

Abstract
The Rab GTPase activating protein (RabGAP), AS160/TBC1D4, is an important substrate of protein kinase B (PKB), and regulates insulin-stimulated trafficking of glucose transporter 4. Besides, AS160/TBC1D4 has also been shown to regulate trafficking of many other membrane proteins including FA translocase/CD36 in cardiomyocytes. However, it is not clear whether it plays any role in regulating heart functions in vivo. Here, we found that PKB-mediated phosphorylation of Thr649 on AS160/TBC1D4 represented one of the major PAS-binding signals in the heart in response to insulin. Mutation of Thr649 to a non-phosphorylatable alanine increased the R-wave amplitude in the AS160Thr649Ala knockin mice. However, this knockin mutation did not affect the heart functions under both normal and infarct conditions. Interestingly, myocardial infarction induced the expression of a related RabGAP, TBC1D1, in the infarct zone as well as in the border zone. Together, these data show that the Thr649 phosphorylation of AS160/TBC1D4 plays an important role in the heart’s electrical conduction system through regulating the R-wave amplitude.
Introduction
Insulin signaling in the heart plays a critical role in regulating cardiac development, postnatal growth, metabolisms and cardiac contractility [1]. Insulin resistance under pathological conditions such as type II diabetes is a risk factor for development of heart failure after myocardial infarction [2]. In the past few decades, genetically-modified mouse models have helped to elucidate the roles of myocardial insulin signaling under physiological conditions, and the contributions of its impairment in the adaptation to myocardial infarction [3–6]. Cardiomyocyte-specific knockout of insulin receptor (IR) in mouse reduces cardiomyocyte size and moderately impairs cardiac performance [3]. The mild decrease in cardiac performance in this mouse model is due to the compensation of insulin-like growth factor 1 receptor (IGF1R) in cardiomyocytes. When both IR and IGF1R are deleted in cardiomyocytes, the double knockout mouse develops dilated cardiomyopathy and eventually dies of heart failure [7]. Phosphoinositide-dependent protein kinase 1 (PDK1), an important protein kinase downstream of both receptor kinases, mediates insulin- or IGF1-stimulated activation of protein kinase B (PKB, also known as Akt). Likewise, deletion of PDK1 in cardiac muscle results in a decrease in cardiomyocyte size, and the mouse develops cardiomyopathy and dies of heart failure [6]. While cardiac-specific knockout PKBβ/Akt2 impairs insulin-stimulated glucose uptake into cardiomyocytes [5], deletion of PKBα/Akt1 results in heart developmental defects and decreased heart function, which can be partially rescued by downregulation of p38α [8, 9]. These studies highlight the importance of insulin-PKB/Akt pathway in regulating heart functions, and its deregulation directly links to the pathogenesis of cardiac diseases. However, factors downstream of PKB/Akt mediating the pathogenesis of these heart diseases are still not very clear.
AS160/TBC1D4 is a Rab GTPase activating protein (RabGAP) that was initially identified as a phosphorylated PKB substrate in adipocytes [10]. A related RabGAP, TBC1D1, can also be phosphorylated by PKB [11, 12]. Both AS160/TBC1D4 and TBC1D1 regulate translocation of glucose transporter 4 (GLUT4) storage vesicles [10, 12]. In addition, AS160/TBC1D4 has been implicated in regulating intracellular trafficking of FA translocase/CD36 [13], aquaporin-2 [14], epithelial sodium channel [15], and Na+/K+-ATPase [16]. Thr649 on AS160/TBC1D4 (numbering according to the mouse protein) is a critical PKB phosphorylation site that binds to the 14-3-3 regulatory proteins [17, 18]. A knockin mouse in which Thr649 on AS160/TBC1D4 is mutated to a non-phosphorylatable alanine develops insulin resistance in skeletal muscle and exhibits an increased ratio of heart to body weight [19].
In this study we investigated the impacts of the AS160-Thr649 phosphorylation on the heart’s electrical conduction system and heart functions under normal and myocardial infarction conditions using the AS160Thr649Ala knockin mouse model.
Materials and Methods
Materials
Recombinant human insulin was from Novo Nordisk (Bagsvaerd, Denmark), microcystin-LR was from Taiwan Algal Science Inc (Taoyuan, Taiwan, China), and Protein G-Sepharose was from GE Healthcare (Little Chalfont, Buckinghamshire, UK). All other chemicals were from Sigma-Aldrich (St. Louis, Missouri, USA) or Sangon Biotech (Shanghai, China).
Antibodies
The sheep antibody against phosphorylated Ser231 on TBC1D1 was previously reported [11]. The flotillin-1 antibody (Cat No. sc-25506) was from Santa Cruz (Dallas, Texas, USA). The antibodies that recognise phosphorylated Ser473 on PKB (Cat No. 9271), total PKB (Cat No. 9272), and total TBC1D1 (Cat No. 4629), and the phospho-Akt substrate (PAS) antibody (Cat No. 9611) were from Cell Signaling Technology. The total AS160/TBC1D4 (Cat No. 07–741) was from Millipore. The antibody that recognises phosphorylated Thr649 on AS160/TBC1D4 (Cat No. 441071G) was from Life Technologies. The GAPDH antibody (Cat No. G8795) was from Sigma, and the cTnT antibody (Cat No. MA5-12960) was from Thermo Scientific.
Mouse husbandry and insulin injection
The Ethics Committee at Model Animal Research Center of Nanjing University approved the mouse procedures used in this study. Mice were maintained under a light/dark cycle of 12 h, and had free access to food and water unless stated. The AS160Thr649Ala knockin mice are as previously described [19, 20]. In total, 90 mice were used in this study.
Intraperitoneal injection of insulin (150 mU insulin per g of body weight) was carried out in anaesthetized mice that had been restricted from food access overnight (16 hours). After insulin injection, mice were kept on a heating pad for 20 min before cervically dislocated. Hearts were immediately removed and snap-frozen in liquid nitrogen.
Heart protein extraction and western blot
Mouse hearts were homogenized in lysis buffer using a Polytron homogenizer (Kinematica, Luzern, Switzerland), and lysed on ice for 30 min as previously described [19]. Protein extracts were obtained after tissue debris was removed via centrifugation. Proteins were electrophoretically separated via SDS-PAGE, and immunoblotted onto nitrocellulose membranes that were subsequently incubated with primary antibodies. Membranes were further incubated with horseradish-peroxidase-conjugated secondary antibodies. Chemiluminescence signals were detected using a gel documentation system (Syngene, UK) after ECL substrates were added (GE Healthcare, UK).
Electrocardiography (ECG)
Mice were anaesthetized with 2% isoflurane mixed with 100% O2 (0.5 L/min), and anesthetized animals were maintained by continuous gaseous isoflurance (1% to 1.5%) mixed with 100% O2 (0.5 L/min). The electrocardiograms were collected with PowerLab 8/30 (AD, ML870) and dual bio amp/stimulator (AD, ML408) system as previously described [21]. The parameters were measured by using the ECG analysis module of labchart software.
Echocardiography (Echo)
Mice were anaesthetized with gaseous isoflurane as afore-mentioned. Echocardiography was performed on anaesthetized mice using a Vevo 770 high-resolution in vivo micro-imaging system (VisualSonics, inc) with a 30MHz RMV-707B ultrasonic probe as previously described [21]. Briefly, mice were shaved on their chests, and then placed on a heating pad in a supine position with their chests covered with ultrasound transmission gel. The ultrasonic probe was immobilized with a 90° angle between the probe and the heart to collect M-mode pictures. The following parameters were measured on the M-mode tracing and averaged from 6 cardiac cycles: left ventricle anterior wall (LVAW), left ventricle posterior wall (LVPW), left ventricle internal dimension (LVID) of systole and diastole. The calculation of ejection fraction (EF) formula is EF% = [(LV Vol;d—LV Vol;s)] x 100%, and fraction shortening (FS) is calculated as FS% = [(LVID;d—LVID;s)/LVID;d] x 100%.
Induction of myocardial infarction
Mice were anaesthetized via intraperitoneal injection of a mixture of ketamine (100 mg per kg of body weight) and xylazine (10 mg per kg of body weight). During thoracotomy, mice were incubated endotracheally and ventilated using a Harvard Mouse Mini-Vent (type 845, Havard Apparatus, Germany) (stroke volume: 0.3 mL; rate: 120 times/min). Thoracotomy on the left side of the chest was performed via the fourth intercostal space to provide access to the beating heart. The pericardium was opened, and the left anterior descending coronary artery (LAD) was ligated with silk suture near its origin between the pulmonary outflow tract and the edge of the left atrium. After artery ligation, the chest was closed, and mice were kept on the heating pad until they became conscious again. The control mice with sham operation underwent the same procedure of thoracotomy and pericardium opening but with no ligation of LAD. After surgery, mice were monitored daily and administered with carprofen if they experienced distress. None of the mice died due to surgery.
Masson’s staining
Hearts were isolated immediately after termination of mice and fixed in 4% PFA overnight at 4°C. Hearts were embedded in paraffin and cut into 5-μm-thick slices for Masson’s staining. Briefly, after deparaffinization and rehydration, the sections were first stained with Biebrich scarlet for 50 sec. The sections were rinsed in distilled water and put in phosphotungstic acid and phosphomolybdic acid for 10 min. Afterwards, they were stained with Aniline blue for 4 min. After covered with coverslips and sealed, the sections were photographed using an Olympus BX53F microscope.
Statistical analysis
Data were analyzed via two-way ANOVA unless stated, and differences were considered statistically significant at p < 0.05.
Results
Expression and phosphorylation of AS160/TBC1D4 in mouse heart
We previously reported that AS160/TBC1D4 is ubiquitously expressed in most mouse tissues and its expression level is relatively high in the heart [22]. A more detailed analysis of expression of AS160/TBC1D4 was first carried out in mouse heart (Fig 1). The expression levels of AS160/TBC1D4 were more than 5-times higher in atria than in ventricles, and its highest expression levels were found in the left atrium (Fig 1A and 1B). This expression pattern of AS160/TBC1D4 was distinct from that of GLUT4 that showed the highest expression level in the left ventricle (Fig 1A and 1C).
Fig 1. The expression of AS160/TBC1D4 and GLUT4 in the four chambers of mouse heart.

A, Immunoblotting analyses of AS160/TBC1D4 and GLUT4 in the lysates (40 μg) of the four chambers of mouse heart. GAPDH was used as a loading control. B, Quantitation of AS160/TBC1D4 expression in the four chambers of mouse heart. n = 6. Asterisk indicates p < 0.05 (atrium versus ventricle). ♯ indicates p < 0.05 (right atrium versus left atrium). C, Quantitation of GLUT4 expression in the four chambers of mouse heart. n = 6. Asterisk indicates p < 0.05 (left ventricle versus the other three chambers).
AS160/TBC1D4 was initially identified as a PAS-binding protein in the adipose tissue [10], and the PAS binding site is the phospho-Thr649 [18]. We found that AS160/TBC1D4 also represented one of the major PAS-binding phospho-proteins upon insulin stimulation in the heart from the wild-type mice (Fig 2). These PAS-binding signals ~160 kDa were diminished in the heart from the AS160Thr649Ala knockin mice in parallel with undetectable phospho-Thr649 signals (Fig 2). The activation of PKB upon insulin stimulation was normal in the heart from these AS160Thr649Ala knockin mice (Fig 2).
Fig 2. Phosphorylation of AS160/TBC1D4 in mouse heart in response to insulin.
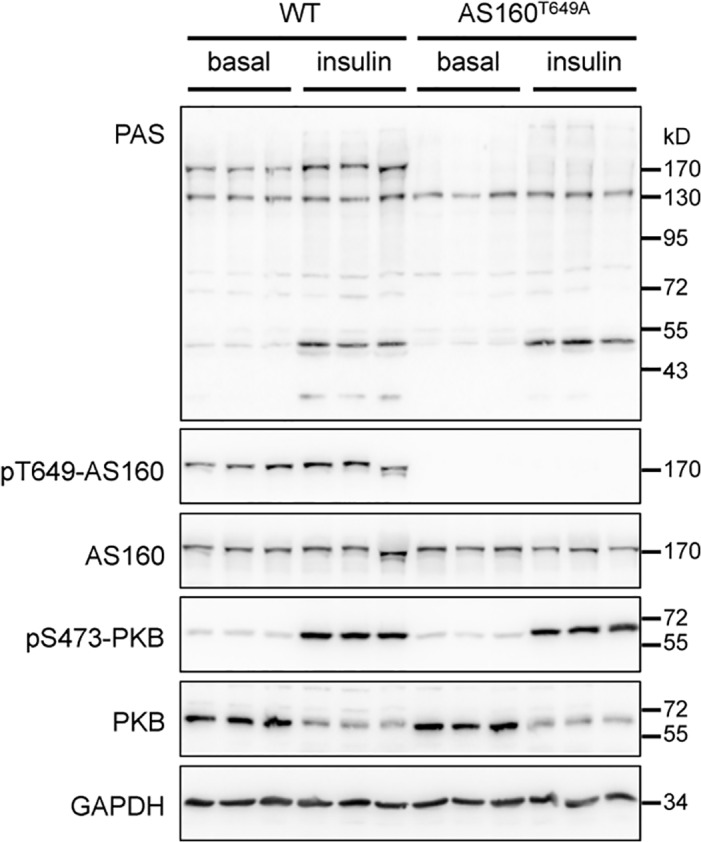
PAS-binding signals, phosphorylation of AS160-Thr649 and PKB-Ser473, and total AS160 and PKB were detected in the lysates (40 μg) of hearts from the wild-type and AS160Thr649Ala knockin mice that were intraperitoneally injected with PBS (basal) or insulin (150 mU per kg of body weight) for 20 min. GAPDH was used as a loading control.
The R-wave amplitude was increased in the hearts from the AS160Thr649Ala knockin mice
We previously reported that the ratio of heart to body weight was increased in the AS160Thr649Ala mice [19]. We next studied the heart’s electrical conduction system of the AS160Thr649Ala mice via electrocardiography (ECG). Interestingly, the R-wave amplitude in the ECG was moderately increased in the heart of the young male AS160Thr649Ala mice (2 month-old) and significantly increased in the heart of the old male AS160Thr649Ala mice (8 month-old) under ambient conditions (Table 1). The rest of ECG parameters appeared normal in the heart of both the young and old male AS160Thr649Ala mice except that the QT interval and QTc were significantly increased in the heart of the old male AS160Thr649Ala mice (Table 1). In the female AS160Thr649Ala mice (2.5 month-old), the amplitudes of the Q-, R- and S-waves were significantly altered while the other ECG parameters remained normal (Table 1). The heart rates were comparable between the wild-type and knockin mice in both genders under these conditions (Table 1).
Table 1. Electrocardiographic parameters of the wild-type and AS160Thr649Ala knockin hearts.
| ECG parameters | Genotype | Young male | Old male | Female | |||
|---|---|---|---|---|---|---|---|
| WT (n = 14) | AS160Thr649Ala (n = 9) | WT (n = 13) | AS160Thr649Ala (n = 6) | WT (n = 9) | AS160Thr649Ala (n = 11) | ||
| Heart rate | 461 ± 13 | 461 ± 20 | 384 ± 9 | 436 ± 26 | 393 ± 11 | 388 ± 10 | |
| RR Interval (ms) | 130.1 ± 4.3 | 132.8 ± 5.4 | 157.2 ± 3.8 | 140.3 ± 8.7 | 153.7 ± 4.4 | 155.7 ± 3.8 | |
| QRS Interval (ms) | 11.5 ± 0.3 | 11.5 ± 0.2 | 12.0 ± 0.1 | 12.3 ± 0.2 | 12.3 ± 0.3 | 12.5 ± 0.2 | |
| QT Interval (ms) | 23.2 ± 0.9 | 22.6 ± 0.7 | 19.9 ± 0.3 | 21.2 ± 0.5* | 24.1 ± 0.3 | 23.5 ± 0.7 | |
| QTc (ms) | 64.9 ± 3.3 | 62.3 ± 2.7 | 49.9 ± 1.1 | 57.1 ± 2.7* | 61.6 ± 1.1 | 59.7 ± 1.9 | |
| JT Interval (ms) | 11.7 ± 1.0 | 11.1 ± 0.8 | 8.3 ± 0.4 | 8.9 ± 0.5 | 11.8 ± 0.3 | 11.0 ± 0.7 | |
| Tpeak Tend Interval (ms) | 9.5 ± 1.0 | 8.9 ± 0.8 | 6.0 ± 0.3 | 6.6 ± 0.3 | 9.1 ± 0.3 | 8.6 ± 0.6 | |
| Q Amplitude (mV) | - 0.041 ± 0.005 | - 0.037 ± 0.005 | - 0.036 ± 0.004 | - 0.043 ± 0.005 | - 0.011 ± 0.004 | - 0.036 ± 0.006* | |
| R Amplitude (mV) | 0.656 ± 0.058 | 0.798 ± 0.076 | 0.661 ± 0.025 | 0.884 ± 0.050* | 0.549 ± 0.032 | 0.734 ± 0.063* | |
| S Amplitude (mV) | - 0.279 ± 0.030 | - 0.345 ± 0.036 | - 0.255 ± 0.040 | - 0.363 ± 0.050 | - 0.226 ± 0.049 | - 0.348 ± 0.042* | |
| ST Height (mV) | 0.094 ± 0.009 | 0.097 ± 0.013 | 0.060 ± 0.006 | 0.057 ± 0.006 | 0.137 ± 0.024 | 0.106 ± 0.009 | |
| T Amplitude (mV) | 0.207 ± 0.017 | 0.208 ± 0.026 | 0.135 ± 0.012 | 0.151 ± 0.032 | 0.230 ± 0.023 | 0.224 ± 0.015 | |
Electrocardiography was performed on the anaesthetized AS160Thr649Ala knockin mice and wild-type littermate controls. The male mice were at age of 2 months (young male) and 8 months (old male), respectively, while the female mice were at age of 2.5 months. Student’s t-test was performed and asterisk indicates p < 0.05.
Heart functions were normal in the AS160Thr649Ala knockin mice
We next studied the heart functions of the AS160Thr649Ala knockin mice via echocardiography (Echo). The heart functions including ejection fraction (EF) and fractional shortening (FS) were normal in the young male AS160Thr649Ala mice (Table 2) and remained unaffected in the old male AS160Thr649Ala mice (Table 2). The volume of left ventricles was significantly decreased in the heart from the young male AS160Thr649Ala mice but was normal in the heart from the old male AS160Thr649Ala mice (Table 2). No cardiac hypertrophy was observed in the heart of the male AS160Thr649Ala mice (Table 2). Similarly, the female AS160Thr649Ala mice also displayed normal heart functions (Table 2).
Table 2. Echocardiographic parameters of the wild-type and AS160Thr649Ala knockin hearts.
| Echo parameters | Genotype | Young male | Old male | Female | |||
|---|---|---|---|---|---|---|---|
| WT (n = 14) | AS160Thr649Ala (n = 9) | WT (n = 13) | AS160Thr649Ala (n = 6) | WT (n = 9) | AS160Thr649Ala (n = 11) | ||
| %EF | 53.78 ± 1.19 | 58.13 ± 2.97 | 45.24 ± 1.02 | 45.22 ± 2.46 | 53.52 ± 1.75 | 52.65 ± 1.75 | |
| %FS | 27.37 ± 0.74 | 30.36 ± 1.93 | 22.26 ± 0.59 | 22.27 ± 1.38 | 26.16 ± 1.23 | 26.47 ± 1.07 | |
| LV Vol; d | 65.14 ± 2.40 | 58.33 ± 1.81* | 75.85 ± 1.52 | 75.82 ± 6.65 | 49.84 ± 1.74 | 51.36 ± 2.59 | |
| LV Vol; s | 30.38 ± 1.74 | 24.58 ± 2.07* | 41.05 ± 1.31 | 42.34 ± 5.34 | 22.49 ± 1.72 | 26.04 ± 2.12 | |
| LVID; d | 3.85 ± 0.06 | 3.70 ± 0.05 | 4.16 ± 0.04 | 4.12 ± 0.16 | 3.47 ± 0.05 | 3.51 ± 0.07 | |
| LVID; s | 2.81 ± 0.07 | 2.58 ± 0.09 | 3.20 ± 0.04 | 3.21 ± 0.18 | 2.50 ± 0.08 | 2.69 ± 0.08 | |
| LVAW; d | 0.73 ± 0.01 | 0.74 ± 0.01 | 0.64 ± 0.00 | 0.64 ± 0.00 | 0.69 ± 0.01 | 0.74 ± 0.01* | |
| LVAW; s | 1.00 ± 0.01 | 1.01 ± 0.03 | 0.85 ± 0.00 | 0.86 ± 0.00* | 0.90 ± 0.02 | 0.91 ± 0.02 | |
| LVPW; d | 0.72 ± 0.02 | 0.72 ± 0.03 | 0.75 ± 0.02 | 0.74 ± 0.03 | 0.66 ± 0.01 | 0.72 ± 0.02 | |
| LVPW; s | 1.01 ± 0.02 | 1.01 ± 0.03 | 0.95 ± 0.02 | 0.96 ± 0.04 | 0.93 ± 0.02 | 0.93 ± 0.03 | |
| LV Mass (AW) | 97.79 ± 2.71 | 91.89 ± 2.80 | 103.15 ± 1.96 | 102.02 ± 7.48 | 78.34 ± 2.08 | 84.07 ± 2.99 | |
| LV Mass (AW) Corrected | 78.23 ± 2.17 | 73.51 ± 2.24 | 82.52 ± 1.56 | 81.62 ± 5.99 | 62.67 ± 1.67 | 67.26 ± 2.39 | |
Echocardiography was performed on the anaesthetized AS160Thr649Ala knockin mice and wild-type littermate controls. The male mice were at age of 2 months (young male) and 8 months (old male), respectively, while the female mice were at age of 2.5 months. Student’s t-test was performed and asterisk indicates p < 0.05.
Myocardial infarction after coronary artery ligation were comparable in the heart from the wild-type and AS160Thr649Ala knockin mice
We next induced myocardial infarction in mice via ligation of the left anterior descending coronary artery, and monitored the heart’s electrical conduction system and heart functions of the mice through ECG and Echo before and after surgery. Though the R amplitude in the ECG was modestly larger in the knockin heart than the wild-type heart before surgery (Table 1), the R amplitude was diminished or even inverted in the heart from both genotypes four weeks after surgery, and no obvious difference in the ECG could be observed between the two genotypes after surgery (Fig 3A). The EF and FS were decreased in the wild-type and AS160Thr649Ala mice to similar levels two and four weeks after surgery (Fig 3B). The infarcted regions were also comparable as revealed by Masson’s staining of the infarct heart sections (Fig 3C and 3D).
Fig 3. Cardiac functions in the wild-type and AS160Thr649Ala knockin mice before and after myocardial infarction.
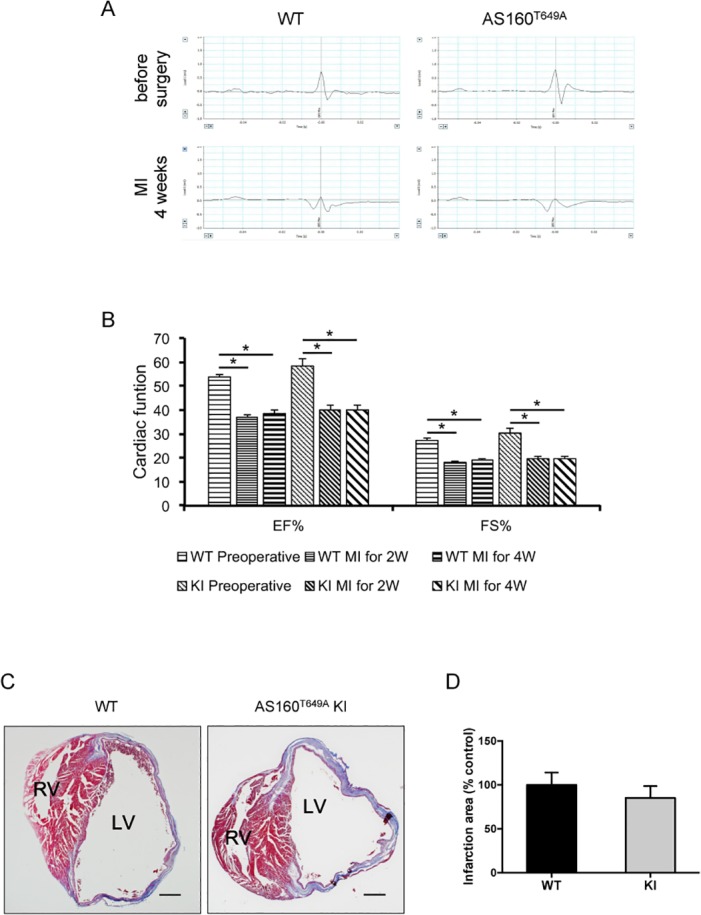
A, ECG was measured before, and 4 weeks after myocardial infarction in the wild-type and AS160Thr649Ala knockin mice. Representative ECG graphs were shown. B, Cardiac functions including EF and FS were measured via echocardiography before, and 2 and 4 weeks after myocardial infarction in the wild-type and AS160Thr649Ala knockin mice. Asterisk indicates p < 0.05. C, Masson’s staining of the infarct heart from the wild-type and AS160Thr649Ala knockin mice 4 weeks after myocardial infarction. D, Quantitation of the infarction areas detected by Masson’s staining. n = 3.
TBC1D1 expression was induced in the infarct heart from the wild-type and AS160Thr649Ala knockin mice
We investigated the molecular changes in the infarct heart from the AS160Thr649Ala knockin mice and wild-type littermate controls (Fig 4). The expression levels of the cardiac troponin T (cTnT) and GLUT4 were decreased in the border and infarct zones of the heart from both genotypes to similar levels. The expression levels of AS160 were decreased in the infarct zone in parallel with the Thr649 phosphorylation of AS160 in the wild-type heart. Similarly, the expression of AS160 was also down-regulated in the infarct zone of the AS160Thr649Ala knockin mice. As expected, the Thr649 phosphorylation of AS160 was not detectable in the lysates of the AS160Thr649Ala knockin heart. Interestingly, the expression of TBC1D1 was strongly induced in the infarct zones and also increased in the border zones of the heart from both genotypes (Fig 4A and 4B).
Fig 4. Expression and phosphorylation of AS160 and TBC1D1 in the infarct heart.
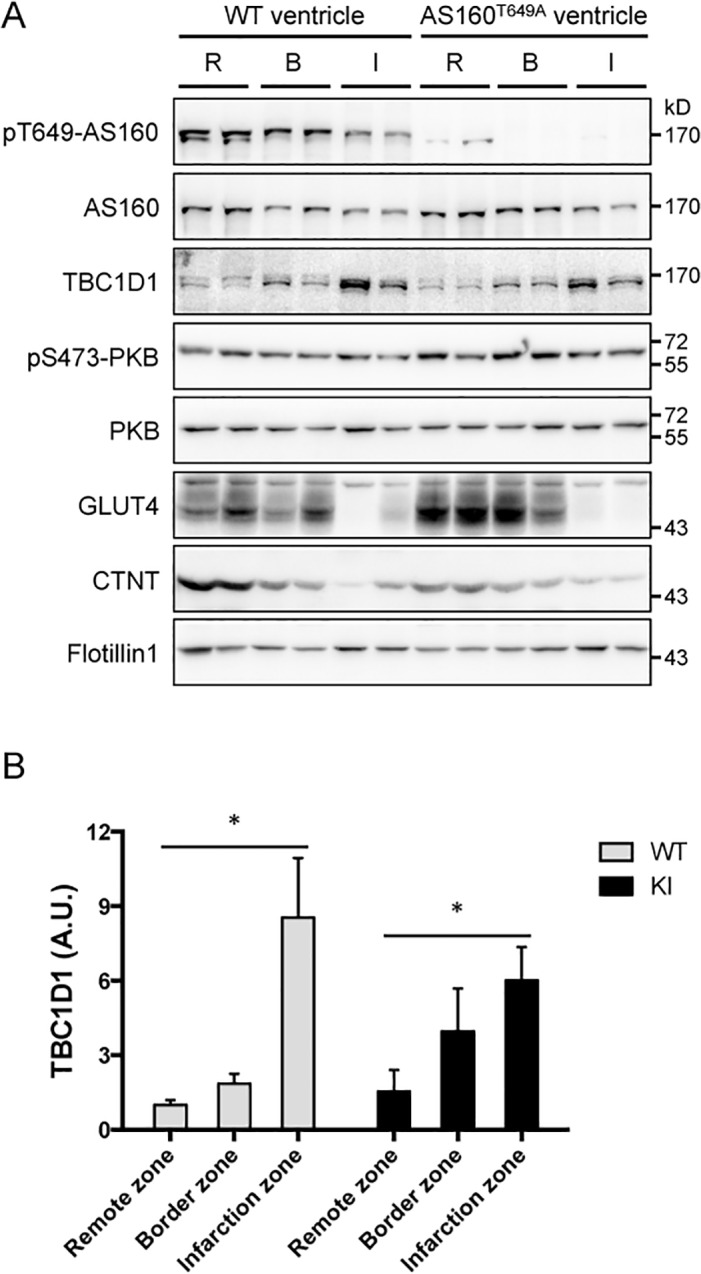
Immunoblotting analyses of phosphorylation of AS160-Thr649 and PKB-Ser473, and total AS160, TBC1D1, PKB, GLUT4 and cTnT in the lysates (40 μg) of the infarct zone (I), border zone (B) and remote zone (R) of mouse heart 4 weeks after myocardial infarction. Flotillin-1 was used as a loading control. A, representative blots. B, quantitation of TBC1D1 signals, n = 4. Asterisk indicates p < 0.05 (infarct zone versus remote zone).
Discussion
In this study, we found that PKB-mediated phosphorylation of Thr649 on AS160/TBC1D4 represented one of the major PAS-binding signals in the heart in response to insulin. Using the AS160Thr649Ala knockin mouse model, we revealed that the Thr649 phosphorylation of AS160/TBC1D4 plays an important role in the heart’s electrical conduction system.
The R-wave amplitude was moderately but not statistically increased in the hearts of the young male AS160Thr649Ala knockin mice, and significantly increased in the hearts of the old male and young female AS160Thr649Ala knockin mice under normal conditions. The increased R-wave amplitude is a diagnostic marker for cardiac hypertrophy [23]. Although the ratio of heart to body weight was increased in the AS160Thr649Ala knockin mice, the hearts showed no hypertrophic symptom as evidenced by the heart weight (data not shown) and the left ventricle mass detected by Echo (Table 2). Since the AS160/TBC1D4 regulates trafficking of epithelial sodium channel and Na+/K+-ATPase in kidney cells [15, 16], it is possible that this RabGAP may regulate ion homeostasis in cardiomyocytes thus affecting the R-wave amplitude. Since the AS160Thr649Ala knockin mutation occurs at the whole-body level, it is also possible that this mutation causes the increased R-wave amplitude indirectly. Apart from the increased R-wave amplitude, the heart of the AS160Thr649Ala knockin mice was normal regarding the heart functions such as the EF and FS. This AS160Thr649Ala knockin mutation also did not severe the heart infarction after artery ligation. It is possible that factors other than Thr649 phosphorylation of AS160/TBC1D4 mediate the pathogenesis of dilated cardiomyopathy and cardiac infarction due to the loss of PKB/Akt. For example, other phosphorylation sites on the AS160/TBC1D4 may have a more important role in maintaining heart functions. Another possibility is that other proteins may play a compensatory role for the AS160Thr649Ala knockin mutation in regulating heart functions. In line with this possibility, the expression of the related RabGAP, TBC1D1, was markedly increased in the infarct heart from both AS160Thr649Ala knockin mice and wild-type littermate controls. TBC1D1 and AS160/TBC1D4 display similar GAP activities towards downstream Rabs including Rab2, Rab8a, Rab10 and Rab14 [12]. TBC1D1 can also be phosphorylated by PKB on Thr590 that is a paralogous site of Thr649 on AS160 [11, 12].
The finding that TBC1D1 is induced in the infarct heart is intriguing and deserved for further investigation in future. The cardiac ischemia due to blockage of the coronary artery causes hypoxia in cardiomyocytes [24]. If the blockage persists, the ischemia will consequently cause cell death of cardiomyocytes but concomitantly trigger fibrosis to maintain the heart functions for a short period. However, this compensatory response may have a deadly consequence that eventually leads to myocardial infarction. It would be interesting in future to find out whether hypoxia or fibrosis in the infarct heart is responsible for the induction of TBC1D1 expression. Another intriguing question is whether the infarction-induced TBC1D1 expression facilitates the decline of heart functions or protects the infarct heart from further damage. Nevertheless, these data demonstrate that TBC1D1 can be used as a novel biomarker to monitor cardiac remodeling during infarction.
In summary, the Thr649 phosphorylation of AS160/TBC1D4 does not regulate heart functions under both normal and infarct conditions, but plays an important role in the heart’s electrical conduction system through regulating the R-wave amplitude.
Data Availability
All relevant data are within the paper.
Funding Statement
This work was supported by the Ministry of Science and Technology of China (Grant No. 2014CB964704; the National Key Scientific Research Program of China to SC), the National Natural Science Foundation of China (Grant No. 31271498 to SC), and the Ministry of Education of China (Grant Nos. 20120091120048 and NCET-13-0270 to SC). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Muniyappa R, Montagnani M, Koh KK, Quon MJ. Cardiovascular actions of insulin. Endocrine reviews 2007; 28: 463–491. [DOI] [PubMed] [Google Scholar]
- 2. Malmberg K, Ryden L, Hamsten A, Herlitz J, Waldenstrom A, Wedel H. Effects of insulin treatment on cause-specific one-year mortality and morbidity in diabetic patients with acute myocardial infarction. DIGAMI Study Group. Diabetes Insulin-Glucose in Acute Myocardial Infarction. European heart journal 1996; 17: 1337–1344. [DOI] [PubMed] [Google Scholar]
- 3. Belke DD, Betuing S, Tuttle MJ, Graveleau C, Young ME, Pham M, et al. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. The Journal of clinical investigation 2002; 109: 629–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sena S, Hu P, Zhang D, Wang X, Wayment B, Olsen C, et al. Impaired insulin signaling accelerates cardiac mitochondrial dysfunction after myocardial infarction. Journal of molecular and cellular cardiology 2009; 46: 910–918. 10.1016/j.yjmcc.2009.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. DeBosch B, Sambandam N, Weinheimer C, Courtois M, Muslin AJ. Akt2 regulates cardiac metabolism and cardiomyocyte survival. The Journal of biological chemistry 2006; 281: 32841–32851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mora A, Davies AM, Bertrand L, Sharif I, Budas GR, Jovanovic S, et al. Deficiency of PDK1 in cardiac muscle results in heart failure and increased sensitivity to hypoxia. The EMBO journal 2003; 22: 4666–4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Laustsen PG, Russell SJ, Cui L, Entingh-Pearsall A, Holzenberger M, Liao R, et al. Essential role of insulin and insulin-like growth factor 1 receptor signaling in cardiac development and function. Molecular and cellular biology 2007; 27: 1649–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. DeBosch B, Treskov I, Lupu TS, Weinheimer C, Kovacs A, Courtois M, et al. Akt1 is required for physiological cardiac growth. Circulation 2006; 113: 2097–2104. [DOI] [PubMed] [Google Scholar]
- 9. Chang Z, Zhang Q, Feng Q, Xu J, Teng T, Luan Q, et al. Deletion of Akt1 causes heart defects and abnormal cardiomyocyte proliferation. Developmental biology 2010; 347: 384–391. 10.1016/j.ydbio.2010.08.033 [DOI] [PubMed] [Google Scholar]
- 10. Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, et al. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. The Journal of biological chemistry 2003; 278: 14599–14602. [DOI] [PubMed] [Google Scholar]
- 11. Chen S, Murphy J, Toth R, Campbell DG, Morrice NA, MacKintosh C. Complementary regulation of TBC1D1 and AS160 by growth factors, insulin and AMPK activators. The Biochemical journal 2008; 409: 449–459. [DOI] [PubMed] [Google Scholar]
- 12. Roach WG, Chavez JA, Miinea CP, Lienhard GE. Substrate specificity and effect on GLUT4 translocation of the Rab GTPase-activating protein Tbc1d1. The Biochemical journal 2007; 403: 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Samovski D, Su X, Xu Y, Abumrad NA, Stahl PD. Insulin and AMPK regulate FA translocase/CD36 plasma membrane recruitment in cardiomyocytes via Rab GAP AS160 and Rab8a Rab GTPase. Journal of lipid research 2012; 53: 709–717. 10.1194/jlr.M023424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim HY, Choi HJ, Lim JS, Park EJ, Jung HJ, Lee YJ, et al. Emerging role of Akt substrate protein AS160 in the regulation of AQP2 translocation. American journal of physiology Renal physiology 2011; 301: F151–161. 10.1152/ajprenal.00519.2010 [DOI] [PubMed] [Google Scholar]
- 15. Liang X, Butterworth MB, Peters KW, Frizzell RA. AS160 modulates aldosterone-stimulated epithelial sodium channel forward trafficking. Molecular biology of the cell 2010; 21: 2024–2033. 10.1091/mbc.E10-01-0042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Comellas AP, Kelly AM, Trejo HE, Briva A, Lee J, Sznajder JI, et al. Insulin regulates alveolar epithelial function by inducing Na+/K+-ATPase translocation to the plasma membrane in a process mediated by the action of Akt. Journal of cell science 2010; 123: 1343–1351. 10.1242/jcs.066464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ramm G, Larance M, Guilhaus M, James DE. A role for 14-3-3 in insulin-stimulated GLUT4 translocation through its interaction with the RabGAP AS160. The Journal of biological chemistry 2006; 281: 29174–29180. [DOI] [PubMed] [Google Scholar]
- 18. Geraghty KM, Chen S, Harthill JE, Ibrahim AF, Toth R, Morrice NA, et al. Regulation of multisite phosphorylation and 14-3-3 binding of AS160 in response to IGF-1, EGF, PMA and AICAR. The Biochemical journal 2007; 407: 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen S, Wasserman DH, MacKintosh C, Sakamoto K. Mice with AS160/TBC1D4-Thr649Ala knockin mutation are glucose intolerant with reduced insulin sensitivity and altered GLUT4 trafficking. Cell metabolism 2011; 13: 68–79. 10.1016/j.cmet.2010.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ducommun S, Wang HY, Sakamoto K, MacKintosh C, Chen S. Thr649Ala-AS160 knock-in mutation does not impair contraction/AICAR-induced glucose transport in mouse muscle. American journal of physiology Endocrinology and metabolism 2012; 302: E1036–1043. 10.1152/ajpendo.00379.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Di R, Wu X, Chang Z, Zhao X, Feng Q, Lu S, et al. S6K inhibition renders cardiac protection against myocardial infarction through PDK1 phosphorylation of Akt. The Biochemical journal 2012; 441: 199–207. 10.1042/BJ20110033 [DOI] [PubMed] [Google Scholar]
- 22. Wang HY, Ducommun S, Quan C, Xie B, Li M, Wasserman DH, et al. AS160 deficiency causes whole-body insulin resistance via composite effects in multiple tissues. The Biochemical journal 2013; 449: 479–489. 10.1042/BJ20120702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mazzaro Cdo L, Costa Fde A, Bombig MT, Luna Filho B, Paola AA, Carvalho AC, et al. Ventricular mass and electrocardiographic criteria of hypertrophy: evaluation of new score. Arquivos brasileiros de cardiologia 2008; 90: 227–231. [DOI] [PubMed] [Google Scholar]
- 24. von Bibra H, St John Sutton M. Impact of diabetes on postinfarction heart failure and left ventricular remodeling. Current heart failure reports 2011; 8: 242–251. 10.1007/s11897-011-0070-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.