

Abstract
Purpose
The vesicular acetylcholine transporter (VAChT) is a specific biomarker for imaging presynaptic cholinergic neurons. The syntheses and C-11 labeling of two potent enantiopure VAChT inhibitors are reported here.
Procedures
Two VAChT inhibitors, (±)-2 and (±)-6, were successfully synthesized. A chiral HPLC column was used to resolve the enantiomers from each corresponding racemic mixture for in vitro characterization. The radiosyntheses of (−)-[11C]2 and (−)-[11C]6 from the corresponding desmethyl phenol precursor was accomplished using [11C]methyl iodide or [11C]methyl triflate, respectively.
Results
The synthesis of (−)-[11C]2 was accomplished with 40–50 % radiochemical yield (decay-corrected), SA>480 GBq/μmol (EOB), and radiochemical purity >99 %. Synthesis of (−)-[11C]6 was accomplished with 5–10 % yield, SA>140 GBq/μmol (EOB), and radiochemical purity >97 %. The radiosynthesis and dose formulation of each tracer was completed in 55–60 min.
Conclusions
Two potent enantiopure VAChT ligands were synthesized and 11C-labeled with good radiochemical yield and specific activity.
Keywords: VAChT, Alzheimer’s disease, PET imaging, Radiotracer, Vesamicol
Introduction
The cholinergic system plays a critical role in the functioning of the central nervous system, and cholinergic dysfunction is believed to be critical in dementia associated with neurodegenerative diseases including Alzheimer’s disease and Parkinson’s disease [1, 2]. Cholinergic neuronal transmission involves synthesis of acetylcholine (ACh) in the cytoplasm, its storage in the presynaptic vesicles, and release into the presynaptic cleft. The vesicular acetylcholine transporter (VAChT), predominantly located in the presynaptic vesicles [3], is responsible for the loading of ACh into the presynaptic vesicles. Extensive research has revealed that VAChT plays a vital role in the release of ACh and physiological functions in the nervous system [4].
Extensive structure-activity relationship studies performed by Parsons et al. laid the foundation for the development of the existing positron emission tomography (PET) and single-photon emission computed tomography (SPECT) radiotracers used to image cholinergic neuron terminals [5–7]. Both modalities permit noninvasive imaging and could be used to quantify VAChT in vivo and study changes in cholinergic function in response to therapy. Until recently, only one radiopharmaceutical, [123I]IBVM, had been used in clinical studies to map cholinergic terminal density at presynaptic regions using SPECT [8–11]. Because PET imaging affords higher sensitivity than SPECT [12], significant efforts have been made since the late 1990s to develop PET radiotracers with selectivity for VAChT over the σ1 receptor [13–16], and several promising PET radiotracers have been reported [17–22]. For example, (−)o-[11C]methylvesamicol was reported to have high affinity for VAChT (Ki=6.7 nM) and five fold selectivity versus the σ1 receptor; however, the target to nontarget ratios at 30 min p.i. were only 1.1-fold and 1.6-fold in rat and in nonhuman primate (NHP), respectively [20]. Koeppe et al. recently reported the first clinical study with the promising PET tracer [18F]FEOBV [22], but unfortunately, slow equilibrium binding was reported in rat, NHP, and also human subjects [14, 16, 22]. Nonetheless, [18F]FEOBV represent the first FDA approved PET radiopharmaceutical for assessing the level of VAChT in clinical studies. By contrast, we have focused on 11C-labeled VAChT tracers since the short half-life (t1/2=20.4 min) results in lower radiation exposure to patients and enables multiple PET studies in the same subject in a single day. Here, we report the successful synthesis and resolution of the racemic compounds and the radiosyntheses of two potent and selective 11C-labeled radiotracers, (−)-[11C]2 (Ki-VAChT =1.6±0.1 nM, selectivity >35- and 1,600-fold for VAChT over σ1 and σ2 receptors) and (−)-[11C]6 (Ki-VAChT =3.5±0.3 nM, selectivity >125-and 650-fold for VAChT versus σ1 and σ2 receptors). These radiotracers provide the basis for our subsequent evaluation in rodents and nonhuman primates which is reported in a companion manuscript [23].
Materials and Methods
General
All solvents and reagents were obtained commercially and used as received, unless specified otherwise. All anhydrous reactions were carried out in oven-dried or flame-dried and nitrogen or argon purged glassware. Anhydrous tetrahydrofuran was dried over sodium sulfate and distilled prior to use. Anhydrous dichloromethane was distilled over calcium hydride prior to use. Reactions were monitored by thin layer chromatography (TLC) using EMD Chemicals Inc. silica gel 60F254 glass plates. Flash column chromatography was performed over silica gel (32–63 μm); HPLC grade solvents were used for chromatography. 1HNMR were recorded on a Varian Mercury-VX 300 MHz spectrometer. The chemical shifts were reported as δ values (ppm) relative to TMS as an internal reference. Varian Prostar 216 system was used for both analytical and semipreparative HPLC. Elemental analyses were determined by Atlantic Microlab, Inc. (Norcross, GA). The specific rotation of the enantiomers was determined on an automatic polarimeter (Autopol 111, Rudolph Research, Flanders, NJ).
[11C]CH3I was produced at our institution from [11C]CO2 using a GE PETtrace MeI Microlab. Up to 51.8 GBq of [11C]CO2 was produced from Washington University’s JSW BC-16/8 cyclotron by irradiating a gas target of 0.2 % O2 in N2 for 15–30 min with a 40 μA beam of 16 MeV protons. The Microlab converts the [11C]CO2 to [11C]CH4 using a nickel catalyst (Shimalite-Ni, Shimadzu, Japan P.N.221-27719) in the presence of hydrogen gas at 360 °C; it is further converted to [11C]CH3I by reaction with iodine that is held in a column in gas phase at 690 °C. Several hundred mCi of gaseous [11C]CH3I were delivered approximately 12 min after the end of bombardment (EOB) to the hot cell where the radiosynthesis was accomplished [24].
((3-Hydroxy-1,2,3,4-Tetrahydronaphthalen-2-yl)Piperidin-4-yl)(4-Methoxyphenyl)Methanone (2)
A 200 ml flask was charged with 2.0 g (15.4 mmol) of 1,4-dihydronaphthalene and flushed with argon; 25 ml dichloromethane was added and the mixture cooled to 0 °C. A solution of 4.1 g (24 mmol) of 77 % m-CPBA in 25 ml dichloromethane was added and the reaction mixture was stirred for 4 h then filtered. The filtrate was washed with saturated aqueous sodium carbonate followed by brine. The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The crude material was purified on a silica gel column (1:15 ethyl acetate in hexane to 1:10 ethyl acetate in hexane) to give 1.24 g (8.52 mmol, 55 %) of 1a,2,7,7a-tetrahydronaphtho[2,3-b]oxirene as a white solid. 1HNMR (300 MHz, CDCl3): δ 7.25–7.11 (m, 2H), 7.07–7.02 (m, 2H), 3.49–3.47 (m, 2H), 3.35–3.16 (m, 4H). Two hundred thirty-four milligrams (1.6 mmol) of 1a,2,7,7a-tetrahydronaphtho[2,3-b]oxirene and 10 ml ethanol was added to a 20 ml round bottom flask followed by 384 mg (1.5 mmol) of (4-methoxyphenyl)(piperidin-4-yl)methanone hydrochloride and 885 mg (6.4 mmol) of potassium carbonate. The reaction mixture was refluxed for 44 h, cooled to rt and filtered. The filtrate was concentrated, re-dissolved in methylene chloride, washed with brine, dried over sodium sulfate, and filtered; solvents were evaporated to yield a brown oil. The crude product was purified on a silica gel column (1:5 to 1:3 to 1:1 ethyl acetate: hexane) to give 90 mg (0.25 mmol, 16.5 %) of (±)-2 as a white solid. 1HNMR (300 MHz, CDCl3): δ 7.95 (d, J=8.7 Hz, 2H), 7.13–7.09 (m, 4 H), 6.96 (d, J= 8.7 Hz, 2H), 3.87 (s, 3H), 3.33–3.01 (m, 2H), 3.00–2.76 (m, 8H), 2.44–2.38 (m, 1H), 2.04–1.80 (m, 5H).
Resolution of Enantiomers of ((3-Hydroxy-1,2,3,4-Tetrahydronaphthalen-2-yl)Piperidin-4-yl)(4-Methoxy-Phenyl)Methanone ((±)-2)
Approximately 70 mg of (±)-2 was dissolved in 10 ml methanol and the enantiomers were resolved using a Chiralcel OD column (250 mm×10 mm) under isocratic conditions (10 % hexane and 20 % isopropanol in methanol), flow rate 4.0 ml/min to give 22 mg of (+)-2 (Rt=15 min; enantiomeric purity >99 %) and 34 mg of (−)-2 (Rt = 31 min; enantiomeric purity >99 %), and UV wavelength 254 nm. The optical rotation of (+)-2 was [α]D =+50.0° at a concentration of 1.4 mg/ml in methanol. The optical rotation of (−)-2 was [α]D=−50.7° at the concentration of 0.8 mg/ml in methanol.
Approximately, 18.7 mg (0.051 mmol) of (+)-2 was placed in a 50 ml flask, and 2.0 ml dichloromethane was added. One equivalent of oxalic acid (4.6 mg, 0.051 mmol) in 0.6 ml ethyl acetate was added and the reaction mixture was stirred at rt overnight. The precipitate was collected by filtration to give 20.5 mg (0.045 mmol, 88 %) of (+)-2 oxalate as a white solid. MP: 224–225 °C; Anal Calcd for C25H29NO7•0.4H2O: C, 64.89; H, 6.49; N, 3.03; Found: C, 64.98; H, 6.27; N, 3.19.
Approximately 25 mg (0.068 mmol) of (−)-2 was placed in a 50 ml flask and 3.0 ml dichloromethane was added. One equivalent of oxalic acid (6.1 mg, 0.068 mmol) in 0.8 ml ethyl acetate was added to the above solution and the reaction mixture was stirred at rt overnight. The precipitate was collected by filtration to give 21.8 mg (0.048 mmol, 70 %) of (−)-2 oxalate as a white solid. MP: 224–225 °C; Anal Calcd for C25H29NO7•0.45H2O: C, 64.77; H, 6.50; N, 3.02; Found: C, 64.77; H, 6.25; N, 3.18.
(4-Hydroxyphenyl)(Piperidin-4-yl)Methanone Hydrochloride (8)
To 0.99 g (3.87 mmol) of (4-methoxyphenyl)(piperidin-4-yl)methanone hydrochloride in a 25 ml reaction vessel was added 0.3 ml water followed by 18 ml concentrated HCl. The reaction vessel was sealed tightly and heated to 100 °C. After 8 days of heating, the reaction mixture was diluted with water and evaporated to dryness to give 0.9 g (3.72 mmol, 96 %) of 8 as a white solid. MP: >250 °C; 1HNMR (300 MHz, D2O): δ 7.72 (dd, J=6.9, 1.8 Hz, 2H), 6.75 (dd, J=6.9, 2.1 Hz, 2H), 3.56–3.49 (m, 1H), 3.31–3.25 (m, 2H), 2.95 (td, J=12.9, 3.2 Hz, 2H), 1.90–1.86 (m, 2H), 1.71–1.56 (m, 2H).
((3-Hydroxy-1,2,3,4-Tetrahydronaphthalen-2-yl)Piperidin-4-yl)(4-Hydroxyphenyl)Methanone (9)
1a,2,7,7a-Tetrahydronaphtho[2,3-b]oxirene (0.33 g, 2.25 mmol) and 20 ml ethanol was added to a reaction flask, then 1.5 g of potassium carbonate was added followed by 0.5 g (2.08 mmol) of 8. The reaction mixture was refluxed for 4 days, cooled to rt and filtered. The filtrate was concentrated and the residue was treated with methylene chloride to give a precipitate. The precipitate which was not soluble in either aqueous or organic layers was removed by filtration. The organic layer was washed with brine, dried over anhydrous sodium sulfate, concentrated under reduced pressure, and purified on a silica gel column (1:3 to 1:1 ethyl acetate/hexane) to give 0.45 g (1.28 mmol, 62 %) of 9 as viscous oil. 1HNMR (300 MHz, CDCl3): δ 7.97–7.89 (m, 2H), 7.19–7.12 (m, 4H), 6.90 (d, J=6.0 Hz, 2H), 3.87 (m, 1H), 3.40–3.20 (m, 2H), 3.01–2.77 (m, 7H), 2.42–2.25 (m, 1H), 2.00–1.80 (m, 4H).
(4-(Benzyloxy)Phenyl)(1-(3-Hydroxy-1,2,3,4-Tetrahydronaphthalen-2-yl)Piperidin-4-yl)Methanone ((±)-10)
(±)-9 (0.45 g, 1.28 mmol), 1.5 g potassium carbonate, and 30 ml (1.32 mmol) benzyl bromide was added dropwise. The reaction mixture was stirred at 45 °C overnight then filtered, concentrated under reduced pressure, and purified on a silica gel column (1:2 to 1:1 ethyl acetate/hexane) to give 0.35 g of (±)-10 (0.79 mmol, 62 %) as a white solid. MP: 167–169 °C. 1HNMR (300 MHz, CDCl3): δ 7.95–7.93 (d, J=8.7 Hz, 2H), 7.45–7.34 (m, 4H), 7.26–7.09 (m, 5H), 7.03–7.01 (d, J=9.0 Hz, 2H), 5.14 (s, 2H), 3.88–3.83 (m, 1H), 3.35–3.27 (m, 1H), 3.01–2.78 (m, 6H), 2.63 (s, 2H), 2.44–2.37 (m, 1H), 2.17–1.80 (m, 5H).
Resolution of Enantiomers of (4-(Benzyloxy)Phenyl)(1-(3-Hydroxy-1,2,3,4-Tetrahydronaphthalen-2-yl)-Piperidin-4-yl)Methanone ((±)-10)
The enantiomers of (±)-10 were resolved using a Chiralcel OD column (250 mm×10 mm) under isocratic conditions (50 % isopropanol in hexane), flow rate 4.0 ml/min, and UV wavelength 254 nm. The retention time of (+)-10 was 22 min and that of (−)-10 was 49 min; enantiomeric purity >95 %.
(−)-((3-Hydroxy-1,2,3,4-Tetrahydronaphthalen-2-yl)Piperidin-4-yl)(4-Hydroxyphenyl)-Methanone ((−)-9)
40 mg (0.09 mmol) of (−)-10 was placed in a Parr hydrogenator flask and 40 ml methanol was added. A catalytic amount of 10 % palladium on activated carbon was added and the mixture was hydrogenated at rt and 70 psi for 21 h. The reaction mixture was then filtered on Celite and the filtrate concentrated to give 29 mg (0.082 mmol, 90 %) viscous yellow oil. The optical rotation of (−)-9 was [α]D=−60.5° at a concentration of 1.2 mg in 2.0 ml methanol. 1HNMR (300 MHz, CDCl3): δ 7.91 (d, J=9.0 Hz, 2H), 7.19–7.05 (m, 4H), 6.90 (d, J=9.0 Hz, 2H), 3.87 (m, 1H), 3.30–3.25 (m, 2H), 3.10–2.75 (m, 8H), 2.00–1.80 (m, 4H).
Tert-Butyl 4-(Methoxy(Methyl)Carbamoyl)Piperidine-1-Carboxylate (4)
In a 500 ml reaction flask was placed 10.0 g (77.5 mmol) isonipecotic acid, 21.4 g (155 mmol) potassium carbonate, and 150 ml water was added. The mixture was cooled to 0 °C. Approximately 16.9 g (77.5 mmol) of diterbutyldicarbonate in THF was added to the above mixture at 0 °C. The reaction mixture was stirred at rt overnight. THF was removed under reduced pressure. The aqueous phase was washed with ether and treated with 1 N HCl until pH=2. The acidic aqueous layer was extracted with ethyl acetate and the ethyl acetate layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to give a white solid which was recrystallized from ethyl acetate/hexane to give 11.7 g (51 mmol, 66 %) of 1-(tert-butoxycarbonyl)piperidine-4-carboxylic acid as a white solid. MP: 150–152 °C 1HNMR (300 MHz, CDCl3): δ 4.00 (m, 2H), 3.00–2.70 (m, 2H), 2.60–2.40 (m, 1H), 2.00–1.80 (m, 2H), 1.70–1.50 (m, 2H), 1.45 (s, 9H). Approximately 3.38 g (14.7 mmol) of 1-(tert-butoxycarbonyl)piperidine-4-carboxylic acid was placed in an argon-purged reaction flask; 1.43 g of N,O-dimethylhydroxylamine hydrochloride (14.7 mmol) and 21.0 ml DMF were added followed by 4.1 ml triethylamine. The mixture was cooled to 0 °C and 3.76 g (14.7 mmol) of BOP-Cl was added at 0 °C, then the reaction mixture was allowed to warm to rt overnight. The crude product was diluted with ethyl acetate and washed with 50 ml water, 50 ml 1 N HCl, 50 ml saturated aqueous Na2CO3, and 50 ml brine and dried over anhydrous sodium sulfate. The organic layer was concentrated under reduced pressure to give 2.16 g (7.94 mmol, 54 %) of tert-butyl-4-(methoxy(methyl)carbamoyl)piperidine-1-carboxylate (4) as a colorless oil. 1HNMR (300 MHz, CDCl3): δ 4.15–4.11 (m, 2H), 3.71 (s, 3H), 3.19 (s, 3H), 2.81–2.76 (m, 4H), 1.72–1.64 (m, 3H), 1.46 (s, 9H).
Tert-Butyl-4-(6-Methoxynicotinoyl)Piperidine-1-Carboxylate (5)
5-Bromo-2-methoxy-pyridine (1.54 ml, 11.92 mmol) and 17 ml freshly distilled THF was placed in a reaction flask. The mixture was cooled to −78 °C and 8.5 ml (1.6 N, 13.63 mmol) n-butyl lithium was added; the mixture was stirred at −78 °C for 75 min. Approximately 2.16 g (7.94 mmol) of tert-butyl-4-(methoxy(methyl)carbamoyl)piperidine-1-carboxylate (4) in 18 ml freshly distilled THF was added dropwise to the reaction mixture at −78 °C which continued to stir at this temperature for 135 min. The reaction was quenched with aqueous ammonium chloride. The organic layer was separated and the aqueous layer was extracted with ethyl acetate. The combined organic fractions were washed with water and brine then dried over anhydrous Na2SO4 and concentrated under reduced pressure to give a brown oil. The crude product was purified on a silica gel column (1:5 to 1:3 ethyl acetate/hexane) to afford 1.91 g (5.96 mmol, 75 %) of tert-butyl-4-(6-methoxynicotinoyl)piperidine-1-carboxylate (5) as a pale yellow solid. MP: 81–84 °C. 1HNMR (300 MHz, CDCl3): δ 8.80 (d, J=2.4 Hz, 1H), 8.13 (dd, J=8.7, 2.4 Hz, 1H), 6.81 (d, J=8.4 Hz, 1H), 4.15–4.10 (m, 2H), 4.01 (s, 3H), 3.30 (m, 1H), 2.80–3.00 (m, 2H), 1.81–1.68 (m, 4H), 1.62 (s, 9H).
((3-Hydroxy-1, 2, 3, 4-Tetrahydronaphthalen-2-yl)Piperidin-4-yl)(6-Methoxypyridin-3-yl)-Methanone (6)
tert-Butyl-4-(6-methoxynicotinoyl)piperidine-1-carboxylate (5, 184 mg, 0.57 mmol) 2.5 ml dichloromethane and 0.5 ml TFA were placed in a 50 ml reaction flask. The reaction mixture was stirred at rt for 2.5 h then concentrated under reduced pressure to give a residue which was neutralized with 2.0 ml 1 N NaOH and extracted with dichloromethane. The organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford 124 mg (0.56 mmol, 98 %) of (6-methoxypyridin-3-yl)(piperidin-4-yl)methanone as a pale yellow solid. 1HNMR (300 MHz, CDCl3): δ 8.80 (d, J=2.4 Hz, 1H), 8.14 (dd, J=8.7, 2.4 Hz, 1H), 6.81 (dd, J=9.0, 0.4 Hz, 1H), 3.98 (s, 3H), 3.33–3.16 (m, 3H), 2.75 (td, J= 12.2, 2.8 Hz, 2H), 1.85–1.63 (m, 5H). MP: 55–58 °C. One hundred twenty-four milligrams (0.56 mmol) of (6-methoxypyridin-3-yl)(piperidin-4-yl)methanone, 91 mg (0.63 mmol) of 1a,2,7,7a-tetrahydronaphtho[2,3-b]oxirene, and 4.0 ml methanol were added to a 50 ml reaction flask followed by 0.5 ml triethylamine. The reaction mixture was heated to 60 °C and stirred under argon at this temperature overnight. The reaction mixture was cooled to rt and diluted with ethyl acetate. The reaction mixture was washed with saturated aqueous NaHCO3, and the organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure to give the crude product which was purified on a silica gel column (1:1 ethyl acetate/hexane) to afford 50 mg (0.136 mmol, 24% ) of (±)-((3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(6-methoxypyridin-3-yl)methanone (±6) as a colorless oil. 1HNMR (300 MHz, CDCl3): δ 8.80 (d, J=2.1 Hz, 1H), 8.15 (dd, J=8.9, 2.7 Hz, 1H), 7.16–7.08 (m, 4H), 7.82 (d, J=8.7 Hz, 1H), 4.00 (s, 3H), 3.88–3.83 (m, 1H), 3.33–3.02 (m, 2H), 3.01–2.76 (m, 7H), 2.41 (td, J=11.2, 2.8 Hz, 1H), 1.95–1.80 (m, 4H).
Resolution of Enantiomers of (±)-((3-Hydroxy-1,2,3,4-Tetrahydronaphthalen-2-yl)Piperidin-4-yl)(6-Methoxypyridin-3-yl)Methanone ((±)-6)
Approximately 40 mg of (±)-6 was resolved using a Chiralcel OD column (250 mm×10 mm, acetonitrile, flow rate 4.0 ml/min, and UV wavelength 254 nm) to give 18.5 mg of (+)-6 (Rt =12 min; enantiomeric purity >95 %) and 18.1 mg of (−)-6 (Rt=18 min; enantiomeric purity >95 %). The optical rotation of (+)-6 was [α]D=+10.1° at a concentration of 9.25 mg/ml in methanol. The optical rotation of (−)-6 was [α]D= −9.7° at a concentration of 9.05 mg/ml in methanol.
Approximately 15 mg (0.041 mmol) of (+)-6 was placed in a 50 ml flask with 2.0 ml dichloromethane. One equivalent of oxalic acid (3.68 mg, 0.041 mmol) in 0.6 ml ethyl acetate was added and the reaction mixture was stirred at rt overnight. The precipitate was collected by filtration to give 14.0 mg (0.031 mmol, 76 %) of (+)-6 oxalate as a white solid. MP: 194–199 °C; Anal Calcd for C24H28N2O7•0.25H2O: C, 62.53; H, 6.23; N, 6.08; Found: C, 62.53; H, 6.23; N, 6.08.
Approximately 18.1 mg (0.049 mmol) of (−)-6 was placed in a 50 ml flask with 2.0 ml dichloromethane. One equivalent of oxalic acid (4.4 mg, 0.049 mmol) in 0.6 ml ethyl acetate was added and the reaction mixture was stirred at rt overnight. The precipitate was collected by filtration to give 13.6 mg (0.03 mmol, 61 %) of (−)-6 oxalate as white solid. MP: 194–200 °C; Anal Calcd for C24H28N2O7•0.53H2O: C, 61.85; H, 6.29; N, 6.01; Found: C, 61.84; H, 6.38; N, 6.17.
(−)-((3-Hydroxy-1,2,3,4-Tetrahydronaphthalen-2-yl)Piperidin-4-yl)(6-Hydroxypyridin-3-yl)Methanone ((−)-7)
Three milliliters of chloroform and 77 mg (0.21 mmol) (−)-6 was placed in a 50 ml reaction flask under argon followed by 63 μl (0.44 mmol) of trimethylsilyl iodide (the colorless solution turned to clear yellow). The reaction mixture was heated to 55 °C for 90 min then cooled to rt, and 0.6 ml methanol was added. The reaction mixture was concentrated under reduced pressure, diluted with ethyl acetate, and quenched with aqueous NaHCO3. The organic layer was separated and the aqueous layer extracted with ethyl acetate. The combined organic fractions were washed with brine and dried over anhydrous Na2SO4 then concentrated under reduced pressure to afford 75 mg (0.21 mmol, 99 %) of (−)-((3-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)piperidin-4-yl)(6-hydroxypyridin-3-yl)methanone ((−)-7) as an off-white solid. MP: 215–219 °C; 1HNMR (CDCl3): δ 8.18 (d, J=5.4 Hz, 1H), 8.08 (dd, J=9.6, 2.1 Hz, 1H), 7.19–7.09 (m, 4H), 6.64 (d, J=9.6 Hz, 1H), 3.96–3.80 (m, 1H), 3.31 (dd, J=16.2, 6.0 Hz, 1H), 3.10–2.65 (m, 8H), 2.45–2.25 (m, 1H), 1.99–1.65 (m, 4H).
Radiosynthesis of (−)-[11C]2
Approximately 1.5 mg of the precursor (−)-9 was placed in a reaction vessel with 0.25 ml of DMF followed by 2.6 μl of 5 N NaOH and thoroughly mixed on a vortex. [11C]CH3I was bubbled into the reaction vessel. The reaction mixture was heated at 90 °C for 5 min and quenched with 1.75 ml of HPLC mobile phase and purified by HPLC. The radioactive product, (−)-[11C]2, was collected between 15 and 19 min in 50 ml sterile water. The aqueous solution was passed through C18 Sep-Pak Plus by applying nitrogen pressure. The vial was rinsed with 10 ml of water which was also passed through the Sep-Pak Plus. The trapped product was eluted with 1.0 ml of ethanol to afford (−)-[11C]2 with 45 % yield and specific activity (SA)>480 GBq/μmol (decay-corrected to EOB). HPLC purification conditions are as follows: Econosil C18 semi-prep column (250 mm×10 mm, 10 μm); mobile phase: 33 % acetonitrile in 0.1 M ammonium formate buffer (pH 4.5), flow rate 4.2 ml/min; UV wavelength 270 nm. The entire process was completed in approximately 1 h. HPLC QC conditions are as follows: Alltech Altima C18 column (4.6×250 mm, 10 μm); mobile phase: 42 % acetonitrile in 0.1 M ammonium formate buffer (pH 4.5), flow rate 1.5 ml/min; UV wavelength 270 nm. Rt= 4.4 min.
Radiosynthesis of (−)-[11C]6
Approximately 1.5 mg of the precursor (−)-7 was placed in a reaction vessel with 0.25 ml DMF followed by 3 μl of 5 N NaOH and thoroughly mixed on a vortex. [11C]CH3I was passed through a silver triflate column to obtain [11C]CH3OTf which was bubbled into the reaction vessel. The reaction mixture was heated at 90 °C for 5 min and quenched by adding 1.75 ml of HPLC mobile phase. The reaction mixture was loaded on to HPLC for purification. Radioactive product was collected between 14 and 16 min into a 100 ml bottle containing ~50 ml sterile water. The aqueous solution was passed through C18 Sep-Pak Plus by applying nitrogen pressure. The collection bottle was rinsed with 10 ml of water which was also passed through the Sep-Pak Plus. The trapped product was eluted with 1.0 ml of ethanol to afford (−)-[11C]6 with SA>140 GBq/μmol and a radiochemical purity of >97 %. HPLC purification conditions are as follows: Econosil C18 column (250 mm×10 mm, 10 μm); mobile phase 33 % acetonitrile in 0.1 M ammonium formate buffer (pH 4.5), flow rate 3.2 ml/min; UV wavelength 270 nm. The entire process was completed in approximately 1 h. HPLC QC conditions are as follows: Grace Altima C18 column (4.6×250 mm, 10 μm); mobile phase: 35 % acetonitrile in 0.1 M ammonium formate buffer (pH 4.5), flow rate 1.5 ml/min; UV wavelength 270 nm. Rt=4.4 min.
Results and Discussion
Chemistry
Synthesis of 2 followed a procedure similar to that previously published by our group [19]. Treatment of the commercially available amine 1 with 1a,2,7,7a-tetrahydronaphtho[2,3-b]oxirene in ethanol in the presence of potassium carbonate as the base (Scheme 1) gave compound 2 as a mixture of enantiomers. 1a,2,7,7a-Tetrahydronaphtho[2,3-b]oxirene was synthesized via the epoxidation of 1,4-dihydronaphthalene with m-CPBA. Based on previously obtained X-ray crystal structure analysis for a similar compound [19, 25], the trans-relative stereochemistry was assigned for the epoxide ring opened product 2.
Scheme 1.
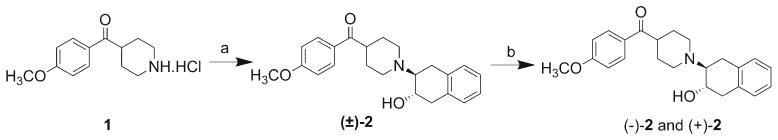
Syntheses of standard compounds (−)-2 and (+)-2. Reagents and conditions: a) 1a,2,7,7a-tetrahydronaphtho[2,3-b]oxirene, K2CO3, EtOH reflux, b) HPLC separation of enantiomers: Chiralcel OD column, mobile phase: 1 % hexane/20 % IPA70% MeOH; flow rate: 4.0 ml/min (+)-2 at 14 min and (−)-2 at 30 min.
Compound 6 was synthesized as shown in Scheme 2. Commercially available isonipecotic acid was treated with Boc2O and converted to Weinreb amide 4 with BOPCl. Weinreb amide 4 was treated with lithiated 2-methoxy-5-bromopyridine to give compound 5. Deprotection of the Boc group in 5 followed by treatment with 1a,2,7,7a-tetrahydronaphtho[2,3-b]oxirene under basic conditions gave the target pyridine 6 as a racemic mixture.
Scheme 2.

Syntheses of enantiomers of (+)-6 and (−)-6, and the desmethyl counterpart (−)-7. Reagents and conditions: a) 1 eq. (BOC)2O, K2CO3, THF/H2O, 0 °C to rt, 66 %; b) N,O-dimethylhydroxylamine hydrochloride, NEt3, BOPCl, DMF, 0 °C to rt, overnight, 54 %; c) 5-bromo-2-methoxypyridine, BuLi, THF, −78 °C, 75 %; d) TFA, CH2Cl2, 98 %; e) 1a,2,7,7a-tetrahydronaphtho[2,3-b]oxirene, NEt3, MeOH, 24 %; f) Separation of enantiomers on chiral HPLC (Chiralcel OD 250×10 mm, mobile phase: 100 % acetonitrile, 4 ml/min), Rt (+)-enantiomer at 12 min and (−)-enantiomer at 18 min; g) TMSI, CHCl3, 55 °C, 99 %.
The racemic compounds 2 and 6 were resolved by HPLC using a Chiralcel OD semipreparative column (250 mm× 10 mm) under normal-phase isocratic conditions. A mixture of hexane, 2-propanol, and methanol was used as the mobile phase to separate the enantiomers of 2. Acetonitrile was used as the mobile phase to separate the enantiomers of the 2-methoxypyridine analogue 6. Optical rotation measurements of both the enantiomers of the compounds 2 and 6 revealed that the (+) enantiomers eluted first and was followed by the (−) counterparts.
The resolved enantiomers were converted to the corresponding oxalate salts by treating with one equivalent of oxalic acid in ethyl acetate. In vitro binding affinity studies were performed to identify the more potent enantiomer. The (−)-enantiomers of both compound 2 and 6 were found to be more potent than the corresponding (+)-enantiomers. Those results are reported in the accompanying article in this journal along with the in vivo evaluation of the radioligands [23].
In order to evaluate the potential of (−)-[11C]2 and (−)-[11C]6 as PET radiotracers to image VAChT, the precursors (−)-9 and (−)-7 were synthesized as shown in Schemes 2 and 3, respectively. Synthesis of (−)-9 began with the commercially available amine 1. Demethylation of 1 under strongly acidic conditions gave the hydroxy compound 8, which was reacted with 1a,2,7,7a-tetrahydronaphtho[2,3-b]oxirene to give precursor 9 as a racemic mixture. Attempts to separate the enantiomers of 9 using chiral HPLC were unsuccessful. Therefore, the hydroxy group of 9 was protected with a benzyl group, enabling a more convenient separation of enantiomers by chiral HPLC. The (−)-enantiomer of the benzyl-protected compound 9 was hydrogenated at rt in the presence of 10 % palladium on activated carbon under 70 psi hydrogen atmosphere to give the precursor (−)-9 for synthesis of (−)-[11C]2. The 2-methoxypyridine (−)-6 was treated with trimethylsilyl iodide in chloroform to afford (−)-7, the precursor for synthesis of (−)-[11C]6.
Scheme 3.
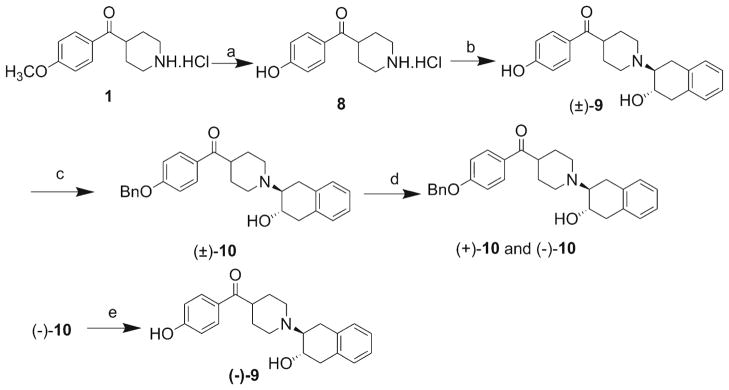
Synthesis of precursor (−)-9. Reagents and conditions: a) Conc. HCl, 100 °C, 8 days, 96 %; b) 1a,2,7,7a-tetrahydronaphtho[2,3-b]oxirene, K2CO3, EtOH, reflux, 4 days, 62 %; c) BnBr, K2CO3, acetone, 45 °C, overnight, 62 %; d) Separation of enantiomers of 10 on HPLC: Chiralcel OD column 250×10 mm; mobile phase: 60 % isopropanol in hexanes; flow rate: 4.0 ml/min; (+)-enantiomer at 22 min and (−)-enantiomer at 49 min; e) (−)-10, 10 % Pd/C, MeOH, 70 psi, 21 h, 90 %.
Radiochemistry
Precursors (−)-9 and (−)-7 were used for 11C-labeling to obtain (−)-[11C]2 and (−)-[11C]6, respectively. Radiosynthesis of (−)-[11C]2 was accomplished by treating (−)-9 in N,N-dimethylformamide with [11C]CH3I in the presence of 5 N NaOH (aq.) at 90 °C for 5 min as shown in Scheme 4. The reaction was quenched with HPLC mobile phase (33 % acetonitrile in 0.1 M ammonium formate buffer pH 4.5) and purified by HPLC (Econosil C18, 250 mm× 10 mm column). The radioactive product was collected in 50 ml sterile water and passed through a C18 Sep-Pak Plus cartridge to trap the product which was eluted with ethanol to afford (−)-[11C]2 with a radiochemical yield of 40–50 % (n>5, decay-corrected to EOB), SA>480 GBq/μmol (at EOB), and a radiochemical purity >99 %.
Scheme 4.
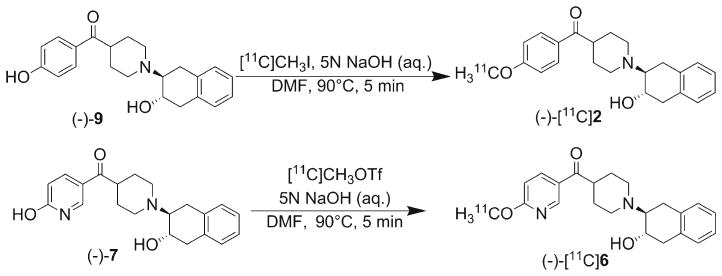
Radiosyntheses of (−)-[11C]2 and (−)-[11C]6.
The radiosynthesis of (−)-[11C]6 was initially attempted under similar conditions, but the yield was insufficient for conducting in vivo studies. Changing the base from NaOH to NaH did not significantly improve the yield. Using tetrabutyl ammonium hydroxide as the base did not yield the desired product. However, the radiosynthesis was successfully accomplished by treating (−)-7 in N,N-dimethylformamide with the more reactive methylating agent, [11C]CH3OTf, which is obtained by passing [11C]CH3I through a silver triflate column. NaOH was used as the base and the reaction was heated at 90 °C for 5 min (Scheme 4). The reaction was quenched with HPLC mobile phase (33 % acetonitrile in 0.1 M ammonium formate buffer pH 4.5), and purified by HPLC (Econosil C18, 250 mm×10 mm column). The radioactive product was collected in 50 ml sterile water, and passed through a C18 Sep-Pak Plus cartridge to trap the product which was eluted with ethanol to afford (−)-[11C]6 with radiochemical yield of 5–10 % (n=3, decay-corrected to EOB), SA>140 GBq/μmol (at EOB), and radiochemical purity >97 %.
Conclusion
Two potent enantiopure VAChT inhibitors were synthesized and successfully 11C-labeled. The radiosynthesis of (−)-[11C]2 and (−)-[11C]6 followed two different approaches. [11C]CH3I was used as the methylating agent for the radiosynthesis of the methoxyphenyl analog (−)-[11C]2 while radiosynthesis of the methoxypyridinyl analog (−)-[11C]6 required more the reactive [11C]CH3OTf. Radiosynthesis of (−)-[11C]2 was successfully accomplished with high radiochemical yield, radiochemical purity, and high specific activity. Although the radiochemical yield of (−)-[11C]6 was lower than that of (−)-[11C]2, it was sufficient to conduct initial in vivo studies. The further evaluation of each tracer required to determine their suitability as PET radiopharmaceuticals for in vivo quantification of VAChT is reported separately.
Acknowledgments
This work was supported by NIH grants NS061025, NS075527, and MH092797. The authors thank William H. Margenau and David Ficke in the Cyclotron Facility for radionuclide production. Optical rotation was determined in the laboratory of Dr. Douglas F. Covey in the Department of Molecular Biology and Pharmacology of Washington University.
Abbreviation
- ACh
Acetylcholine
- Anal
Analysis
- BBB
Brain–blood barrier
- BOC
t-butoxycarbonyl
- BOPCl
Bis(2-oxo-3-oxazolidinyl)-phosphinic chloride
- Bq
Becquerel
- Calcd
Calculated
- m-CPBA
m-chloroperoxybenzoic acid
- DMF
N,N-dimethylformamide
- DMSO
Dimethylsulfoxide
- EOB
End of bombardment
- EOS
End of synthesis
- HPL
CHigh-performance liquid chromatography
- IBVM
5-Iodobenzovesamicol
- PET
Positron emission tomography
- rt
Room temperature
- SA
Specific activity
- SPECT
Single-photon emission computed tomography
- TLC
Thin layer chromatography
- TMS
Tetramethylsilane
- TFA
Trifluoroacetic acid
- UV
Ultraviolet
- VAChT
Vesicular acetylcholine transporter
- Vesamicol
Trans-2-(4-phenylpiperidino)cyclohexanol
Footnotes
Conflict of Interest. The authors declare that they have no conflict of interest.
References
- 1.Schliebs R, Arendt T. The significance of the cholinergic system in the brain during aging and in Alzheimer’s disease. J Neural Transm. 2006;113:1625–1644. doi: 10.1007/s00702-006-0579-2. [DOI] [PubMed] [Google Scholar]
- 2.Hilker R, Thomas AV, Klein JC, et al. Dementia in Parkinson disease: functional imaging of cholinergic and dopaminergic pathways. Neurology. 2005;65:1716–1722. doi: 10.1212/01.wnl.0000191154.78131.f6. [DOI] [PubMed] [Google Scholar]
- 3.Gilmor ML, Nash NR, Roghani A, et al. Expression of the putative vesicular acetylcholine transporter in rat brain and localization in cholinergic synaptic vesicles. J Neurosci. 1996;16:2179–2190. doi: 10.1523/JNEUROSCI.16-07-02179.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prado VF, Martins-Silva C, de Castro BM, et al. Mice deficient for the vesicular acetylcholine transporter are myasthenic and have deficits in object and social recognition. Neuron. 2006;51:601–612. doi: 10.1016/j.neuron.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 5.Rogers GA, Parsons SM, Anderson DC, et al. Synthesis, in vitro acetylcholine-storage-blocking activities, and biological properties of derivatives and analogues of trans-2-(4-phenylpiperidino)cyclohexanol (vesamicol) J Med Chem. 1989;32:1217–1230. doi: 10.1021/jm00126a013. [DOI] [PubMed] [Google Scholar]
- 6.Widen L, Eriksson L, Ingvar M, et al. Positron emission tomographic studies of central cholinergic nerve terminals. Neurosci Lett. 1992;136:1–4. doi: 10.1016/0304-3940(92)90633-i. [DOI] [PubMed] [Google Scholar]
- 7.Bravo D, Parsons SM. Microscopic kinetics and structure-function analysis in the vesicular acetylcholine transporter. Neurochem Int. 2002;41:285–289. doi: 10.1016/s0197-0186(02)00058-x. [DOI] [PubMed] [Google Scholar]
- 8.Kuhl DE, Koeppe RA, Fessler JA, et al. In vivo mapping of cholinergic neurons in the human brain using SPECT and IBVM. J Nucl Med. 1994;35:405–410. [PubMed] [Google Scholar]
- 9.Kuhl DE, Minoshima S, Fessler JA, et al. In vivo mapping of cholinergic terminals in normal aging, Alzheimer’s disease, and Parkinson’s disease. Ann Neurol. 1996;40:399–410. doi: 10.1002/ana.410400309. [DOI] [PubMed] [Google Scholar]
- 10.Albin RL, Cross D, Cornblath WT, et al. Diminished striatal [123I]iodobenzovesamicol binding in idiopathic cervical dystonia. Ann Neurol. 2003;53:528–532. doi: 10.1002/ana.10527. [DOI] [PubMed] [Google Scholar]
- 11.Mazere J, Prunier C, Barret O, et al. In vivo SPECT imaging of vesicular acetylcholine transporter using [123I]IBVM in early Alzheimer’s disease. Neuroimaging. 2008;40:280–288. doi: 10.1016/j.neuroimage.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 12.Meikle SR, Kench P, Kassiou M, et al. Small animal SPECT and its place in the matrix of molecular imaging technologies. Phys Med Biol. 2005;50:R45–R61. doi: 10.1088/0031-9155/50/22/R01. [DOI] [PubMed] [Google Scholar]
- 13.Jung YW, Frey KA, Mulholland GK, et al. Vesamicol receptor mapping of brain cholinergic neurons with radioiodine-labeled positional isomers of benzovesamicol. J Med Chem. 1996;39:3331–3342. doi: 10.1021/jm9507486. [DOI] [PubMed] [Google Scholar]
- 14.Mulholland GK, Wieland DM, Kilbourn MR, et al. [18F]fluoroethoxy-benzovesamicol, a PET radiotracer for the vesicular acetylcholine transporter and cholinergic synapses. Synapse. 1998;30:263–274. doi: 10.1002/(SICI)1098-2396(199811)30:3<263::AID-SYN4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 15.Efange SM, Khare AB, von Hohenberg K, et al. Synthesis and in vitro biological evaluation of carbonyl group-containing inhibitors of vesicular acetylcholine transporter. J Med Chem. 2010;53:2825–2835. doi: 10.1021/jm9017916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giboureau N, Som IM, Boucher-Arnold A, et al. PET radioligands for the vesicular acetylcholine transporter (VAChT) Curr Top Med Chem. 2010;10:1569–1583. doi: 10.2174/156802610793176846. [DOI] [PubMed] [Google Scholar]
- 17.Kilbourn MR, Hockley B, Lee L, et al. Positron emission tomography imaging of (2R,3R)-5-[18F]fluoroethoxybenzovesamicol in rat and monkey brain: a radioligand for the vesicular acetylcholine transporter. Nucl Med Biol. 2009;36:489–493. doi: 10.1016/j.nucmedbio.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mach RH, Voytko ML, Ehrenkaufer RL, et al. Imaging of cholinergic terminals using the radiotracer [18F](+)-4-fluorobenzyltrozamicol: in vitro binding studies and positron emission tomography studies in nonhuman primates. Synapse. 1997;25:368–380. doi: 10.1002/(SICI)1098-2396(199704)25:4<368::AID-SYN8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 19.Tu Z, Efange SM, Xu J, et al. Synthesis and in vitro and in vivo evaluation of 18F-labeled positron emission tomography (PET) ligands for imaging the vesicular acetylcholine transporter. J Med Chem. 2009;52:1358–1369. doi: 10.1021/jm8012344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawamura K, Shiba K, Tsukada H, et al. Synthesis and evaluation of vesamicol analog (−)-O-[11C]methylvesamicol as a PET ligand for vesicular acetylcholine transporter. Ann Nucl Med. 2006;20:417–424. doi: 10.1007/BF03027377. [DOI] [PubMed] [Google Scholar]
- 21.Kilbourn MR, Jung YW, Haka MS, et al. Mouse brain distribution of a carbon-11 labeled vesamicol derivative: presynaptic marker of cholinergic neurons. Life Sci. 1990;47:1955–1963. doi: 10.1016/0024-3205(90)90408-j. [DOI] [PubMed] [Google Scholar]
- 22.Petrou M, Frey KA, Kilbourn MR, et al. In vivo imaging of human cholinergic nerve terminals with (−)-5-18F-fluoroethoxybenzovesamicol: biodistribution, dosimetry, and tracer kinetic analyses. J Nucl Med. 2014;55:396–404. doi: 10.2967/jnumed.113.124792. [DOI] [PubMed] [Google Scholar]
- 23.Padakanti PK, Zhang X, Jin H, et al. In vitro and in vivo characterization of two C-11 labeled PET tracers for vesicular acetylcholine transporter. Mol Imaging Biol. 2014 doi: 10.1007/s11307-014-0749-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li J, Zhang X, Zhang Z, et al. Heteroaromatic and aniline derivatives of piperidines as potent ligands for vesicular acetylcholine transporter. J Med Chem. 2013;56:6216–6233. doi: 10.1021/jm400664x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang W, Cui J, Lu X, et al. Synthesis and in vitro biological evaluation of carbonyl group-containing analogues for Sigma-1 receptors. J Med Chem. 2011;54:5362–5372. doi: 10.1021/jm200203f. [DOI] [PMC free article] [PubMed] [Google Scholar]