6:240–250. doi: 10.1007/s10024-002-0039-4. [DOI] [PubMed] [Google Scholar]
- Medawar PB. Transplantation of tissues and organs: introduction. Br Med Bull. 1965;21:97–99. [Google Scholar]
- Lagaaij EL, Cramer-Knijnenburg GF, van Kemenade FJ, van Es LA, Bruijn JA, van Krieken JH. Endothelial cell chimerism after renal transplantation and vascular rejection. Lancet. 2001;357:33–37. doi: 10.1016/S0140-6736(00)03569-8. [DOI] [PubMed] [Google Scholar]
- Hruban RH, Beschrner WE, Baumgartner WA, Augustine SM, Ren H, Reitz BA, Hutchins GM. Accelerated arteriosclerosis in heart transplant recipients is associated with a T-lymphocyte-mediated endothelialitis. Am J Pathol. 1990;137:871–882. [PMC free article] [PubMed] [Google Scholar]
- Taylor PM, Rose ML, Yacoub MH, Pigott R. Induction of vascular adhesion molecules during rejection of human cardiac allografts. Transplantation. 1992;54:451–457. doi: 10.1097/00007890-199209000-00013. [DOI] [PubMed] [Google Scholar]
- Brockmeyer C, Schendel DJ, Weiss EH, Hillebrand G, Burkhardt K, Land W, Gokel MJ, Riethmuller G, Feucht HE. Distribution of cell adhesion molecules (ICAM-1, VCAM-1, ELAM-1) in renal tissue during allograft rejection. Transplantation. 1993;55:610–615. doi: 10.1097/00007890-199303000-00027. [DOI] [PubMed] [Google Scholar]
- Rose ML. Endothelial as antigen-presenting cells: role in human transplant rejection. Cell Mol Life Sci. 1998;54:965–978. doi: 10.1007/s000180050226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo R, Resau JH, Halverson D, Hudson EA, Dambach M, Powell D, Wasserman K, Oppenheim JJ. Differential expression and responsiveness of chemokine receptors (CXCR1-3) by human microvascular endothelial cells and umbilical vein endothelial cells. FASEB J. 2000;14:2055–2064. doi: 10.1096/fj.99-0963com. [DOI] [PubMed] [Google Scholar]
- Auerbach R, Lewis R, Shinners B, Kubai L, Akhtar N. Angiogenesis assays: a critical overview. Clin Chem. 2003;49:32–40. doi: 10.1373/49.1.32. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Matsui A, Inao M, Mochida S, Fujiwara K. ERK/MAPK-dependent PI3K/Akt phosphorylation through VEGFR-1 after VEGF stimulation in activated hepatic stellate cells. Hepatol Res. 2003;26:232–236. doi: 10.1016/S1386-6346(03)00112-8. [DOI] [PubMed] [Google Scholar]
- Huot J, Houle F, Rousseau S, Deschesnes RG, Shah GM, Landry J. SAPK2/p38-dependent F-actin reorganization regulates early membrane blebbing during stress-induced apoptosis. J Cell Biol. 1998;143:1361–1373. doi: 10.1083/jcb.143.5.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura M, Sebastian S, Yang S, Gurates B, Fang Z, Okamura K, Bulun SE. Induction of cyclooxygenase-2 in human endometrial stromal cells by malignant endometrial epithelial cells: evidence for the involvement of extracellularly regulated kinases and CCAAT/enhancer binding proteins. J Mol Endocrinol. 2003;31:95–104. doi: 10.1677/jme.0.0310095. [DOI] [PubMed] [Google Scholar]
- Shariat SF, Matsumoto K, Kim J, Ayala GE, Zhou JH, Jian W, Benedict WF, Lerner SP. Correlation of cyclooxygenase-2 expression with molecular markers, pathological features and clinical outcome of transitional cell carcinoma of the bladder. J Urol. 2003;170:985–989. doi: 10.1097/01.ju.0000080401.85145.ee. [DOI] [PubMed] [Google Scholar]
- De Leng WW, Westerman AM, Weterman MA, De Rooij FW, Dekken Hv H, De Goeij AF, Gruber SB, Wilson JH, Offerhaus GJ, Giardiello FM, Keller JJ. Cyclooxygenase 2 expression and molecular alterations in Peutz-Jeghers hamartomas and carcinomas. Clin Cancer Res. 2003;9:3065–3072. [PubMed] [Google Scholar]
- Armesilla AL, Lorenzo E, Gomez del Arco P, Martinez-Martinez S, Alfranca A, Redondo JM. Vascular Endothelial Growth Factor Activates Nuclear Factor of Activated T-Cells in Human Endothelial Cells: a Role for Tissue Factor Gene Expression. Mol Cell Biol. 1999;19:2032–2043. doi: 10.1128/mcb.19.3.2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez GL, Volpert O, Iniguez MA, Lorenzo E, Martinez-Martinez S, Grau R, Fresno M, Redondo JM. Selective inhibition of vascular endothelial growth factor-mediated angiogenesis by cyclosporin A: roles of the nuclear factor of activated T cells and cyclooxygenase 2. J Exp Med. 2001;193:607–620. doi: 10.1084/jem.193.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner L, Hoey J, Erdely A, Boegehold MA, Baylis C. The nitric oxide pathway is amplified in venular vs arteriolar cultured rat mesenteric endothelial cells. Microvasc Res. 2001;62:401–409. doi: 10.1006/mvre.2001.2359. [DOI] [PubMed] [Google Scholar]
- Mason JC, Yarwood H, Sugars K, Haskard DO. Human umbilical vein and dermal microvascular endothelial cells show heterogeneity in response to PKC activation. Am J Physiol. 1997;273:c1233–c1240. doi: 10.1152/ajpcell.1997.273.4.C1233. [DOI] [PubMed] [Google Scholar]
- Garlanda C, Dejana E. Heterogeneity of endothelial cells. Specific markers. Atheroscler Thromb Vasc Biol. 1997;17:1193–1102. doi: 10.1161/01.atv.17.7.1193. [DOI] [PubMed] [Google Scholar]
- Petzelbauer P, Bender JR, Wilson J, Pober JS. Heterogeneity of dermal microvascular endothelial cell antigen expression and cytokine responsiveness in situ and in cell culture. J Immunol. 1993;151:5062–5072. [PubMed] [Google Scholar]
- Page C, Rose M, Yacoub M, Pigott R. Antigenic heterogeneity of vascular endothelium. Am J Pathol. 1992;141:673–683. [PMC free article] [PubMed] [Google Scholar]
- Binion DG, West GA, Ina K, Ziats NP, Emancipator SN, Fiocchi C. Enhanced leukocyte binding by intestinal microvascular endothelial cells in inflammatory bowel disease. Gastroenterology. 1997;112:1895–1807. doi: 10.1053/gast.1997.v112.pm9178682. [DOI] [PubMed] [Google Scholar]
- Heidemann J, Ogawa H, Dwinell MB, Rafiee P, Maaser C, Gockel HR, Otterson MF, Ota DM, Lugering N, Domschke W, Binion DG. Angiogenic effects of interleukin 8 (CXCL8) in human intestinal microvascular endothelial cells are mediated by CXCR2. J Biol Chem. 2003;278:8508–8515. doi: 10.1074/jbc.M208231200. [DOI] [PubMed] [Google Scholar]
- Rafiee P, Johnson CP, Li MS, Ogawa H, Heidemann J, Fisher PJ, Lamirand TH, Otterson MF, Wilson KT, Binion DG. Cyclosporine A enhances leukocyte binding by human intestinal microvascular endothelial cells through inhibition of p38 MAPK and iNOS. Paradoxical proinflammatory effect on the microvascular endothelium. J Biol Chem. 2002;277:35605–35615. doi: 10.1074/jbc.M205826200. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6:389–397. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- Tibbles LA, Woodgett JR. The stress-activated protein kinase pathways. Cell Mol Life Sci. 1999;55:1230–1254. doi: 10.1007/s000180050369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau S, Houle F, Kotanides H, Witte L, Waltenberger J, Landry J, Huot J. Vascular endothelial growth factor (VEGF)-driven actin-based motility is mediated by VEGFR2 and requires concerted activation of stress-activated protein kinase 2 (SAPK2/p38) and geldanamycin-sensitive phosphorylation of focal adhesion kinase. J Biol Chem. 2000;275:10661–10672. doi: 10.1074/jbc.275.14.10661. [DOI] [PubMed] [Google Scholar]
- Seetharam L, Gotoh N, Maru Y, Neufeld G, Yamaguchi S, Shibuya M. A unique signal transduction from FLT tyrosine kinase, a receptor for vascular endothelial growth factor VEGF. Oncogene. 1995;10:135–147. [PubMed] [Google Scholar]
- Tanaka K, Abe M, Sato Y. Roles of extracellular signal-regulated kinase 1/2 and p38 MAPK in the signal transduction of basic fibroblast groeth factor in endothelial cells during angiogenesis. Jpn J Cancer Res. 1999;90:647–654. doi: 10.1111/j.1349-7006.1999.tb00796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova TV, Makinen T, Alitalo K. Signaling via vascular endothelial growth factor receptors. Exp Cell Res. 1999;253:117–130. doi: 10.1006/excr.1999.4707. [DOI] [PubMed] [Google Scholar]
- Loh C, Shaw KT, Carew J, Viola JP, Luo C, Perrino BA, Rao A. Calcineurin binds the transcription factor NFAT1 and reversibly regulates its activity. J Biol Chem. 1996;271:10884–10891. doi: 10.1074/jbc.271.18.10884. [DOI] [PubMed] [Google Scholar]
- Shaw KT, Ho AM, Raghavan A, Kim J, Jain J, Park J, Sharma S, Rao A, Hogan PG. Immunosuppressive drugs prevent a rapid dephosphorylation of transcription factor NFAT1 in stimulated immune cells. Proc Natl Acad Sci U S A. 1995;92:11205–11209. doi: 10.1073/pnas.92.24.11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber SL, Crabtree GR. The mechanism of action of cyclosporin A and FK506. Immunol Today. 1992;13:136–142. doi: 10.1016/0167-5699(92)90111-J. [DOI] [PubMed] [Google Scholar]
- Northrop JP, Ho SN, Chen L, Thomas DJ, Timmerman LA, Nolan GP, Admon A, Crabtree GR. NF-AT components define a family of transcription factors targeted in T-cell activation. Nature. 1994;369:497–502. doi: 10.1038/369497a0. [DOI] [PubMed] [Google Scholar]
- Garcia JE, de Cabo MR, Rodriguez FM, Losada JP, Lopez AJ, Arellano JL. Effect of cyclosporin A on inflammatory cytokine production by U937 monocyte-like cells. Mediators Inflamm. 2000;9:169–174. doi: 10.1080/09629350020008682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S, Ono M, Shono T, Izumi H, Ishibashi T, Suzuki H, Kuwano M. Involvement of Interleukin-8, Vascular Endothelial Growth Factor, and Basic Fibroblast Growth Factor in Tumor Necrosis Factor Alpha-Dependent Angiogenesis. Mol Cell Biol. 1997;17:4015–4023. doi: 10.1128/mcb.17.7.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, DuBois RN. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell. 1998;93:705–716. doi: 10.1016/S0092-8674(00)81433-6. [DOI] [PubMed] [Google Scholar]
- Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J Biol Chem. 1996;271:33157–33160. doi: 10.1074/jbc.271.52.33157. [DOI] [PubMed] [Google Scholar]
- Iniguez MA, Punzon C, Fresno M. Induction of cyclooxygenase-2 on activated T lymphocytes: regulation of T cell activation by cyclooxygenase-2 inhibitors. J Immunol. 1999;163:111–119. [PubMed] [Google Scholar]
- Inoue H, Yokoyama C, Hara S, Tone Y, Tanabe T. Transcriptional regulation of human prostaglandin-endoperoxide synthase-2 gene by lipopolysaccharide and phorbol ester in vascular endothelial cells. Involvement of both nuclear factor for interleukin-6 expression site and cAMP response element. J Biol Chem. 1995;270:24965–24971. doi: 10.1074/jbc.270.42.24965. [DOI] [PubMed] [Google Scholar]
- Yu Y, Sato JD. MAP kinases, phosphatidylinositol 3-kinase, and p70 S6 kinase mediate the mitogenic response of human endothelial cells to vascular endothelial growth factor. J Cell Physiol. 1999;178:235–246. doi: 10.1002/(SICI)1097-4652(199902)178:2<235::AID-JCP13>3.3.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Vinals F, Chambard JC, Pouyssegur J. p70 S6 kinase-mediated protein synthesis is a critical step for vascular endothelial cell proliferation. J Biol Chem. 1999;274:26776–26782. doi: 10.1074/jbc.274.38.26776. [DOI] [PubMed] [Google Scholar]
- Bancroft CC, Chen Z, Yeh J, Sunwoo JB, Yeh NT, Jackson S, Jackson C, Van Waes C. Effects of pharmacologic antagonists of epidermal growth factor receptor, PI3K and MEK signal kinases on NF-kappaB and AP-1 activation and IL-8 and VEGF expression in human head and neck squamous cell carcinoma lines. Int J Cancer. 2002;99:538–548. doi: 10.1002/ijc.10398. [DOI] [PubMed] [Google Scholar]
- Minet E, Michel G, Mottet D, Piret JP, Barbieux A, Raes M, Michiels C. c-JUN gene induction and AP-1 activity is regulated by a JNK-dependent pathway in hypoxic HepG2 cells. Exp Cell Res. 2001;265:114–124. doi: 10.1006/excr.2001.5180. [DOI] [PubMed] [Google Scholar]
- Strieter RM, Polverini PJ, Kunkel SL, Arenberg DA, Burdick MD, Kasper J, Dzuiba J, Van Damme J, Walz A, Marriott D, Chan S, Rocziak S, Shanafelt AB. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J Biol Chem. 1995;270:27348–57. doi: 10.1074/jbc.270.45.27348. [DOI] [PubMed] [Google Scholar]
- Denes L, Jednakovits A, Hargitai J, Penzes Z, Balla A, Talosi L, Krajcsi P, Csermely P. Pharmacologically activated migration of aortic endothelial cells is mediated through p38 SAPK. Br J Pharma. 2002;136:597–503. doi: 10.1038/sj.bjp.0704738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvucci O, Yao L, Villalba S, Sajewicz A, Pittaluga S, Tosato G. Regulation of endothelial cell branching morphogenesis by endogenous chemokine stromal-derived factor-1. Blood. 2002;99:2703–2711. doi: 10.1182/blood.V99.8.2703. [DOI] [PubMed] [Google Scholar]
- Rafiee P, Lee JK, Leung CC, Raffin TA. TNF-a induces tyrosine phosphorylation of mitogen-activated protein kinase in adherent human neutrophils. J Immunol. 1995;154:4785–4792. [PubMed] [Google Scholar]