

Abstract
Background
Excitotoxicity (the toxic overstimulation of neurons by the excitatory transmitter Glutamate) is a central process in widespread neurodegenerative conditions such as brain ischemia and chronic neurological diseases. Many mechanisms have been suggested to mediate excitotoxicity, but their significance across diverse excitotoxic scenarios remains unclear. Death Associated Protein Kinase (DAPK), a critical molecular switch that controls a range of key signaling and cell death pathways, has been suggested to have an important role in excitotoxicity. However, the molecular mechanism by which DAPK exerts its effect is controversial. A few distinct mechanisms have been suggested by single (sometimes contradicting) studies, and a larger array of potential mechanisms is implicated by the extensive interactome of DAPK.
Results
Here we analyze a well-characterized model of excitotoxicity in the nematode C. elegans to show that DAPK is an important mediator of excitotoxic neurodegeneration across a large evolutionary distance. We further show that some proposed mechanisms of DAPK’s action (modulation of synaptic strength, involvement of the DANGER-related protein MAB-21, and autophagy) do not have a major role in nematode excitotoxicity. In contrast, Pin1/PINN-1 (a DAPK interaction-partner and a peptidyl-prolyl isomerase involved in chronic neurodegenerative conditions) suppresses neurodegeneration in our excitotoxicity model.
Conclusions
Our studies highlight the prominence of DAPK and Pin1/PINN-1 as conserved mediators of cell death processes in diverse scenarios of neurodegeneration.
Keywords: Ischemia, Glutamate, Excitotoxicity, Neurodegeneration, Death-Associated protein kinase, Autophagy, Peptidyl prolyl isomerase Pin1
Background
Excitotoxicity is a neurodegenerative process believed to be the central mediator of brain damage in acute conditions such as brain ischemia and traumatic injury, and an important contributor to a range of chronic neurodegenerative diseases [1-4]. In excitotoxicity, the malfunction of Glutamate (Glu) Transporters (GluTs) [5-7] causes accumulation of Glu in excitatory synapses and exaggerated stimulation of postsynaptic Glu receptors (GluRs) [8]. The excessive influx of ions (especially Ca2+) into the postsynaptic neurons leads to their cell death via a spectrum of mechanisms that range from necrosis (at the core of the ischemic damage) to apoptosis or even recovery (at the penumbra). Despite our familiarity with the first few steps in excitotoxicity, our understanding of the steps following Ca2+ influx is very limited. Clinical trials using GluRs antagonists ended with disappointment, and recent data suggests that using antagonists to block GluR functions might be counterproductive, because Glu signaling includes both neurotoxic and pro-survival cascades [9,10]. These complications emphasize the need to illuminate cell-death-specific signaling cascades in excitotoxicity downstream of GluRs. A considerable number of such excitotoxic mechanisms have been suggested, but in many cases the data that supports a given suggestion is limited to specific excitotoxic paradigms.
One suggested mediator of excitotoxicity implicated in multiple experimental setups is the CaM-dependent Death Associated Protein Kinase (DAPK) [11-14]. Originally identified by an unbiased screen for mediators of interferon-induced cell death [15], DAPK was later recognized as a molecular switch that controls the choice between cell death processes such as apoptosis and autophagy [16,17]. Moreover, an elaborate web of biochemical interactions and functional connections has been revealed, placing DAPK in a key position at the center of many critical signaling cascades [11,18]. A considerable body of evidence suggests that DAPK also contributes to cell death in excitotoxicity [19], and its inhibition reduces neuronal loss in models of brain ischemia [20]. Several mechanisms have been suggested to explain DAPK’s involvement in excitotoxicity, but the issue remains controversial. One possible mechanism involves DAPK’s regulation of autophagy [17], a process that modulates some neurodegenerative conditions [21]. Alternatively, an intriguing study suggests the involvement of DANGER, a protein that contains a region of homology to the nematode protein MAB-21 and functions as a regulator of the IP3R [22]. Indeed DANGER was found to also interact with and inhibit the activity of DAPK [23]. The most cited suggestion in the field attributes the involvement of DAPK in excitotoxicity to the potentiation of Ca2+ currents through NR2B/GluN2B subunit-containing complexes of the NMDA- receptor (NMDA-Rs) family of GluRs [24]. This suggestion fits well with a proposed leading role for extrasynaptic NR2B/GluN2B –containing NMDA-Rs in excitotoxicity [25]. However, recently the proposed unique significance of NR2B/GluN2B –containing extrasynaptic NMDA-Rs in excitotoxicity has been brought into question [26-28]. Moreover, an earlier study suggests that in some mammalian neurons DAPK knockout provides protection from excitotoxicity that is not dependent on NMDA-Rs [29]. Indeed, some cases of excitotoxicity are mediated by another family of GluRs, the Ca2+ -Permeable AMPA Receptors (CPARs) [30-32]. These observations suggest that if DAPK is widely involved in excitotoxicity, including in cases where NR2B/GluN2B is not a main determinant of neurodegeneration, it might act through additional mechanisms to exert its effect.
We hypothesize that many signaling cascades might be involved in specific cases of excitotoxicity, depending on the exact scenario being used to induce it. However, the key features that constitute the core of the excitotoxic process might be conserved across differences in cell death scenarios and large evolutionary distances, as is the case in apoptosis [33,34] and autophagy [35,36]. We therefore set out to study the role of DAPK in excitotoxicity in our model of CPAR-mediated neurodegeneration in C. elegans [37], where GluR-dependent necrosis of central neurons postsynaptic to Glu connections is triggered by knockout (ko) of the GluT gene glt-3 [38] in a sensitized background (nuIs5 [39]). Indeed, this model has proven effective in identifying core processes that are conserved between nematode and mammalian excitotoxicity [37,40,41]. DAPK is particularly well conserved in C. elegans (in 52% sequence homology, presenting all of DAPK’s functional domains, and in its involvement in a number of signaling cascades [42-44]). The nematode DAPK-1 is widely expressed (including in neurons [42]), allowing us to test its involvement in nematode excitotoxicity and to study its mechanism of action. In this study we establish the central role of DAPK in Glu-triggered neurodegeneration in C. elegans, suggesting that its function is conserved across evolution and excitotoxic scenarios. We find little or no support for the views that DAPK’s regulation of excitotoxicity is mediated through the modulation of synaptic strength, MAB-21, or autophagy. Instead, we identify PINN-1, the nematode homolog of the DAPK-interaction-partner and phosphorylation-dependent peptidyl-prolyl isomerase Pin1, as an important factor in nematode excitotoxicity.
Results
DAPK-1 has a central role in nematode excitotoxicity
To test the involvement of DAPK in nematode excitotoxicity we combined our excitotoxicity strain (glt-3;nuIs5) with a deletion allele of the nematode homolog of DAPK, dapk-1(gk219), a gene knockout that was used to confirm the effect of the (ubiquitously expressed) nematode DAPK in autophagy and innate immunity [42,45]. In our previously described model of neurodegeneration we use the sensitizing transgenic modification nuIs5, where hyperactive Gαs and GFP are expressed under the glr-1 promoter in ~30 neurons [39] and cause GluR-independent stochastic degeneration of ~1 of these at-risk neurons per animal. When we add the KO of the GluT gene glt-3 we observe that more of these at-risk neurons degenerate [37]. The GluT-KO-triggered exacerbated necrosis in glt-3;nuIs5 is GluR-dependent, and therefore qualifies as nematode excitotoxicity. Nematode excitotoxicity causes neuronal swelling and death that is manifested with characteristic kinetics as gradually and stochastically appearing vacuole-like structures in some of the at-risk postsynaptic neurons. These vacuole-like structures become more abundant during larval development as the Glu signaling system matures (usually reaching up to ~4.5 head neurons/animal at L3), and then decline due to removal of cell corpses by engulfment [37]. We now observe that adding dapk-1 ko to this excitotoxicity strain causes a strong and statistically significant suppression of neurodegeneration throughout development (Figure 1A, an additional independent cross gave very similar results, not shown). To further confirm the contribution of dapk-1 to nematode excitotoxicity, we overexpressed the wt dapk-1 cDNA from an extra-chromosomal transgenic construct under a heat-shock promoter [42]. Since heat-shock might affect susceptibility to neurodegeneration, we took special care to compare an exact match of treated animals, without or with the dapk-1 overexpression transgene. To that end we took advantage of the fact that the random and partial segregation of the non-integrated overexpression construct allows us to compare transgenic and non-transgenic animals on the same plate exposed to the same conditions. We observed that dapk-1 overexpression resulted in a strong and statistically significant potentiation of necrotic neurodegeneration in postsynaptic neurons in all developmental stages (Figure 1B). Together, our data indicate that DAPK is an important mediator of excitotoxicity in C. elegans, suggesting that diverse scenarios of excitotoxicity share a common mechanism that assigns a central role to DAPK.
Figure 1.
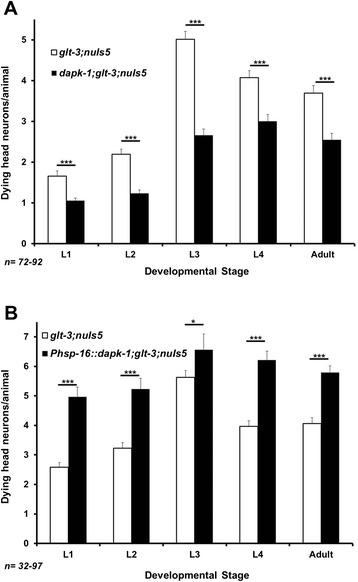
dapk-1 is an important mediator of nematode excitotoxicity. A) Dynamics of neurodegeneration in nematode excitotoxicity during development (using the glt-3;nuIs5 excitotoxicity model). dapk-1 ko mutation suppresses neurodegeneration in all developmental stages, but does not bring it down to background levels. B) Overexpression of wt dapk-1 from a heat-shock promoter enhances neurodegeneration compared to matched controls. In all bar graphs, error bars represent SE. Statistical significance is calculated using z score. *p < 0.05; ***p < 0.01.
dapk-1 ko does not alter presynaptic release or postsynaptic Glu response
We used two main guidelines in trying to track the mechanism by which dapk-1 regulates excitotoxic neurodegeneration in the nematode: 1) we looked at previous reports suggesting specific mechanisms for DAPK’s involvement in mammalian excitotoxicity; 2) we inferred from the general map of DAPK’s connectome in other cell processes [18] which additional proteins are plausible candidates for mediating DAPK’s effect in nematode excitotoxicity. One line of evidence suggests that DAPK interacts with, and may regulate the function of, Syntaxin 1A [46]. Syntaxin is part of the general mechanism of vesicular neurotransmitter release, a mechanism that is shared among all neurotransmitters [47]. Therefore, an effect of dapk-1 ko on any component of the synaptic vesicle release could lead to DAPK-mediated changes in excitotoxicity levels. However, given the ubiquitous expression of this gene, such an effect of DAPK on the common synaptic vesicle release mechanism will affect the dynamics of neurotransmitter release in all synapses. The study of synaptic vesicle release is very well developed in the nematode, and aldicarb is routinely used in C. elegans to identify mutations that cause even modest changes to the general synaptic release mechanism [48]. As aldicarb suppresses the degradation of Acetylcholine in the neuro-muscular junction, it causes animal paralysis with a typical dynamics. Mutations that reduce the activity of the general, common vesicle release mechanism (such as rab-3 [48]) cause a pronounced resistance to aldicarb (shifting the time-dependent paralysis curve to the right, Figure 2A), while mutations that enhance synaptic release (such as the ko of cpx-1, which encodes the synaptic vesicle release regulator complexin [49]) cause increased sensitivity to aldicarb (shifting the paralysis curve to the left, Figure 2A). We observed that the sensitivity of dapk-1 animals to aldicarb is indistinguishable from that of WT animals, suggesting that dapk-1 ko does not modify the synaptic release mechanism, as would have been expected of an effect on syntaxin (Figure 2A).
Figure 2.
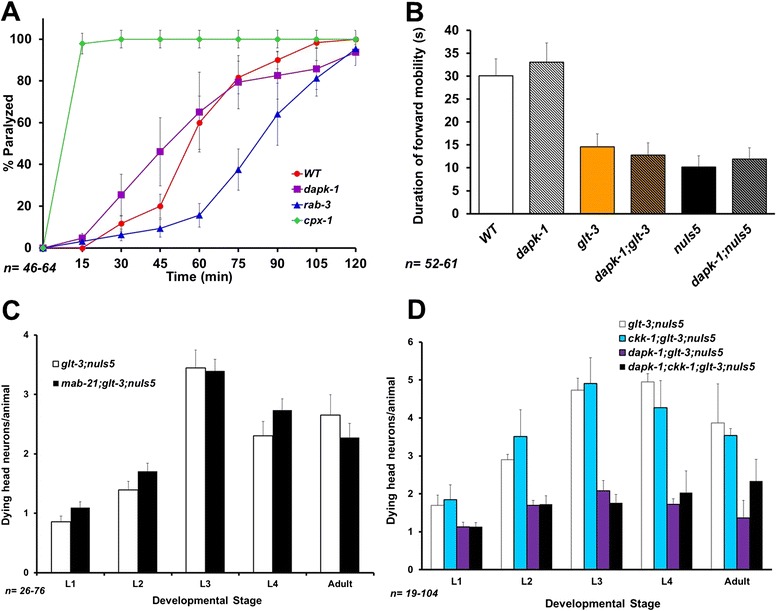
The effect of dapk-1 does not correlate with several proposed mechanisms. A) Aldicarb assay measures the overall activity of the common mechanism of synaptic vesicle release. Complexin (cpx-1) mutant is used as example of an oversensitive mutant (where synaptic release is exaggerate), while rab-3 serves as an example of aldicarb resistant mutant (where synaptic release is abnormally low). dapk-1 mutants do not show oversensitivity or resistance, suggesting their level of synaptic release is roughly normal. B) Duration of forward runs during spontaneous mobility is a sensitive reporter of the overall strength of Glu synapses that control this parameter. Even small changes in the activity or number of GluRs typically modify this behavior. dapk-1 mutants are not different from normal counterparts in either a WT background, in an excessive Glu stimulation background (glt-3), or in the sensitized background (nuIs5) used in our excitotoxicity model. These three conditions by themselves do not cause excessive command neuron loss, a loss that might otherwise render the assay uninformative as it is suppressible by dapk-1. C) A mutation in the gene encoding the DANGER-related protein MAB-21 does not affect nematode excitotoxicity. D) A mutation in the gene encoding the nematode homolog of CaMKK does not affect nematode excitotoxicity.
We next asked if DAPK modifies the extent of postsynaptic response in the glutamatergic synapses where excitotoxicity occurs (as reported in mammals, where DAPK was suggested to modify synaptic strength [24]). Two behavioral assays have proven very effective in detecting even small changes in the activity level of glutamatergic synapses in C. elegans. These are the Nose Touch (NOT) assay, and the duration of spontaneous forward mobility assay. Therefore, if dapk-1 ko causes changes in the specific Glu packaging mechanism in synaptic vesicles (e.g., by affecting vGluTs), or changes the number or activity-level of GluRs in the synapse, this should be reflected in behavioral changes in these assays. In the particular case of dapk-1, a secondary phenotype of this mutation, namely overgrowth of cuticle on the animal’s nose [42], makes the nose-touch assay less informative. However, the duration of spontaneous forward mobility depends on the internal balance between forward and backward circuits, and should not be affected by the cuticle aberration. This assay is very reliable in detecting both under-activity and over-activity of the relevant Glu synapses [50-52], allowing us to use it as a proxy measure for Glu synaptic strength. We did not observe any changes in spontaneous mobility triggered by dapk-1 ko, either in WT background or in the background of each of the two components (glt-3 or nuIs5) used in our excitotoxicity model to trigger the intensified Glu signaling (Figure 2B). These observations suggest that dapk-1 has no strong effect on synaptic release or the overall strength of signaling in Glu synapses in C. elegans.
The DANGER-related protein MAB-21 and the CaMKK CKK-1 do not exert strong regulation of nematode excitotoxicity
The characterization of DAPK as a CaM-dependent kinase is particularly intriguing to us, since Ca2+ signaling is critical to excitotoxicity in both mammals and nematodes. We therefore examined DAPK-partners that might also be involved in Ca2+ signaling. DANGER is a mammalian DAPK inhibitor that regulates Ca2+ release from the ER [22,23]. There is no direct, full-length homolog of DANGER in the worm genome, but the core of the mammalian protein is thought to be homologous to the nematode protein MAB-21 [53]. It is therefore reasonable to assume that mab-21 will be essential to the role of a hypothetic DANGER-like complex in C. elegans. However, a mutation in mab-21 had no effect on nematode excitotoxicity (Figure 2C). Another important protein that binds mammalian DAPK and is involved in Ca2+ signaling is CaMKK. However, again, a mutation in the only CaMKK gene in the worm, ckk-1 [54], had no effect on excitotoxicity (Figure 2D). These results suggest that these two putative Ca2+ signaling regulators and DAPK-interaction-partners do not have a large contribution to nematode excitotoxicity.
We also considered the many other proteins that are known to interact with DAPK [18] as possible modulators of excitotoxicity, but we could not assign to many of them high priority because either there was no immediate obvious connection to neurodegeneration (e.g., tropomyosin), there is no clear nematode homolog (e.g., NFκB), the nematode homolog is not known to be active in C. elegans neurons (e.g., p53), or the process in which this protein is involved has been shown by us to be not involved in nematode excitotoxicity (e.g., apoptosis [41]). However, DAPK is also known to interact with Beclin1, a key regulator of the evolutionary conserved process of autophagy, suggesting that autophagy-mediated cell death could potentially be an avenue for DAPK to regulate nematode excitotoxicity.
Autophagy has a minor role in nematode excitotoxicity
The interaction between mammalian DAPK and Beclin1 is considered a major avenue for DAPK’s ability to regulate autophagy [11,16,17,55]. Autophagy has been suggested to be an important factor in some neurodegenerative conditions [21,56,57], and has also been shown to be a major contributor to degenerin-mediated neurodegeneration in the nematode [58-60] (where necrotic neurodegeneration is triggered by a constitutively open channel of the DEG/ENaC family [61]). Moreover, dapk-1 regulates autophagy in C. elegans [43]. We therefore set out to determine the role of autophagy in nematode excitotoxicity, using some of the same reagents used to demonstrate autophagy’s role in degenerin-induced neurodegeneration in the worm [58,60,62]. One such reagent is the red fluorescent labeled LC3 –homolog LGG-1 (originally created by the Tavernarakis lab [62], for the current study assigned the strain name IMN21). This reporter indicates elevated autophagy by both the appearance of intracellular puncta and by an overall increase in cellular fluorescence. We crossed IMN21 with our excitotoxicity strain glt-3;nuIs5. Since the red fluorescence can appear in any body cell that triggers autophagy, we used the Pglr-1::GFP marker expressed in our excitotoxicity strains to focus our attention on the postsynaptic neurons that are at risk for neurodegeneration. If autophagy is a strong component of excitotoxicity, we would expect at-risk neurons (labeled with GFP) to express autophagy marker (DsRed) upon exposure to high concentrations of Glu (triggered by the glt-3 ko). We counted the number of at-risk neurons that show LGG-1 puncta and measured the overall intensity of DsRed::LGG-1 signal in these neurons. Since autophagy was reported to have a role in the low-level neurodegeneration caused by nuIs5 alone [60], we concentrated on the added effect of excitotoxicity by comparing DsRed::LGG-1 signals in animals that express only the sensitizing construct nuIs5 to those of animals in which neurons are subjected to the full excitotoxic insult (glt-3;nuIs5). We found that excitotoxicity resulted in only a small and statistically-insignificant increase in LGG-1 puncta, and no change in overall LGG-1 intensity in at-risk neurons (Figure 3A and B).
Figure 3.
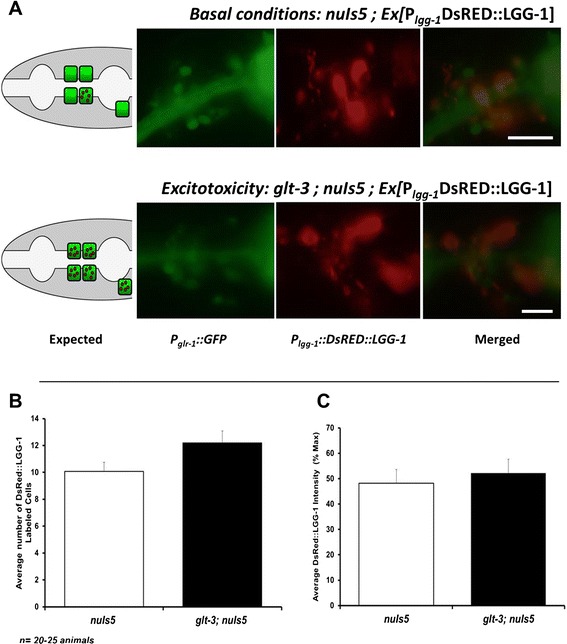
A DsRed::LGG-1 reporter of autophagy does not provide convincing evidence for triggering of autophagy in at-risk neurons exposed to the excitotoxic insult. A) Representative images showing at-risk neurons in green (expressing P glr-1 ::GFP) and DsRed::LGG-1 expressing cells in red. Lateral view, anterior left, dorsal up, illustration on the left describing the results expected from a putative involvement of autophagy in excitotoxicity. Expression of green labeling in the pharynx comes from the co-injection marker for the DsRed::LGG-1 label, expressing P myo-2 ::GFP. B) Analysis of images taken from the two groups shows a similar number of at-risk neurons (green cells) showing DsRed::LGG-1 puncta. The observed small difference is not statistically significant. (t test used here) C) The average intensity of the DsRed::LGG-1 signal in at-risk neurons (green) in very similar in the two groups.
To further study the possible involvement of autophagy in nematode excitotoxicity we examined the effect of inhibiting autophagy by genetic and chemical means. While a number of mutations and drugs have been used in the past, not all of them are available to us here. For example, bec-1 ko was previously used to monitor the requirement of this autophagy regulator for mec-4(d) –induced necrosis in early development [58]. However, the lethal effect of this mutation in later development, when most of the nematode excitotoxic necrosis occurs, prevents us from using this approach. Similarly, the vacuolar-type ATPase inhibitor bafilomycin is commonly used to block autophagy by elevating lysosomal pH [63]. However, the same V-ATPase is used in neurons to acidify synaptic vesicles as a means to provide the driving force for neurotransmitter loading [64-66], and therefore using bafilomycin can be expected to reduce neurotransmitter release. We therefore turned to use other means of intervention that are more compatible with our system: a mutation in the autophagy regulator unc-51 (using the e369 allele) and the chemical inhibitor 3MA (both used previously to show that degenerin-triggered neurodegeneration in C. elegans depends strongly on autophagy [58,60]). We noticed only a moderate effect for these two factors, evident in some developmental stages (Figure 4A and B). Such a moderate effect is in line with the reported effect of unc-51 on nuIs5 alone [60] (see discussion). We then used an independent set of experiments and epistasis analysis to determine if this moderate effect works independently of dapk-1 or in the same pathway. We noticed that the effect of blocking autophagy on the extent of excitotoxicity is reproducible only in one developmental stage (L3). Trying to determine if dapk-1 and 3MA work in the same pathway, we compared their observed combined effect (in the dapk-1 + 3MA combination) to the calculated expected effect if these two processes were completely independent. However, given the moderate size of the 3MA effect and the inherent variability in our experiments, it is currently difficult to determine if dapk-1 and autophagy work in the same pathway or independently. Nonetheless, the fact that the effect of autophagy is much more limited in size and duration than that of dapk-1 supported a continued search for other mechanisms by which dapk-1 might regulate excitotoxicity.
Figure 4.
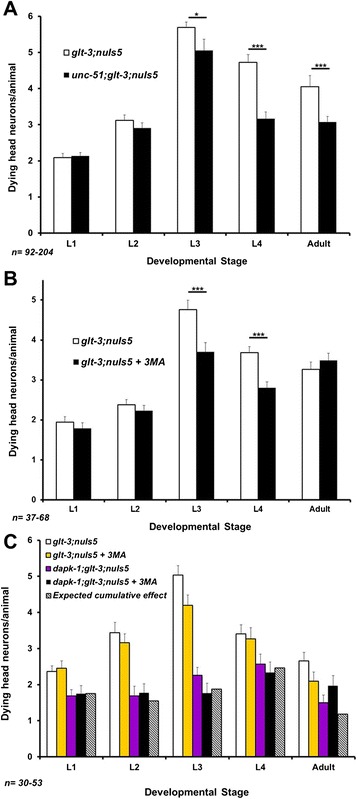
Treatments that block autophagy (and dramatically reduce neurodegeneration in other forms of necrotic neurodegeneration in C. elegans) have a reproducible but small effect in nematode excitotoxicity. A) A mutation in unc-51 shows a moderate effect on nematode excitotoxicity. *p < 0.05 ; ***p < 0.01 B) Treatment with the autophagy-blocking drug 3MA has a moderate effect on nematode excitotoxicity. ***p < 0.01 C) Independent repetition of the experiment shown in B with the addition of epistasis analysis. The data shows that the only reproducible effect of autophagy blockade is in L3. Although at this stage the combined effect of autophagy blockade and dapk-1 correlates with a model of independent action of these two factors, the moderate extent of effects limits the strength of such a conclusion.
The DAPK interaction-partner and phosphorylation-dependent peptidyl-prolyl isomerase Pin1/PINN-1 is a conserved suppressor of neurodegeneration
Pin1 is an isomerase that changes the conformation of proline residues located next to phosphorylated Ser or Thr residues, thus changing overall protein conformation and controlling the activity of many phosphoproteins [67,68]. In recent years this protein has gained recognition as a major regulator of many signaling cascades, involved in both normal cell physiology, pathology, and in neurodegenerative diseases [67,69-71]. Pin1 is expressed in dendrites, its activity is modulated by Glu signaling, and it regulates PKCζ and PKMζ [72,73]. Pin1 is also known for its regulation of neuronal cytoskeleton and Tau protein phosphorylation, and for modulating neurodegeneration [69,74,75]. Recently, Pin1 was shown to functionally interact with DAPK [76]. We find that pinn-1 ko [77] causes increased neurodegeneration in nematode excitotoxicity (Figure 5). Like dapk-1 ko, the effect of pinn-1 ko is seen in all developmental stages (Figure 5A), and does not seem to involve a change in Glu synaptic strength (Figure 5B and C), suggesting that they influence cell-death processes subsequent to- (and not at the level of-) GluR.
Figure 5.
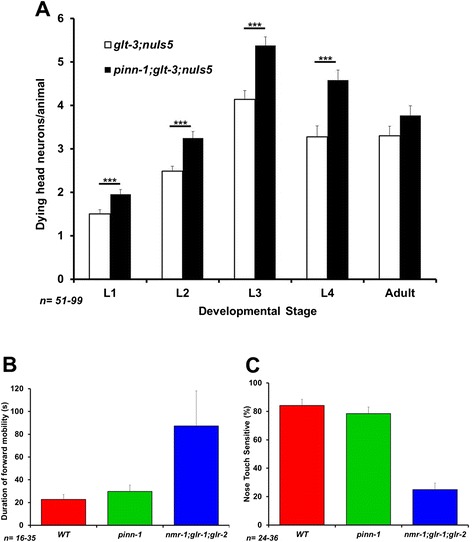
pinn-1 is an important suppressor of nematode excitotoxicity that does not affect basic synaptic strength. A) pinn-1 mutation enhances excitotoxicity throughout development. **p < 0.01 B & C) pinn-1 does not affect the duration of spontaneous forward mobility or nose touch sensitivity, two sensitive measures of Glu synaptic strength.
We wished to confirm that the effects of dapk-1 and pinn-1 on the dynamics of vacuolar appearance (as seen in Figures 1A and 5A) translate to ultimate survival of specific neurons in adult animals. Given the stochastic nature of neurodegeneration in glt-3;nuIs5 animals, it is usually difficult to identify which of the ~30 at-risk head neurons are degenerating in different animals. To circumvent this difficulty, we address the survival of specific neurons by focusing on the easily identifiable RIG neurons. The RIG neurons are part of the group of at-risk head neurons, they show only very minor levels of neurodegeneration in nuIs5 alone, and they are sensitive to all the treatments that modify total head neuron degeneration analyzed in our previous studies [37,40,41]. Indeed, RIG neurons exhibit inverse correlation between the number of vacuole-like structures they show during development (as observed by Nomarski) and the number of GFP-labeled RIG neurons surviving in the adult [37]. We now confirm that the number of degenerating RIG neurons during development is reduced by dapk-1 ko and increased by pinn-1 ko (data not shown). Importantly, we find that the number of GFP-labeled RIG neurons that survive to adulthood is increased by dapk-1 ko and decreased by pinn-1 ko (Figure 6B). These observations suggest that the effects of dapk-1 and pinn-1 on vacuolar appearance during development indeed translate to changes in ultimate survival of identified neurons.
Figure 6.
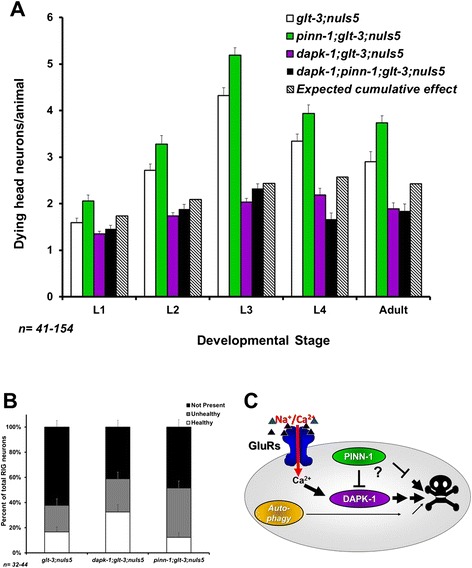
dapk-1 might cooperate with pinn-1 to regulate nematode excitotoxicity. A) Epistasis analysis testing the involvement of dapk-1 and pinn-1 in nematode excitotoxicity. Since the combined mutant shows strong suppression of neurodegeneration compared to the starting strain, these observations strongly suggest that pinn-1 is not downstream of dapk-1 (though the data is not conclusive enough to choose between the possibilities of pinn-1 acting upstream or independently of dapk-1). B) Verification that the effects of dapk-1 and pinn-1 mutations on the dynamics of overall neurodegeneration in head neurons translates to survival of identified neurons in the adult (counting surviving GFP-labeled RIG neurons in young adult animals). C) One of the likely models that can account for our observations on the role of DAPK-1, PINN-1, and autophagy in nematode excitotoxicity.
Finally, we wanted to determine if dapk-1 (where a KO causes decreased neurodegeneration) and pinn-1 (where a KO causes increased neurodegeneration) work in the same or separate pathways. Mammalian studies suggest that DAPK is acting upstream of Pin1 to inhibit its function [76], so that KO of pinn-1 could be expected to exert its death-stimulating effect regardless of whether dapk-1 is present or not. Instead, using epistasis analysis (Figure 6A), we observe that the double knockout dapk-1 ; pinn-1 exhibited suppression of excitotoxicity. We tried to further determine if the effect of dapk-1; pinn-1 double knockout reflects an intermediate outcome (in line with an independent, parallel effect of these two factors) or a dapk-1 ko –only-like outcome (in line with an obligatory sequential effect, where the dapk-1 mutation-induced decrease in neurodegeneration completely masks the ability of pinn-1 mutation to increase neurodegeneration). To that end we calculated what would be the effect of these two factors acting in parallel, to predict their cumulative independent effect (Figure 6A). Unfortunately, the small difference between the observed effect of dapk-1 alone and the calculated independent cumulative effects of dapk-1 and pinn-1, together with the variability in our data, do not allow us to discriminate with confidence between these two options. We therefore limit our conclusion to say that dapk-1 acts either downstream or in parallel to- (but not upstream of-) pinn-1 in nematode excitotoxicity.
Discussion
Mammalian studies have generated a plethora of proposed mechanisms in excitotoxicity, and some of these studies include suggested pathways for the involvement of DAPK in critical cell death events. However, the significance of these proposed mechanisms across divergent excitotoxic conditions remains unclear. In our study we focused on a glutamate-dependent neuronal death in C. elegans and examined a set of candidate mechanisms to define those that might be conserved through a large evolutionary distance. We previously found that some core constitutes of excitotoxicity are well conserved in our nematode model of GluT KO –triggered and CPAR-mediated neuronal necrosis. These include both death-promoting factors (such as release of Ca2+ from the ER [37]), and neuroprotective factors (such as cell stress resistance to insults, resistance that is conferred by FoxO/DAF-16 [40,41]). We now report that DAPK is also a highly conserved regulator of excitotoxicity (Figure 1A and B), though we note that dapk-1 ko does not bring neurodegeneration all the way down to background levels. We find no evidence for dapk-1 –mediated regulation of synaptic strength, as defined by the spontaneous mobility assay (Figure 2B), suggesting that the effect of DAPK on mammalian NR2B/GluN2B, though probably very important, does not extend to all forms of excitotoxicity. We also found no role for the DANGER-related protein MAB-21 in nematode excitotoxicity (though DANGER is a much larger protein than MAB-21, suggesting it might have additional functions not tested here).
Although dapk-1 is an important regulator of autophagy in C. elegans, we find only a minor role for autophagy in the neurodegenerative condition we study in glt-3;nuIs5 animals. We find that blocking autophagy has a relatively small neurodegeneration-reducing effect (~0.5-1 dying neuron/animal, Figure 4). This effect is smaller than the neurodegeneration-suppressing effect of dapk-1 ko (a decrease of ~2.5 dying neurons/animal, three independent isolates counted in Figures 1A, 4C, and 6A). Therefore, even if related, autophagy cannot account for the majority of DAPK’s effect in glt-3;nuIs5 animals. The minor effect of autophagy on neurodegeneration in glt-3;nuIs5 is in sharp contrast to the major effect that autophagy has on degenerin-mediated neurodegeneration, where it suppresses neurodegeneration by 75-90% [60]. We further note that these previous studies have demonstrated that blocking autophagy decreases the extent of low-level neurodegeneration triggered by nuIs5 alone [60]. Since neurodegeneration by nuIs5 alone (independent of excitotoxicity) is part of the total number of degenerating neurons we count in our assay (accounting for ~1 dying neuron/animal [37]), the small effect of autophagy seen in the current study can be attributed to its documented effect of nuIs5 alone. Indeed, we see no significant difference in labeling of fluorescent autophagy reporter when comparing nuIs5 to glt-3;nuIs5 animals (Figure 3). The results of epistasis analysis are not conclusive, but put together with the difference in size and timing of the effects of dapk-1 and autophagy on nematode excitotoxicity, our observations cause us to favor one of the possible models in which autophagy has only a minor role in nematode excitotoxicity in parallel to the role of DAPK-1 (Figure 6C). We emphasize that the parallel action of DAPK and autophagy is preferred by us in the specific scenario of nematode excitotoxic stress, while other stresses might include DAPK and autophagy in other pathway configurations.
The observation that the DAPK-interaction-partner Pin1/PINN-1 is a significant regulator of excitotoxicity is particularly intriguing and novel. In addition to its involvement in cancer [71] and stress response [78], Pin1 has been shown to be a critical regulator of dendritic Glu responses [72,73] and of neurodegeneration in Alzheimer disease [69,74,75]. The results of our epistasis analysis do not seem to support a role for Pin1/PINN-1 as an obligatory step downstream of DAPK/DAPK-1, as might be inferred from mammalian cancer studies [76]. Instead, our data supports that PINN-1 acts in parallel or upstream of DAPK-1 (Figure 6A and C).
Conclusions
Put together, our study suggests that nematode excitotoxicity can be an important tool to sift through many proposed mechanisms of excitotoxicity, allowing us to identify conserved mechanisms that might be at the core of neurodegenerative processes common across divergent excitotoxic scenarios. Furthermore, our studies now illuminate DAPK/DAPK-1 and Pin1/PINN-1 as two such factors, important for excitotoxicity mechanisms across a large evolutionary distance. We can now further use this system to decipher DAPK-1 and PINN-1 –related mechanisms in nematode excitotoxicity, with the hope that such understanding might help us to continue uncovering conserved core mechanisms in this important form of neurodegeneration.
Methods
Strains
The following C. elegans strains were obtained from the C. elegans Genetic Center (CGC), from the Japanese National Bioresource Project (NBSP), or from the original producers: WT: Bristol N2 (RRID:CGC_N2 (ancestral)); Nematode excitotoxicity model: ZB1102: glt-3(bz34) IV; nuIs5 V; (RRID:CGC_ZB1102) dapk-1: VC432: dapk-1(gk219) I; (RRID:CGC_VC432) dapk-1 over-expression: CZ9277:frIs7[Pnlp-29::GFP; Pcol-12:DsRed] (IV); juEx1933[Phsp16::DAPK-1; Pttx-3::RFP] (the frIs7 insertion was later eliminated during our cross); Aldicarb assay control strains: sv-hyper-releasing/aldicarb-oversensitive RB1367: cpx-1(ok1552) (RRID:CGC_RB1367); sv-under-releasing/aldicarb-resistant NM791: rab-3(js49) (RRID:CGC_NM791): Autophagy modulatorunc-51: CB369: unc-51(e369) V (RRID:CGC_CB369); Autophagy label, DsRed::LGG-1: (originally created by the Tavernarakis lab [62], strain naming here as per BMC policy) IMN21: N2; Ex [Plgg-1:: DsRed:: LGG-1; Pmyo-2:: GFP]; Glu-regulated mobility defective control: VM1268: nmr-1(ak4) II; glr-2(ak10) glr-1(ky176) III;DANGER-related protein MAB-21: EM128: mab-21(bx53) III (Q203* , CAG -- > UAG) (RRID:CGC_EM128).CaMKK-like KO: ckk-1(ok1033) III (RRID:CGC_VC691);Pin1-like KO:pinn-1(tm2235) II (this 364 bp deletion runs the ORF into a stop codon after 8 codons). Most crosses were followed by PCR analysis to detect deletions, and by monitoring nuIs5’s GFP expression in glr-1-expressing interneurons using a high power fluorescence dissecting scope. dapk-1 over-expressing construct juEx1933 was followed by its coinjection marker, which produces RFP expression on AIY neurons. The unc-51 mutation was followed by uncoordinated phenotype and sequencing. DsRed::LGG-1 was followed by DsRed expression in cells throughout the body and by GFP expression in the pharynx (from the myo-2 promoter). We constructed the following strains: dapk-1 in excitotoxicity (by crossing VC432 and ZB1102) IMN26: dapk-1(gk219) I; glt-3(bz34) IV; nuIs5 V;DAPK-1 overexpression in excitotoxicity (by crossing ZB1102 and CZ9277) IMN25: glt-3(bz34) IV; nuIs5 V; juEx1933 [Phsp16::DAPK-1; Pttx-3::RFP]; Autophagy suppression in excitotoxicity (by crossing ZB1102 and CB369) IMN22: glt-3(bz34) IV; nuIs5 , unc-51(e369) V;Autophagy reporter in excitotoxicity IMN24: glt-3(bz34) IV; nuIs5 V; Ex[Plgg-1:: DsRed:: LGG-1; Pmyo-2:: GFP];CaMKK deletion in excitotoxicity:ckk-1 (ok1033) III; glt-3(bz34) IV; nuIs5 V.DANGER-like deletion in excitotoxicity: (by crossing EM128 and ZB1102): mab-21(bx53) III; glt-3(bz34) IV; nuIs5 V; Pin1-like deletion in excitotoxicity: IMN30: pinn-1(tm2235) II; glt-3(bz34) IV ; nuIs5 V (± dapk-1(gk219) I).
Neurodegeneration analysis
The level of necrotic neurodegeneration in head neurons was monitored using an inverted scope and Nomarski Differential Interference Contrast (DIC). Animals with no anesthetics were examined on a fresh chunk of agar of nematode culture flipped upside down over a slide. Swollen “vacuolated-looking” cells located in the area of the nerve ring were counted as head neurons undergoing necrosis, as described previously [37,41]. Briefly, we use freshly growing mixed-stage animals, and we record the developmental stage and the number of “vacuolated” dying neurons of each animal that we see while scanning through the agar chunk, thus creating a “snap-shot” of the transitory number of dying neurons exhibited by the population of animals at the time of analysis. As mentioned above, in the excitotoxicity strain the number of “vacuolated” cells observed at each developmental stage typically goes up until L3 (with the maturation of Glu signaling in the worm), and then declines with the engulfment of cell copses. This analysis is repeated several times, with several isolates of each strain. For confirmation of suspected effects in new mutant combinations, a representative part of the analysis is done blindly by independent observers. Similar numbers of control and test animals are recorded in each session. Records from all these sessions is pooled together to calculate average neurodegeneration from large number of animals in each strain and each stage (50–200 in each data bar). We occasionally confirmed these dying neurons as neurons postsynaptic to Glu connections by verifying their labeling with the GFP co-expressed in nuIs5 animals under the glr-1 promoter. For heat-shock-induced overexpression of DAPK-1, animals carrying [Phsp16::DAPK-1; Pttx-3::RFP] were placed in a 35°C incubator for 2 hours to induce activation of heat shock promoter, left to rest for about 30 minutes and scored regularly for two days for the extent of swollen degenerating neurons (protocol & strains coordinated with the Chisholm group, UCSD [42]). Experimental animals (animals with extrachromosomal array) and control animals (animals lacking extrachromosomal array) were obtained, identified and scored on the same day from the same pool of heat-shocked animals. For epistasis analysis between given excitotoxicity-modifying mutations X and Y, we calculated the effect of each mutation for each developmental stage as: Fold effect of X = (the average number of dying head neuron in the excitotoxicity strain in the presence of mutation X)/(the average number of dying head neuron in the starting excitotoxicity strain), with a similar calculation for mutation Y. The expected cumulative fold effect of mutations X and Y when working independently in parallel pathways is (fold effect of X) * (fold effect of Y). The calculated expected number of dying neurons presented on the graphs in Figures 4 and 6 is (the average number of dying head neuron in the starting excitotoxicity strain) * (fold effect of X) * (fold effect of Y). Error bars represent SE. Statistical significance of difference between control groups and experimental groups was analyzed using z-test score (as the significance of these differences is calculated for large populations of ~100 animals in each group).
Fluorescence microscopy analysis of autophagy
Animals carrying Ex [Plgg-1:: DsRed:: LGG-1; Pmyo-2:: GFP]; glt-3(bz34) IV; nuIs5 V and Ex [Plgg-1:: DsRed:: LGG-1; Pmyo-2:: GFP]; nuIs5 V were analyzed for the extent to which glr-1 expressing command interneuron (labeled with GFP) were undergoing autophagy (i.e., show DsRed punca or increased DsRed intensity). Animals were mounted on a 2% agar pad. To compare average DsRed intensity between worms we identified the cell with the highest DsRed intensity as 100% and then compared the other neurons to this cell (following the procedure used by the Tavernarakis lab, who developed this DsRed::LGG-1 marker [62]). A few cells were analyzed in each animal and the average intensity from a total from 20–25 animals was calculated. We also counted the average of the number of green-labeled neurons showing DsRed puncta as another indication of neurons undergoing autophagy and compared control (nuIs5 only) versus experimental (glt-3; nuIs5) groups.
Blockade of autophagy with 3MA
For 3-methyladenine (3MA) treatment, mixed stage animals from the excitotoxicity strain (glt-3; nuIs5) or the DAPK-inhibited excitotoxicity strain (dapk-1; glt-3; nuIs5) were incubated overnight with 10 mM 3-MA (Sigma, M9281) dissolved in 1% DMSO (experimental group) or 1% DMSO alone (control). Plates were supplemented with E. coli OP50. The extent of neurodegeneration was recorded 20–24 h after treatment.
Behavioral assays
Behavioral assays on nematode locomotion and worm paralysis were performed blindly following standard methods of behavioral analysis. For duration of Glu-regulated spontaneous forward mobility we followed the protocol of the Maricq group (U Utah) [79]. We did not measure mobility in strains that show excitotoxic neurodegeneration because the degeneration or rescue of command interneurons can change the results of this assay. For aldicarb assays we followed the Nonet lab protocol (Washington U) [48]. Briefly, we soaked worm plates with aldicarb to a final concentration of 0.5 mM and added food. We placed ~30 freshly growing young adult animal onto these plates. The percentages of paralyzed worms were recorded every 15 minutes for a period of 2 hours.
Acknowledgments
We thank Dr. Monica Driscoll, Dr. Christine Li, and all members of the Mano & Li labs for their advice and support. We thank Drs. Andrew Chisholm, Andres Villu Maricq, Jeremy Dittman, King-Lau Chow, Nektarios Tavernarakis, and Alicia Melendez for reagents & advice on associated assays, Dr. S. Mitani (Tokyo Women’s Medical University School of Medicine), the C. elegans Gene Knockout Consortium, and the C. elegans Genetic Center for providing deletion alleles.
This project was supported by the Sinsheimer Foundation (P60134007) and the American Heart Association, National Center (0635367 N) to I.M. The Mano lab is also supported by the Research Centers in Minority Institutions (RCMI) grants 5G12RR003060-26 & 8G12MD7603-28 and by the National Science Foundation (IOS 1022281). J.D.R. was supported by NIH’s RISE program at CCNY. Strain collections facilities are funded by: CGC - NIH Office of Research Infrastructure Programs (P40 OD010440); NBRP - Ministry of Education, Culture, Science, Sports and Technology, Japan.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contribution
JDR, KGF & IM designed the project and the experiments, analyzed the data, and wrote the paper. JDR, KGF, TA, UA, BK, JA, TM, MS, SM, and DA run the experiments. All authors read and approved the final manuscript.
Contributor Information
John S Del Rosario, Email: delrosariojohns@gmail.com.
Katherine Genevieve Feldmann, Email: kfeldmann@gc.cuny.edu.
Towfiq Ahmed, Email: niloyny945@yahoo.com.
Uzair Amjad, Email: uzairamjad210@yahoo.com.
BakKeung Ko, Email: bko0901@gmail.com.
JunHyung An, Email: joonimay@gmail.com.
Tauhid Mahmud, Email: tmahmud03@gmail.com.
Maha Salama, Email: msalama035@yahoo.com.
Shirley Mei, Email: shirlmei@yahoo.com.
Daniel Asemota, Email: dasem111@yahoo.com.
Itzhak Mano, Email: imano@ccny.cuny.edu.
References
- 1.Rothman SM, Olney JW. Glutamate and the pathophysiology of hypoxic–ischemic brain damage. Ann Neurol. 1986;19(2):105–11. doi: 10.1002/ana.410190202. [DOI] [PubMed] [Google Scholar]
- 2.Choi DW. Excitotoxic Cell Death. J Neurobiol. 1992;23(9):1261–76. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- 3.Moskowitz MA, Lo EH, Iadecola C. The Science of Stroke: Mechanisms in Search of Treatments. Neuron. 2010;67(2):181–98. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tymianski M. Emerging mechanisms of disrupted cellular signaling in brain ischemia. Nat Neurosci. 2011;14(11):1369–73. doi: 10.1038/nn.2951. [DOI] [PubMed] [Google Scholar]
- 5.Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65(1):1–105. doi: 10.1016/S0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 6.Rossi DJ, Oshima T, Attwell D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature. 2000;403(6767):316–21. doi: 10.1038/35002090. [DOI] [PubMed] [Google Scholar]
- 7.Tzingounis AV, Wadiche JI. Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat Rev Neurosci. 2007;8(12):935–47. doi: 10.1038/nrn2274. [DOI] [PubMed] [Google Scholar]
- 8.Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62(3):405–96. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ikonomidou C, Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002;1(6):383–6. doi: 10.1016/S1474-4422(02)00164-3. [DOI] [PubMed] [Google Scholar]
- 10.Lai TW, Zhang S, Wang YT. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog Neurobiol. 2014;115:157–88. doi: 10.1016/j.pneurobio.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 11.Bialik S, Kimchi A. The death-associated protein kinases: structure, function, and beyond. Annu Rev Biochem. 2006;75:189–210. doi: 10.1146/annurev.biochem.75.103004.142615. [DOI] [PubMed] [Google Scholar]
- 12.Shiloh R, Bialik S, Kimchi A. The DAPK family: a structure-function analysis. Apoptosis. 2014;19(2):286–97. doi: 10.1007/s10495-013-0924-5. [DOI] [PubMed] [Google Scholar]
- 13.Nair S, Hagberg H, Krishnamurthy R, Thornton C, Mallard C. Death associated protein kinases: molecular structure and brain injury. Int J Mol Sci. 2013;14(7):13858–72. doi: 10.3390/ijms140713858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fujita Y, Yamashita T. Role of DAPK in neuronal cell death. Apoptosis. 2014;19(2):339–45. doi: 10.1007/s10495-013-0917-4. [DOI] [PubMed] [Google Scholar]
- 15.Deiss LP, Feinstein E, Berissi H, Cohen O, Kimchi A. Identification of a novel serine/threonine kinase and a novel 15-kD protein as potential mediators of the gamma interferon-induced cell death. Genes Dev. 1995;9(1):15–30. doi: 10.1101/gad.9.1.15. [DOI] [PubMed] [Google Scholar]
- 16.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8(9):741–52. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 17.Bialik S, Kimchi A. Lethal weapons: DAP-kinase, autophagy and cell death DAP-kinase regulates autophagy. Curr Opin Cell Biol. 2009;22(2):199–205. doi: 10.1016/j.ceb.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 18.Bialik S, Kimchi A. The DAP-kinase interactome. Apoptosis. 2014;19(2):316–28. doi: 10.1007/s10495-013-0926-3. [DOI] [PubMed] [Google Scholar]
- 19.Shamloo M, Soriano L, Wieloch T, Nikolich K, Urfer R, Oksenberg D. Death-associated protein kinase is activated by dephosphorylation in response to cerebral ischemia. J Biol Chem. 2005;280(51):42290–9. doi: 10.1074/jbc.M505804200. [DOI] [PubMed] [Google Scholar]
- 20.Velentza AV, Wainwright MS, Zasadzki M, Mirzoeva S, Schumacher AM, Haiech J, et al. An aminopyridazine-based inhibitor of a pro-apoptotic protein kinase attenuates hypoxia-ischemia induced acute brain injury. Bioorg Med Chem Lett. 2003;13(20):3465–70. doi: 10.1016/S0960-894X(03)00733-9. [DOI] [PubMed] [Google Scholar]
- 21.Rami A. Autophagy in neurodegeneration: firefighter and/or incendiarist? Neuropathol App Neurobiol. 2009;35(5):449–61. doi: 10.1111/j.1365-2990.2009.01034.x. [DOI] [PubMed] [Google Scholar]
- 22.van Rossum DB, Patterson RL, Cheung KH, Barrow RK, Syrovatkina V, Gessell GS, et al. DANGER, a novel regulatory protein of inositol 1,4,5-trisphosphate-receptor activity. J Biol Chem. 2006;281(48):37111–6. doi: 10.1074/jbc.M608760200. [DOI] [PubMed] [Google Scholar]
- 23.Kang BN, Ahmad AS, Saleem S, Patterson RL, Hester L, Dore S, et al. Death-associated protein kinase-mediated cell death modulated by interaction with DANGER. J Neurosci. 2010;30(1):93–8. doi: 10.1523/JNEUROSCI.3974-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tu W, Xu X, Peng L, Zhong X, Zhang W, Soundarapandian MM, et al. DAPK1 Interaction with NMDA Receptor NR2B Subunits Mediates Brain Damage in Stroke. Cell. 2010;140(2):222–34. doi: 10.1016/j.cell.2009.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11(10):682–96. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wroge CM, Hogins J, Eisenman L, Mennerick S. Synaptic NMDA receptors mediate hypoxic excitotoxic death. J Neurosci. 2012;32(19):6732–42. doi: 10.1523/JNEUROSCI.6371-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou X, Hollern D, Liao J, Andrechek E, Wang H. NMDA receptor-mediated excitotoxicity depends on the coactivation of synaptic and extrasynaptic receptors. Cell Death Dis. 2013;4:e560. doi: 10.1038/cddis.2013.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14(6):383–400. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- 29.Schori H, Yoles E, Wheeler LA, Raveh T, Kimchi A, Schwartz M. Immune-related mechanisms participating in resistance and susceptibility to glutamate toxicity. Eur J Neurosci. 2002;16(4):557–64. doi: 10.1046/j.1460-9568.2002.02134.x. [DOI] [PubMed] [Google Scholar]
- 30.Cull-Candy S, Kelly L, Farrant M. Regulation of Ca2+ −permeable AMPA receptors: synaptic plasticity and beyond. Curr Opin Neurobiol. 2006;16(3):288–97. doi: 10.1016/j.conb.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 31.Kwak S, Weiss JH. Calcium-permeable AMPA channels in neurodegenerative disease and ischemia. Curr Opin Neurobiol. 2006;16(3):281–7. doi: 10.1016/j.conb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 32.Liu SJ, Zukin RS. Ca2+ −permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007;30(3):126–34. doi: 10.1016/j.tins.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 33.Lettre G, Hengartner MO. Developmental apoptosis in C. elegans: a complex CEDnario. Nat Rev Mol Cell Biol. 2006;7(2):97–108. doi: 10.1038/nrm1836. [DOI] [PubMed] [Google Scholar]
- 34.Conradt B. Genetic control of programmed cell death during animal development. Annu Rev Genet. 2009;43:493–523. doi: 10.1146/annurev.genet.42.110807.091533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samara C, Tavernarakis N. Autophagy and cell death in Caenorhabditis elegans. Curr Pharm Des. 2008;14(2):97–115. doi: 10.2174/138161208783378770. [DOI] [PubMed] [Google Scholar]
- 36.Melendez A, Levine B. WormBook.org. 2009. Autophagy in C. elegans; pp. 1–26. [DOI] [PubMed] [Google Scholar]
- 37.Mano I, Driscoll M. C. elegans Glutamate Transporter Deletion Induces AMPA-Receptor/Adenylyl Cyclase 9-Dependent Excitotoxicity. J Neurochem. 2009;108(6):1373–84. doi: 10.1111/j.1471-4159.2008.05804.x. [DOI] [PubMed] [Google Scholar]
- 38.Mano I, Straud S, Driscoll M. Caenorhabditis elegans Glutamate Transporters Influence Synaptic Function and Behavior at Sites Distant from the Synapse. J Biol Chem. 2007;282(47):34412–9. doi: 10.1074/jbc.M704134200. [DOI] [PubMed] [Google Scholar]
- 39.Berger AJ, Hart AC, Kaplan JM. Galphas-induced neurodegeneration in Caenorhabditis elegans. J Neurosci. 1998;18(8):2871–80. doi: 10.1523/JNEUROSCI.18-08-02871.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mojsilovic-Petrovic J, Nedelsky N, Boccitto M, Mano I, Georgiades SN, Zhou W, et al. FOXO3a is broadly neuroprotective in vitro and in vivo against insults implicated in motor neuron diseases. J Neurosci. 2009;29(25):8236–47. doi: 10.1523/JNEUROSCI.1805-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tehrani N, Del Rosario J, Dominguez M, Kalb R, Mano I. The Insulin/IGF Signaling Regulators Cytohesin/GRP-1 and PIP5K/PPK-1 Modulate Susceptibility to Excitotoxicity in C. elegans. PLoS One. 2014;9(11):e113060. doi: 10.1371/journal.pone.0113060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tong A, Lynn G, Ngo V, Wong D, Moseley SL, Ewbank JJ, et al. Negative regulation of Caenorhabditis elegans epidermal damage responses by death-associated protein kinase. Proc Natl Acad Sci U S A. 2009;106(5):1457–61. doi: 10.1073/pnas.0809339106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang C, Avery L. Death-associated protein kinase (DAPK) and signal transduction: fine-tuning of autophagy in Caenorhabditis elegans homeostasis. FEBS J. 2010;277(1):66–73. doi: 10.1111/j.1742-4658.2009.07413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chuang M, Chisholm A. Insights into the functions of the death associated protein kinases from C. elegans and other invertebrates. Apoptosis. 2014;19(2):392–7. doi: 10.1007/s10495-013-0943-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kang C, You YJ, Avery L. Dual roles of autophagy in the survival of Caenorhabditis elegans during starvation. Genes Dev. 2007;21(17):2161–71. doi: 10.1101/gad.1573107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tian JH, Das S, Sheng ZH. Ca2+ −dependent phosphorylation of syntaxin-1A by the death-associated protein (DAP) kinase regulates its interaction with Munc18. J Biol Chem. 2003;278(28):26265–74. doi: 10.1074/jbc.M300492200. [DOI] [PubMed] [Google Scholar]
- 47.Südhof Thomas C. Neurotransmitter Release: The Last Millisecond in the Life of a Synaptic Vesicle. Neuron. 2013;80(3):675–90. doi: 10.1016/j.neuron.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mahoney TR, Luo S, Nonet ML. Analysis of synaptic transmission in Caenorhabditis elegans using an aldicarb-sensitivity assay. Nat Protocols. 2006;1(4):1772–7. doi: 10.1038/nprot.2006.281. [DOI] [PubMed] [Google Scholar]
- 49.Martin JA, Hu Z, Fenz KM, Fernandez J, Dittman JS. Complexin Has Opposite Effects on Two Modes of Synaptic Vesicle Fusion. Curr Biol. 2011;21(2):97–105. doi: 10.1016/j.cub.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zheng Y, Brockie PJ, Mellem JE, Madsen DM, Maricq AV. Neuronal control of locomotion in C. elegans is modified by a dominant mutation in the GLR-1 ionotropic glutamate receptor. Neuron. 1999;24(2):347–61. doi: 10.1016/S0896-6273(00)80849-1. [DOI] [PubMed] [Google Scholar]
- 51.Burbea M, Dreier L, Dittman JS, Grunwald ME, Kaplan JM. Ubiquitin and AP180 Regulate the Abundance of GLR-1 Glutamate Receptors at Postsynaptic Elements in C. elegans. Neuron. 2002;35(1):107–20. doi: 10.1016/S0896-6273(02)00749-3. [DOI] [PubMed] [Google Scholar]
- 52.Chang HC, Rongo C. Cytosolic tail sequences and subunit interactions are critical for synaptic localization of glutamate receptors. J Cell Sci. 2005;118(Pt 9):1945–56. doi: 10.1242/jcs.02320. [DOI] [PubMed] [Google Scholar]
- 53.Chow KL, Hall DH, Emmons SW. The mab-21 gene of Caenorhabditis elegans encodes a novel protein required for choice of alternate cell fates. Development. 1995;121(11):3615–26. doi: 10.1242/dev.121.11.3615. [DOI] [PubMed] [Google Scholar]
- 54.Kimura Y, Corcoran EE, Eto K, Gengyo-Ando K, Muramatsu MA, Kobayashi R, et al. A CaMK cascade activates CRE-mediated transcription in neurons of Caenorhabditis elegans. EMBO Rep. 2002;3(10):962–6. doi: 10.1093/embo-reports/kvf191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, et al. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009;10(3):285–92. doi: 10.1038/embor.2008.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90(4):1383–435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 58.Toth ML, Simon P, Kovacs AL, Vellai T. Influence of autophagy genes on ion-channel-dependent neuronal degeneration in Caenorhabditis elegans. J Cell Sci. 2007;120(6):1134–41. doi: 10.1242/jcs.03401. [DOI] [PubMed] [Google Scholar]
- 59.Vellai T, Toth ML, Kovacs AL. Janus-faced autophagy: a dual role of cellular self-eating in neurodegeneration? Autophagy. 2007;3(5):461–3. doi: 10.4161/auto.4282. [DOI] [PubMed] [Google Scholar]
- 60.Samara C, Syntichaki P, Tavernarakis N. Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ. 2008;15(1):105–12. doi: 10.1038/sj.cdd.4402231. [DOI] [PubMed] [Google Scholar]
- 61.Driscoll M, Gerstbrein B. Dying for a cause: invertebrate genetics takes on human neurodegeneration. Nat Rev Genet. 2003;4(3):181–94. doi: 10.1038/nrg1018. [DOI] [PubMed] [Google Scholar]
- 62.Maiuri MC, Le Toumelin G, Criollo A, Rain J-C, Gautier F, Juin P, et al. Functional and physical interaction between Bcl-XL and a BH3-like domain in Beclin-1. EMBO J. 2007;26(10):2527–39. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6(4):304–12. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- 64.Sudhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–47. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- 65.Edwards RH. The Neurotransmitter Cycle and Quantal Size. Neuron. 2007;55(6):835–58. doi: 10.1016/j.neuron.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 66.Saroussi S, Nelson N. Vacuolar H+ −ATPase—an enzyme for all seasons. Pflugers Arch - Eur J Physiol. 2009;457(3):581–7. doi: 10.1007/s00424-008-0458-9. [DOI] [PubMed] [Google Scholar]
- 67.Wulf G, Finn G, Suizu F, Lu KP. Phosphorylation-specific prolyl isomerization: is there an underlying theme? Nat Cell Biol. 2005;7(5):435–41. doi: 10.1038/ncb0505-435. [DOI] [PubMed] [Google Scholar]
- 68.Lu KP, Finn G, Lee TH, Nicholson LK. Prolyl cis-trans isomerization as a molecular timer. Nat Chem Biol. 2007;3(10):619–29. doi: 10.1038/nchembio.2007.35. [DOI] [PubMed] [Google Scholar]
- 69.Lu KP, Zhou XZ. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol. 2007;8(11):904–16. doi: 10.1038/nrm2261. [DOI] [PubMed] [Google Scholar]
- 70.Keune WJ, Jones DR, Divecha N. PtdIns5P and Pin1 in oxidative stress signaling. Ad Biol Regulation. 2013;53(2):179–89. doi: 10.1016/j.jbior.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 71.Lu Z, Hunter T. Prolyl isomerase Pin1 in cancer. Cell Res. 2014;24(9):1033–49. doi: 10.1038/cr.2014.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Westmark PR, Westmark CJ, Wang S, Levenson J, O’Riordan KJ, Burger C, et al. Pin1 and PKMzeta sequentially control dendritic protein synthesis. Sci Signal. 2010;3(112):ra18. doi: 10.1126/scisignal.2000451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sacktor TC. PINing for things past. Sci Signal. 2010;3(112):e9. doi: 10.1126/scisignal.3112pe9. [DOI] [PubMed] [Google Scholar]
- 74.Liou YC, Sun A, Ryo A, Zhou XZ, Yu ZX, Huang HK, et al. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature. 2003;424(6948):556–61. doi: 10.1038/nature01832. [DOI] [PubMed] [Google Scholar]
- 75.Pastorino L, Sun A, Lu P-J, Zhou XZ, Balastik M, Finn G, et al. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-[beta] production. Nature. 2006;440(7083):528–34. doi: 10.1038/nature04543. [DOI] [PubMed] [Google Scholar]
- 76.Lee Tae H, Chen C-H, Suizu F, Huang P, Schiene-Fischer C, Daum S, et al. Death-Associated Protein Kinase 1 Phosphorylates Pin1 and Inhibits Its Prolyl Isomerase Activity and Cellular Function. Mol Cell. 2011;42(2):147–59. doi: 10.1016/j.molcel.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fasseas MK, Dimou M, Katinakis P. The Caenorhabditis elegans parvulin gene subfamily and their expression under cold or heat stress along with the fkb subfamily. Biochem Biophys Res Commun. 2012;423(3):520–5. doi: 10.1016/j.bbrc.2012.05.157. [DOI] [PubMed] [Google Scholar]
- 78.Keune WJ, Jones DR, Bultsma Y, Sommer L, Zhou XZ, Lu KP, et al. Regulation of phosphatidylinositol-5-phosphate signaling by Pin1 determines sensitivity to oxidative stress. Sci Signal. 2012;5(252):ra86. doi: 10.1126/scisignal.2003223. [DOI] [PubMed] [Google Scholar]
- 79.Brockie PJ, Mellem JE, Hills T, Madsen DM, Maricq AV. The C. elegans glutamate receptor subunit NMR-1 is required for slow NMDA-activated currents that regulate reversal frequency during locomotion. Neuron. 2001;31(4):617–30. doi: 10.1016/S0896-6273(01)00394-4. [DOI] [PubMed] [Google Scholar]