

Abstract
The ketone body β-hydroxybutyrate (βOHB) is a convenient carrier of energy from adipocytes to peripheral tissues during fasting or exercise. However, βOHB is more than just a metabolite, having important cellular signaling roles as well. βOHB is an endogenous inhibitor of histone deacetylases (HDACs) and a ligand for at least two cell surface receptors. In addition, the downstream products of βOHB metabolism including acetyl-CoA, succinyl-CoA, and NAD+ (nicotinamide adenine dinucleotide) themselves have signaling activities. These regulatory functions of βOHB serve to link the outside environment to cellular function and gene expression, and have important implications for the pathogenesis and treatment of metabolic diseases including type 2 diabetes.
Keywords: ketone bodies, low carbohydrate, acetylation, HDAC, epigenetics
Metabolites as signals
The landscape of 21st century health is increasingly defined by the environment into which we place our bodies - and which elements of that environment we place into our bodies. The prevalence of type 2 diabetes is rising rapidly throughout the developed world, and is largely attributed to changes in interrelated environmental factors such as obesity, diet, and lifestyle [1]. On a molecular level, we are increasingly understanding precisely how environmental factors like diet can, for example, signal into our cells’ nuclei to regulate gene expression and chromatin structure. Dietary intake of he amino acid threonine affects cellular levels of the methyl donor S-adenosylmethionine, which in turn promotes histone methylation and regulates stem cell function [2]. Lipid-burning states, such as fasting, increase both acetyl-CoA production and levels of histone acetylation [3]. The overall cellular energy balance controls the NAD+/NADH ratio [4], which in turn regulates the activity of sirtuin enzymes in diseases of aging [5].
The ketone body β-hydroxybutyrate (βOHB) is also more than just a metabolite. Long viewed as a simple carrier of energy from the liver to peripheral tissues during prolonged fasting or exercise, βOHB also possesses a variety of signaling functions that might provide for broad regulation of cellular functions with implications for metabolic disease and diabetes. Perhaps most intriguing is that βOHB is an endogenous inhibitor of HDACs [3], joining a small but growing list of metabolic intermediaries that affect gene expression via chromatin modifications [6]. Here, we review the signaling functions of βOHB as an HDAC inhibitor and as ligand for cell surface receptors, as well as the indirect effects of βOHB metabolism on other metabolites with signaling functions including acetyl-CoA, succinyl-CoA and NAD+. This ketone body may have a broad regulatory role in metabolic disease through alterations in histone acetylation and gene expression, post-translational protein function, and cell surface receptor activation.
Metabolism, regulation and function of ketone bodies
Ketone bodies are small lipid-derived molecules that serve as a circulating energy source for tissues in times of fasting or prolonged exercise [7]. Produced in the liver from fatty acids mobilized from adipocytes, they are distributed via the circulation to metabolically active tissues, such as muscle or brain, where they are converted to acetyl-CoA [7]. In humans, basal serum levels of βOHB are in the low micromolar range, but can reach 1–2 mM after 2 days of fasting [8, 9], and over 2 mM with a ketogenic diet that is almost devoid of carbohydrates [10]. The pathways of synthesis and metabolism of ketone bodies are reviewed in [11].
Synthesis of ketone bodies in the liver is controlled by at least two hormone- and nutrient-responsive pathways that regulate transcription of the rate-limiting enzyme, mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase 2 (HMGCS2) [12]. First, insulin and glucagon opposingly regulate acetylation of the forkhead box transcription factor FOXA2. insulin signaling leads to inactivation of FOXA2 via phosphorylation and nuclear export [13], while glucagon activates FOXA2 via p300 acetylation [14], after which FOXA2 binds to the HMGCS2 promoter and activates transcription [15]. FOXA2 deacetylation is controlled by the NAD-dependent enzyme SIRT1, working in cooperation with class I or II HDACs [14]. Second, HMGCS2 transcription is negatively regulated by mTORC1, one of the human target of rapamycin (TOR) complexes that is activated by insulin signaling or abundant glucose (via AMPK) or amino acids (via Rag) [16]. The mTORC1 complex suppresses PPARα [17], and in turn PPARα is required to induce FGF21 [18] in order to activate ketogenesis. Therefore, inhibition of mTOR can promote ketogenesis. The activity of HMGCS2 is also post-translationally regulated by succinylation [19] and acetylation [20]. Interestingly, many of the enzymes involved in the generation of ketone bodies from lipids are both acetylated and succinylated, and are targets for removal of these nutrient-sensitive modifications by the mitochondrial NAD-dependent deacylases SIRT3 [21] and SIRT5 [22]. The post-translational regulation of mitochondrial enzymes by acyl-CoA derived lysine acylations remains an active area of investigation.
Signaling functions of βOHB
The ketone body βOHB is increasingly understood to have a broad range of signaling and regulatory effects, both through a growing panoply of its own actions as well as through the knock-on effects of its metabolism in target tissues. On its own account, βOHB is an endogenous inhibitor of many protein deacetylases, and a ligand for at least two cell surface receptors. In addition, metabolism of βOHB in target tissues can alter the intracellular balance of acetyl-CoA, succinyl-CoA, and NAD, all of which have their own emerging regulatory functions.
βOHB binds to and inhibits class I HDACs
βOHB was recently found to be an endogenous inhibitor of histone deacetylaes (HDACs) [23]. HDACs are a family of proteins that broadly regulate gene expression and may have specific roles in glucose metabolism and diabetes. Histone hyperacetylation is generally associated with activation of gene expression, and so deacetylation of lysine residues on histones by HDACs often suppresses transcription (reviewed in [24-26]). Inhibition of HDACs therefore usually activates gene expression. Although histones were the first known deacetylation targets, many non-histone proteins are also subject to HDAC-mediated deacetylation [27], such as FOXA2 above. HDACs are grouped into four classes; βOHB is so far known to inhibit classes I and IIa. Class I HDACs (e.g., HDACs 1, 2, 3 and 8) are small nuclear proteins that are usually found in multi-protein complexes; Class IIa HDACs (e.g., HDAC4, 5, 7 and 9) are larger proteins with extensive regulatory domains in their N-terminus, that shuttle between the nucleus and cytoplasm. Class IIb HDACs are cytoplasmic proteins, of which the best understood is HDAC6, the tubulin deacetylase. Class IV includes only one poorly understood HDAC. Class III HDACs, the sirtuins, are a structurally distinct group of NAD-dependent deacylases which βOHB is not known to directly regulate. [24].
βOHB inhibits HDACs 1, 3 and 4 (class I and IIa) in vitro with an IC50 of 2–5 mM, suggesting that significant HDAC inhibition occurs even at the 1-2 mM serum concentrations that can be reached with fasting or ketogenic diet. Treating cultured cells with βOHB induces dose-dependent histone hyperacetylation, as does infusing βOHB into mice via osmotic pump. Interestingly, the histone hyperacetylation seen in mouse tissues with βOHB treatment is similar to that seen after fasting or with calorie restriction. Consistent with HDAC inhibition, βOHB induces specific changes in gene expression, including induction of forkhead box O3 (Foxo3), the mammalian ortholog of the stress-responsive transcriptional factor DAF16 that regulates lifespan in worms [28]. Induction of Foxo3 appears to be a direct effect of HDAC inhibition, as HDAC1 and HDAC2 are found on its promoter, knockdown of both relieves the HDAC mediated Foxo3 repression, and βOHB causes hyperacetylation of histones at the Foxo3 promoter that results in increased Foxo3 expression [3].
Consequences of βOHB HDAC inhibition
HDAC inhibition by βOHB might affect the pathogenesis of type 2 diabetes in at least two ways: through direct regulation of HDAC-dependent glucose metabolism, and by promoting resistance to oxidative stress. Studies in knockout mice have shown that class I HDACs have a key role in regulating metabolic disease. HDAC3 regulates expression of gluconeogenic genes [29] and HDAC3 knockout mice have reduced fasting glucose and insulin levels [30-32]. In fact, chronic treatment with butyrate, a broad HDAC inhibitor that might be expected to phenocopy loss of function of HDAC3, keeps mice essentially metabolically normal on a high-fat diet [33]. Butyrate treatment is associated with lower glucose and insulin levels, better glucose tolerance, prevention of weight gain, and improved respiratory efficiency; butyrate also provides some of these benefits even to mice already obese from being fed a high-fat diet [33]. Similarly, the class I HDAC inhibitor SAHA (but not a class II HDAC inhibitor) increases mitochondrial biogenesis, improves insulin sensitivity, and increases metabolic rate and oxidative metabolism in a mouse diabetes model [34]. The mechanism for these metabolic benefits of class I HDAC inhibition may be up-regulation of PGC1α in in a variety of tissues by relief of HDAC3-mediated transcriptional repression [33, 34]. Transcription of FGF21 is similarly up-regulated via butyrate inhibition of HDAC3, activating ketogenesis in obese mice [35]. Several single nucleotide polymorphisms in HDAC3 have been associated with an elevated risk of type 2 diabetes in a Chinese population [36]. βOHB may have similar effects on mitochondrial function, glucose homeostasis, and obesity through endogenous inhibition of HDAC3. Effects of the other class I HDACs on metabolic or metabolic disease have not been systematically studied, but it is notable that HDAC2 knockout mice are 25% smaller than normal, with impaired IGF-1 signaling [37].
The microvascular and macrovascular complications of type 2 diabetes, such as cardiovascular disease, are thought to be due in part to increased oxidative stress brought on through several pathways including polyols, protein kinase C, hexoasamine, and advanced glycosylation end products [38]. In this context, the emerging role of βOHB in suppression of oxidative stress may be relevant to the management of diabetic complications. As described above, βOHB activates transcription of FOXO3 and downstream antioxidant gene via HDAC inhibition, and this is associated with protection of the mouse kidney from oxidative damage [3]. Other studies have previously suggested a role for both βOHB and HDAC inhibitors in protection from oxidative or ischemic stress. Perhaps most relevant to diabetic complications, long-term treatment with an HDAC inhibitor attenuates kidney injury in a mouse model of diabetic nephropathy, probably though transcriptional regulation of endothelial nitric oxide synthase [39]. The relevant class I HDAC may be HDAC2, whose activity is aberrantly increased in the diabetic mouse kidney by oxidative stress through TGFβ [40]. Interestingly, this is similar to the role of aberrant HDAC2 activation in neurodegenerative disease [41]. Butyrate, another HDAC inhibitor, has broad cytoprotective effects including improving overall survival in a rat sepsis model [42], and reducing lung injury after lipopolysaccharide infusion in mice [43]. βOHB protects cultured neurons from hypoxic or hypoglycemic insults [44, 45], as well as from oxidative stress from hydrogen peroxide [46]. In animals, βOHB reduces anoxic lung injury [47, 48], as well as ischemic myocardial damage [49]; a ketogenic diet is also protective from ischemic neuronal death [50, 51]. Induction of protective genes via HDAC inhibition may be the mechanism by which βOHB exerts a protective antioxidant effect under these stresses.
The functional consequences to the cell and organism of HDAC inhibition by βOHB are only just beginning to be cataloged. Given the widespread nature of gene regulation by HDACs, and the important role of HDACs in regulating dynamic changes of gene expression in response to stimuli, we may have only scratched the surface of the clinical and pathophysiological consequences of this signaling function of βOHB.
βOHB receptors
Many G-protein coupled receptors (GPCRs) bind to fatty acid ligands, and have important roles in metabolism and metabolic disease [52, 53]. At least two GPCRs that bind short-chain fatty acids also bind βOHB. HCAR2 (also known as HCA2, PUMA-G, or Gpr109), is a Gi/o-coupled GPCR that was first identified as a nicotinic acid receptor [54]. Recently HCAR2 was shown to bind and be activated by βOHB [55]. HCAR2 activation by βOHB (or other ligands) reduces lipolysis in adipocytes [55, 56]. For βOHB, this might perhaps represent a feedback mechanism to regulate availability of the fatty acid precursors of ketone body metabolism. However, elevated free fatty acids in plasma from dysregulated adipocytes are thought to contribute to insulin resistance through a variety of mechanisms including proinflammatory cytokines, oxidative stress, and ER stress [57]. Pharmacological agonists of HCAR2 reduce both plasma free fatty acids and plasma glucose in humans with type 2 diabetes [58]. HCAR2 also mediates the antiatherosclerotic effects of nicotinic acid in mouse models [59], probably through modulation of macrophage function [60]. Thus, activation of HCAR2 by βOHB may contribute to improved glucose control as well as ameliorating some of the macrovascular complications of type 2 diabetes.
βOHB is also a ligand for the free fatty acid receptor 3 (FFAR3, also known as GPR41). FFAR3 is another Gi/o-protein coupled receptor that is highly expressed in sympathetic ganglions. FFAR3 knockout mice have reduced basal oxygen consumption and body temperature, but are then insensitive to the usual further sympathetic depression seen during prolonged fasting [61]. Antagonism of FFAR3 by βOHB suppresses sympathetic tone and heart rate, and may be responsible for sympathetic depression during fasting [61]. However, an electrophysiological study in rats later reported that βOHB acts as an agonist of FFAR3 [62]. The physiological details of this adrenergic modulation by βOHB, and its effects on glucose homeostasis or cardiovascular disease, remain to be determined.
Regulatory activities of βOHB metabolites
βOHB is only one of a growing list of metabolic intermediaries that have direct regulatory roles, linking environmental factors such a nutrition to changes in cellular function such as epigenetic regulation of gene expression [63]. As another example, intake of threonine affects the levels of the methyl donor S-adenosylmethionine, which in turn promotes histone methylation [2]. Several elements of the metabolism of βOHB in peripheral tissues might themselves have broad regulatory functions: acetyl-CoA, succinyl-CoA, and NAD. The first step in βOHB utilization in peripheral tissues is its conversion to acetoacetate by mitochondrial β-hydroxybutyrate dehydrogenase (BDH1), which uses NAD as a cofactor and generates NADH. Next, acetoacetate is converted to acetoacetyl-CoA by succinyl-CoA:3-ketoacid coenzyme A transferase (OXCT1, also known as SCOT), which uses succinyl-CoA as a CoA donor, releasing free succinate. Acetoacetyl-CoA is then split into two acetyl-CoA that are burned in the TCA cycle to generate ATP in peripheral tissues. Utilization of βOHB therefore might increases acetyl-CoA levels, decrease succinyl-CoA levels, and alter the NAD/NADH ratio in peripheral tissues such as muscle.
Increasing the intracellular pools of acetyl-CoA should indirectly increase protein acetylation. This effect is complementary to HDAC inhibition by βOHB, but may have broader effects in multiple cellular compartments. Increasing the acetyl-CoA substrate for both enzymatic and non-enzymatic protein acetylation should drive these reaction equilibria towards acetylation. This may particularly increase protein acetylation in mitochondria. For example, CR, fasting and high-fat diets, states associated with increased lipid utilization and therefore high acetyl-CoA throughput, all cause increased mitochondrial protein acetylation—even though neither acetyltransferases nor the HDACs that are inhibited by βOHB are known to enter mitochondria [64]. Acetylation – and deacetylation by SIRT3 – is very widespread in mitochondria {Rardin, 2013 #176} and regulates the function of several mitochondrial enzymes including HMGCS2 [20] and the long-chain acyl-CoA dehydrogenase (LCAD) [65]. Increased acetyl-CoA pools also affect nuclear protein acetylation. Mitochondrial acetylcarnitine is known to be a source of acetyl-CoA for histone acetylation [66]. Export of acetyl-CoA from the mitochondria is accomplished via a citrate shuttle mediated by citrate synthase inside mitochondria and ATP citrate lyase in the cytoplasm [67]. ATP citrate lyase is a key enzyme in fatty acid biosynthesis, but its role in facilitating acetyl-CoA export from mitochondria is also required for the increase in histone acetylation that occurs with growth factor stimulation [67]. An alternative pathway for acetyl-CoA export from mitochondria is via the enzymes carnitine acetyltransferase (CAT) and carnitine/acylcarnitine translocase [68]. Indeed, a muscle-specific knockout of CAT in mice compromises glucose tolerance and decreases metabolic flexibility [68], demonstrating the importance of intracellular acetyl-CoA transport to overall metabolic health.
Consumption of succinyl-CoA by OXCT1 during utilization of βOHB in peripheral tissues may also have broad regulatory consequences on cellular metabolism. Lysine succinylation is very widespread in mitochondria [22], and present across diverse organisms [69]. A substantial fraction of these succinylation sites are regulated by the mitochondrial desuccinylase, the sirtuin SIRT5 [22]. HMGCS2 has long known to be succinylated, a modification that reduces its activity [19]; HMGCS2 activity in the liver is restored by desuccinylation by SIRT5. The mechanism of lysine succinylation is not clearly understood; an enzymatic succinyl-transferase is not known to exist in mammalian cells, and as both liver succinyl-CoA abundance and succinylation of HMGCS2 are reduced after treatment of rats with glucagon [19, 70], it is possible that succinylation is primarily a non-enzymatic process dependent upon local concentrations of succinyl-CoA. HMGCS2 is not expressed in peripheral tissues, which do not engage in ketogenesis in any event; but the enzymes in many key mitochondrial pathways are heavily succinylated and regulated by SIRT5, including fatty acid oxidation, branched-chain amino acid catabolism, and the TCA cycle. By analogy with acetylation (and the effect of succinylation on HMGCS2), these pathways may be activated by a reduction in succinylation. Consumption of succinyl-CoA during βOHB utilization and consequent reduction in mitochondrial protein succinylation may therefore regulate many of these crucial mitochondrial pathways in peripheral tissues.
Cellular NAD balance is emerging as a crucial mediator of metabolic disease and aging [71]. NAD utilization by βOHB differs from that of glucose in two important respects. First, βOHB consumes fewer NAD+ per acetyl-CoA produced. Metabolism of one molecule of glucose to two molecules of acetyl-CoA involves conversion of four molecules of NAD+ into NADH. Two of these are converted in the cytosol during glycolysis, and two in the mitochondrion by pyruvate decarboxylase. The cytosolic NADH are shuttled into mitochondria, potentially leading to depletion of the cytoplasmic NAD pool with high glucose utilization. By contrast, metabolism of one βOHB molecule into the same two molecules of acetyl-CoA involves conversion of only two molecules of NAD+ into NADH, both in the mitochondrion by BDH1 and thereby preserving the cytoplasmic NAD pool [7]. The cytoplasmic and mitochondrial NAD pools are relatively distinct, so the preservation of cytoplasmic NAD+ by βOHB may have important cellular effects. NAD+ is a cofactor for sirtuin deacylases (such as nuclear/cytoplasmic SIRT1) as well as poly-ADP-ribose polymerase (PARP) [71]. Consumption of NAD+ by PARP or overproduction of NADH may promote age-related diseases by inhibiting the activity of sirtuins [5]. Conversely, repletion of NAD+ by exogenous feeding with nicotinadmide mononucleotide improves glucose tolerance in both high-fat diet-fed and aged mice [72]. The relative sparing of NAD+ by utilization of βOHB vis a vis glucose may therefore have important consequences for metabolic diseases and diabetes.
βOHB and carbohydrate-restricted diets
Energy-restricted metabolic states, such as CR or intermittent fasting, have metabolic health benefits in mammals [73]. These states are almost inextricably associated with elevations in ketone bodies, whether consistent and modest as in CR or periodic and substantial as in intermittent fasting. Interestingly, the metabolic benefits are not necessarily associated with reduced caloric intake; for example, mice fed only during 8 hours at night are resistant to diet-induced obesity [74] despite identical caloric intake to controls. This suggests that some of the various effects of fasting or CR are regulated by specific elements of the fasting state, perhaps including βOHB. Ketogenic diets that are very low in carbohydrate content are one means of delivering βOHB without fasting, and have been used for decades as a clinical therapy for intractable epilepsies [75]. There is increasing interest in the possible use of therapeutic ketogenic diets in adult type 2 diabetes as well [76].
Much of our understanding of the biological effects of ketogenic diets come from studies in rodents. Ketogenic diets are a high-fat, high-energy state and so some of their effects on metabolism similar to a typical high-fat or Western diet. Lean mice started on a ketogenic diet show increased fasting leptin [77], and hyperglycemia with insulin resistance, although basal insulin levels are lower [77-81]. Ketogenic diets promote liver endoplasmic reticulum (ER) stress, similar to that seen in diabetic mice [81]. There is strong induction of hepatic genes involved in fatty acid oxidation, including acyl coA dehydrogenases and trifunctional enzyme components [81-83]. However, the ketogenic diet is unusual in that it simultaneously activates “fasting” pathways as well. For example, ketogenic diets suppress liver expression of fatty acid synthesis enzymes including stearoyl-CoA desaturase-1 [82, 83], whose induction by a high fat diet is important for development of metabolic syndrome [84]. Instead, the “fasting regulator” genes PPARα and FGF21 [81, 83, 85] are strongly induced, as is peroxisome proliferator-activated receptor gamma co-activator 1-alpha (PGC1α), a master regulator of mitochondrial function [85, 86]. In addition, ketogenic diets are associated with low insulin levels [77-81], reduced IGF signaling [87-89] and Foxo3 induction [3], AMP-activated protein kinase (AMPK) activation [82, 89], mTOR repression [89], and induction of antioxidant genes [3]. Perhaps as a net result of these effects, obese or diabetic mice show improved glucose tolerance and insulin sensitivity on ketogenic diets [46, 83, 87], along with reversal of diabetic nephropathy [46].
There is limited data to date on the use of ketogenic diets in adult humans for treatment of obesity or diabetes, and these diets should only be used under physician supervision or in the context of a clinical trial. But such data as so far exists suggests that very-low carbohydrate and ketogenic diets may provide disproportionate benefit in weight loss and improved insulin resistance [90-92]. For example, intermittent severe energy restriction, presumably ketogenic, is more effective than daily energy restriction at improving insulin sensitivity and promoting weight loss [93] and a recent clinical trial of a ketogenic diet found that obese humans with diabetes lost more weight with greater improvement in glucose control than a lower-calorie low-fat diet [94]. Nonetheless, these dramatic dietary interventions are not without side effects [95], and their efficacy should spur greater efforts to understand the molecular mechanisms of their effects.
Concluding Remarks and Future Perspectives
Ketone bodies are emerging as crucial regulators of metabolic health. More than just a metabolite, the ketone body βOHB can regulate cellular processes directly via HDAC inhibition and binding to cell surface receptors, and indirectly by altering the levels of other regulatory metabolites including acetyl-CoA, succinyl-CoA, and NAD+. The ability of βOHB to regulate HDAC activity and thereby epigenetic gene regulation is particularly notable for potentially implicating a wide variety of genes as regulatory targets of βOHB. βOHB is already known to induce resistance to oxidative stress via HDAC inhibition, and other HDAC inhibitors regulate gluconeogenesis. The unique effects of βOHB may help explain the therapeutic benefit of low-carbohydrate and ketogenic diets. However, teasing apart the specific role of βOHB in the effects of a ketogenic diet is a challenging task. Ketogenic diets inextricable combine reduced carbohydrate consumption, reduced glucose utilization, reliance on beta-oxidation of lipids for energy, reduced insulin signaling, and increased glucagon signaling, along with increased ketone body levels [10]. New experimental tools are required to permit the safe manipulation of βOHB levels outside of the confines of a ketogenic diet in order to understand the full spectrum of action of βOHB in amelioration of metabolic disease.
Figure 1. Cellular signaling functions of the ketone body βOHB.
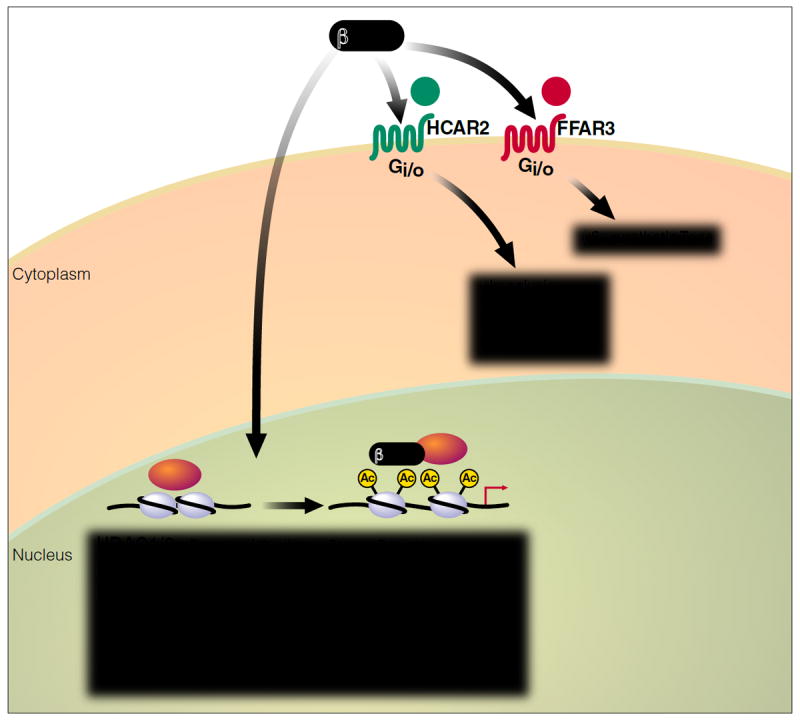
βOHB itself is an endogenous inhibitor of histone deacetylase enzymes, thereby altering gene expression to regulate resistance to oxidative stress and possibly many other cellular functions. βOHB is also a ligand for at least two cell-surface G-protein-coupled receptors that modulate lipolysis, sympathetic tone, and metabolic rate.
Figure 2. Cellular signaling functions of βOHB downstream metabolites.
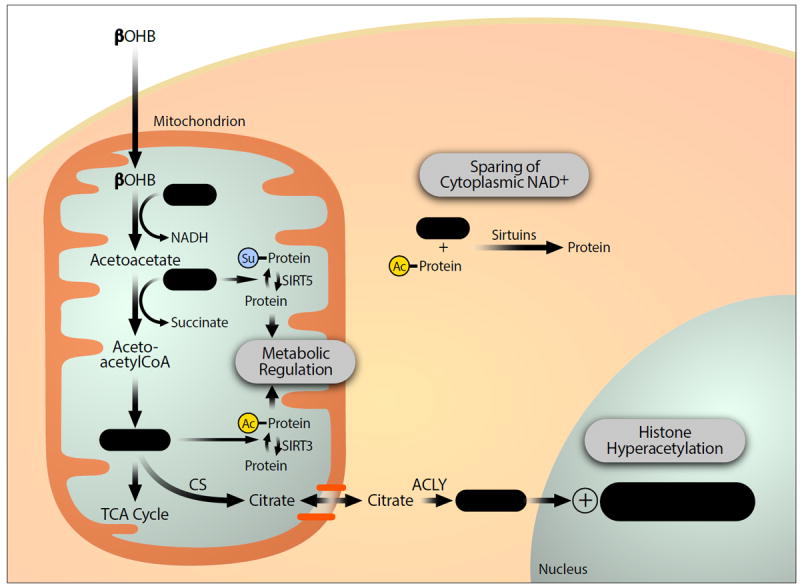
Metabolism of βOHB increases cellular levels of acetyl-CoA and reduces levels of succinyl-CoA and NAD+. These secondary effects can further increase mitochondrial protein acetylation and reduce mitochondrial protein succinylation, potentially regulating the function of many metabolic enzymes. The relative sparing of cytoplasmic NAD levels with utilization of BOHB rather than glucose can alter the activity of NAD-dependent enzymes such as sirtuins. Finally, acetyl-CoA generated in mitochondria can be transported into the nucleus via the citrate shuttle to serve as substrate for histone acetyltransferases, a secondary mechanism by which BOHB might increase histone acetylation and alter gene expression.
Acknowledgments
We thank John Carroll for artistic assistance. EV is supported by funds from NIH/NIDDK and the Gladstone Institutes. JCN is supported by funds from the Larry L. Hillblom Foundation, the John A. Hartford Foundation, the Glenn Foundation for Medical Research, and by NIH/NIA training grant 5-T32-AG000212.
Footnotes
Conflicts of Interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zimmet PZ, Magliano DJ, Herman WH, Shaw JE. Diabetes: a 21st century challenge. The lancet Diabetes & endocrinology. 2014;2:56–64. doi: 10.1016/S2213-8587(13)70112-8. [DOI] [PubMed] [Google Scholar]
- 2.Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, Ratanasirintrawoot S, et al. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science. 2013;339:222–6. doi: 10.1126/science.1226603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339:211–4. doi: 10.1126/science.1227166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stein LR, Imai S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol Metab. 2012;23:420–8. doi: 10.1016/j.tem.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Imai SI, Guarente L. NAD and sirtuins in aging and disease. Trends in cell biology. 2014 doi: 10.1016/j.tcb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gut P, Verdin E. The nexus of chromatin regulation and intermediary metabolism. Nature. 2013;502:489–98. doi: 10.1038/nature12752. [DOI] [PubMed] [Google Scholar]
- 7.Berg JM, Tymoczko JL, Stryer L. Biochemistry. 7. New York: W.H. Freeman; 2012. [Google Scholar]
- 8.Cahill GF, Jr, Herrera MG, Morgan AP, Soeldner JS, Steinke J, Levy PL, et al. Hormone-fuel interrelationships during fasting. J Clin Invest. 1966;45:1751–69. doi: 10.1172/JCI105481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson AM, Williamson DH. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol Rev. 1980;60:143–87. doi: 10.1152/physrev.1980.60.1.143. [DOI] [PubMed] [Google Scholar]
- 10.Kim do Y, Rho JM. The ketogenic diet and epilepsy. Curr Opin Clin Nutr Metab Care. 2008;11:113–20. doi: 10.1097/MCO.0b013e3282f44c06. [DOI] [PubMed] [Google Scholar]
- 11.Newman JC, Verdin E. Ketone bodies as signaling metabolites. Trends Endocrinol Metab. 2014;25:42–52. doi: 10.1016/j.tem.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hegardt FG. Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase: a control enzyme in ketogenesis. Biochem J. 1999;338(Pt 3):569–82. [PMC free article] [PubMed] [Google Scholar]
- 13.Wolfrum C, Besser D, Luca E, Stoffel M. Insulin regulates the activity of forkhead transcription factor Hnf-3beta/Foxa-2 by Akt-mediated phosphorylation and nuclear/cytosolic localization. Proc Natl Acad Sci U S A. 2003;100:11624–9. doi: 10.1073/pnas.1931483100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.von Meyenn F, Porstmann T, Gasser E, Selevsek N, Schmidt A, Aebersold R, et al. Glucagon-induced acetylation of Foxa2 regulates hepatic lipid metabolism. Cell Metab. 2013;17:436–47. doi: 10.1016/j.cmet.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 15.Wolfrum C, Asilmaz E, Luca E, Friedman JM, Stoffel M. Foxa2 regulates lipid metabolism and ketogenesis in the liver during fasting and in diabetes. Nature. 2004;432:1027–32. doi: 10.1038/nature03047. [DOI] [PubMed] [Google Scholar]
- 16.Howell JJ, Manning BD. mTOR couples cellular nutrient sensing to organismal metabolic homeostasis. Trends Endocrinol Metab. 2011;22:94–102. doi: 10.1016/j.tem.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sengupta S, Peterson TR, Laplante M, Oh S, Sabatini DM. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature. 2010;468:1100–4. doi: 10.1038/nature09584. [DOI] [PubMed] [Google Scholar]
- 18.Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007;5:426–37. doi: 10.1016/j.cmet.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 19.Quant PA, Tubbs PK, Brand MD. Glucagon activates mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase in vivo by decreasing the extent of succinylation of the enzyme. Eur J Biochem. 1990;187:169–74. doi: 10.1111/j.1432-1033.1990.tb15291.x. [DOI] [PubMed] [Google Scholar]
- 20.Shimazu T, Hirschey MD, Hua L, Dittenhafer-Reed KE, Schwer B, Lombard DB, et al. SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 2010;12:654–61. doi: 10.1016/j.cmet.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proceedings of the National Academy of Sciences. 2013 doi: 10.1073/pnas.1302961110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, Danielson SR, et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 2013;18:920–33. doi: 10.1016/j.cmet.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 24.Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9:206–18. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mihaylova MM, Shaw RJ. Metabolic reprogramming by class I and II histone deacetylases. Trends Endocrinol Metab. 2013;24:48–57. doi: 10.1016/j.tem.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.New M, Olzscha H, La Thangue NB. HDAC inhibitor-based therapies: can we interpret the code? Mol Oncol. 2012;6:637–56. doi: 10.1016/j.molonc.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 28.Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–12. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- 29.Mihaylova MM, Vasquez DS, Ravnskjaer K, Denechaud PD, Yu RT, Alvarez JG, et al. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell. 2011;145:607–21. doi: 10.1016/j.cell.2011.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knutson SK, Chyla BJ, Amann JM, Bhaskara S, Huppert SS, Hiebert SW. Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. Embo J. 2008;27:1017–28. doi: 10.1038/emboj.2008.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fajas L, Egler V, Reiter R, Hansen J, Kristiansen K, Debril MB, et al. The retinoblastoma-histone deacetylase 3 complex inhibits PPARgamma and adipocyte differentiation. Dev Cell. 2002;3:903–10. doi: 10.1016/s1534-5807(02)00360-x. [DOI] [PubMed] [Google Scholar]
- 32.Bhaskara S, Knutson SK, Jiang G, Chandrasekharan MB, Wilson AJ, Zheng S, et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell. 2010;18:436–47. doi: 10.1016/j.ccr.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes. 2009;58:1509–17. doi: 10.2337/db08-1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galmozzi A, Mitro N, Ferrari A, Gers E, Gilardi F, Godio C, et al. Inhibition of class I histone deacetylases unveils a mitochondrial signature and enhances oxidative metabolism in skeletal muscle and adipose tissue. Diabetes. 2013;62:732–42. doi: 10.2337/db12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Gao Z, Zhang J, Ye X, Xu A, Ye J, et al. Sodium butyrate stimulates expression of fibroblast growth factor 21 in liver by inhibition of histone deacetylase 3. Diabetes. 2012;61:797–806. doi: 10.2337/db11-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeng Z, Liao R, Yao Z, Zhou W, Ye P, Zheng X, et al. Three single nucleotide variants of the HDAC gene are associated with type 2 diabetes mellitus in a Chinese population: a community-based case-control study. Gene. 2014;533:427–33. doi: 10.1016/j.gene.2013.09.123. [DOI] [PubMed] [Google Scholar]
- 37.Zimmermann S, Kiefer F, Prudenziati M, Spiller C, Hansen J, Floss T, et al. Reduced body size and decreased intestinal tumor rates in HDAC2-mutant mice. Cancer Res. 2007;67:9047–54. doi: 10.1158/0008-5472.CAN-07-0312. [DOI] [PubMed] [Google Scholar]
- 38.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circulation research. 2010;107:1058–70. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Advani A, Huang Q, Thai K, Advani SL, White KE, Kelly DJ, et al. Long-term administration of the histone deacetylase inhibitor vorinostat attenuates renal injury in experimental diabetes through an endothelial nitric oxide synthase-dependent mechanism. The American journal of pathology. 2011;178:2205–14. doi: 10.1016/j.ajpath.2011.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noh H, Oh EY, Seo JY, Yu MR, Kim YO, Ha H, et al. Histone deacetylase-2 is a key regulator of diabetes- and transforming growth factor-beta1-induced renal injury. American journal of physiology Renal physiology. 2009;297:F729–39. doi: 10.1152/ajprenal.00086.2009. [DOI] [PubMed] [Google Scholar]
- 41.Graff J, Tsai LH. Histone acetylation: molecular mnemonics on the chromatin. Nature reviews Neuroscience. 2013;14:97–111. doi: 10.1038/nrn3427. [DOI] [PubMed] [Google Scholar]
- 42.Zhang LT, Yao YM, Lu JQ, Yan XJ, Yu Y, Sheng ZY. Sodium butyrate prevents lethality of severe sepsis in rats. Shock. 2007;27:672–7. doi: 10.1097/SHK.0b013e31802e3f4c. [DOI] [PubMed] [Google Scholar]
- 43.Ni YF, Wang J, Yan XL, Tian F, Zhao JB, Wang YJ, et al. Histone deacetylase inhibitor, butyrate, attenuates lipopolysaccharide-induced acute lung injury in mice. Respir Res. 2010;11:33. doi: 10.1186/1465-9921-11-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Masuda R, Monahan JW, Kashiwaya Y. D-beta-hydroxybutyrate is neuroprotective against hypoxia in serum-free hippocampal primary cultures. J Neurosci Res. 2005;80:501–9. doi: 10.1002/jnr.20464. [DOI] [PubMed] [Google Scholar]
- 45.Samoilova M, Weisspapir M, Abdelmalik P, Velumian AA, Carlen PL. Chronic in vitro ketosis is neuroprotective but not anti-convulsant. J Neurochem. 2010;113:826–35. doi: 10.1111/j.1471-4159.2010.06645.x. [DOI] [PubMed] [Google Scholar]
- 46.Poplawski MM, Mastaitis JW, Isoda F, Grosjean F, Zheng F, Mobbs CV. Reversal of diabetic nephropathy by a ketogenic diet. PLoS One. 2011;6:e18604. doi: 10.1371/journal.pone.0018604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klein AH, Wendroth SM, Drewes LR, Andrews MT. Small-volume d-beta-hydroxybutyrate solution infusion increases survivability of lethal hemorrhagic shock in rats. Shock. 2010;34:565–72. doi: 10.1097/SHK.0b013e3181e15063. [DOI] [PubMed] [Google Scholar]
- 48.Mulier KE, Lexcen DR, Luzcek E, Greenberg JJ, Beilman GJ. Treatment with beta-hydroxybutyrate and melatonin is associated with improved survival in a porcine model of hemorrhagic shock. Resuscitation. 2012;83:253–8. doi: 10.1016/j.resuscitation.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 49.Zou Z, Sasaguri S, Rajesh KG, Suzuki R. dl-3-Hydroxybutyrate administration prevents myocardial damage after coronary occlusion in rat hearts. Am J Physiol Heart Circ Physiol. 2002;283:H1968–74. doi: 10.1152/ajpheart.00250.2002. [DOI] [PubMed] [Google Scholar]
- 50.Puchowicz MA, Zechel JL, Valerio J, Emancipator DS, Xu K, Pundik S, et al. Neuroprotection in diet-induced ketotic rat brain after focal ischemia. J Cereb Blood Flow Metab. 2008;28:1907–16. doi: 10.1038/jcbfm.2008.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tai KK, Nguyen N, Pham L, Truong DD. Ketogenic diet prevents cardiac arrest-induced cerebral ischemic neurodegeneration. J Neural Transm. 2008;115:1011–7. doi: 10.1007/s00702-008-0050-7. [DOI] [PubMed] [Google Scholar]
- 52.Blad CC, Tang C, Offermanns S. G protein-coupled receptors for energy metabolites as new therapeutic targets. Nat Rev Drug Discov. 2012;11:603–19. doi: 10.1038/nrd3777. [DOI] [PubMed] [Google Scholar]
- 53.Layden BT, Angueira AR, Brodsky M, Durai V, Lowe WL., Jr Short chain fatty acids and their receptors: new metabolic targets. Transl Res. 2013;161:131–40. doi: 10.1016/j.trsl.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 54.Tunaru S, Kero J, Schaub A, Wufka C, Blaukat A, Pfeffer K, et al. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat Med. 2003;9:352–5. doi: 10.1038/nm824. [DOI] [PubMed] [Google Scholar]
- 55.Taggart AK, Kero J, Gan X, Cai TQ, Cheng K, Ippolito M, et al. (D)-beta-Hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J Biol Chem. 2005;280:26649–52. doi: 10.1074/jbc.C500213200. [DOI] [PubMed] [Google Scholar]
- 56.Offermanns S, Colletti SL, Lovenberg TW, Semple G, Wise A, AP IJ. International Union of Basic and Clinical Pharmacology. LXXXII: Nomenclature and Classification of Hydroxy-carboxylic Acid Receptors (GPR81, GPR109A, and GPR109B) Pharmacol Rev. 2011;63:269–90. doi: 10.1124/pr.110.003301. [DOI] [PubMed] [Google Scholar]
- 57.Boden G. Obesity, insulin resistance and free fatty acids. Current opinion in endocrinology, diabetes, and obesity. 2011;18:139–43. doi: 10.1097/MED.0b013e3283444b09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dobbins RL, Shearn SP, Byerly RL, Gao FF, Mahar KM, Napolitano A, et al. GSK256073, a selective agonist of G-protein coupled receptor 109A (GPR109A) reduces serum glucose in subjects with type 2 diabetes mellitus. Diabetes, obesity & metabolism. 2013;15:1013–21. doi: 10.1111/dom.12132. [DOI] [PubMed] [Google Scholar]
- 59.Lukasova M, Malaval C, Gille A, Kero J, Offermanns S. Nicotinic acid inhibits progression of atherosclerosis in mice through its receptor GPR109A expressed by immune cells. J Clin Invest. 2011;121:1163–73. doi: 10.1172/JCI41651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zandi-Nejad K, Takakura A, Jurewicz M, Chandraker AK, Offermanns S, Mount D, et al. The role of HCA2 (GPR109A) in regulating macrophage function. Faseb J. 2013;27:4366–74. doi: 10.1096/fj.12-223933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kimura I, Inoue D, Maeda T, Hara T, Ichimura A, Miyauchi S, et al. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41) Proc Natl Acad Sci U S A. 2011;108:8030–5. doi: 10.1073/pnas.1016088108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Won YJ, Lu VB, Puhl HL, 3rd, Ikeda SR. beta-Hydroxybutyrate modulates N-type calcium channels in rat sympathetic neurons by acting as an agonist for the G-protein-coupled receptor FFA3. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:19314–25. doi: 10.1523/JNEUROSCI.3102-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Katada S, Imhof A, Sassone-Corsi P. Connecting threads: epigenetics and metabolism. Cell. 2012;148:24–8. doi: 10.1016/j.cell.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 64.He W, Newman JC, Wang MZ, Ho L, Verdin E. Mitochondrial sirtuins: regulators of protein acylation and metabolism. Trends Endocrinol Metab. 2012;23:467–76. doi: 10.1016/j.tem.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 65.Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–5. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Madiraju P, Pande SV, Prentki M, Madiraju SR. Mitochondrial acetylcarnitine provides acetyl groups for nuclear histone acetylation. Epigenetics : official journal of the DNA Methylation Society. 2009;4:399–403. doi: 10.4161/epi.4.6.9767. [DOI] [PubMed] [Google Scholar]
- 67.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–80. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Muoio DM, Noland RC, Kovalik JP, Seiler SE, Davies MN, DeBalsi KL, et al. Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metab. 2012;15:764–77. doi: 10.1016/j.cmet.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weinert BT, Scholz C, Wagner SA, Iesmantavicius V, Su D, Daniel JA, et al. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell reports. 2013;4:842–51. doi: 10.1016/j.celrep.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 70.Quant PA, Tubbs PK, Brand MD. Treatment of rats with glucagon or mannoheptulose increases mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase activity and decreases succinyl-CoA content in liver. Biochem J. 1989;262:159–64. doi: 10.1042/bj2620159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stein LR, Imai S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol Metab. 2012;23:420–8. doi: 10.1016/j.tem.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–36. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fontana L. Excessive adiposity, calorie restriction, and aging. Jama. 2006;295:1577–8. doi: 10.1001/jama.295.13.1577. [DOI] [PubMed] [Google Scholar]
- 74.Hatori M, Vollmers C, Zarrinpar A, DiTacchio L, Bushong EA, Gill S, et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab. 2012;15:848–60. doi: 10.1016/j.cmet.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hartman AL, Vining EP. Clinical aspects of the ketogenic diet. Epilepsia. 2007;48:31–42. doi: 10.1111/j.1528-1167.2007.00914.x. [DOI] [PubMed] [Google Scholar]
- 76.Mobbs CV, Mastaitis J, Isoda F, Poplawski M. Treatment of diabetes and diabetic complications with a ketogenic diet. Journal of child neurology. 2013;28:1009–14. doi: 10.1177/0883073813487596. [DOI] [PubMed] [Google Scholar]
- 77.Borghjid S, Feinman RD. Response of C57Bl/6 mice to a carbohydrate-free diet. Nutr Metab (Lond) 2012;9:69. doi: 10.1186/1743-7075-9-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bainbridge HW. The reduced sensitivity to insulin of rats and mice fed on a carbohydrate-free, excess-fat diet. J Physiol. 1925;60:293–300. doi: 10.1113/jphysiol.1925.sp002248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Burcelin R, Crivelli V, Dacosta A, Roy-Tirelli A, Thorens B. Heterogeneous metabolic adaptation of C57BL/6J mice to high-fat diet. Am J Physiol Endocrinol Metab. 2002;282:E834–42. doi: 10.1152/ajpendo.00332.2001. [DOI] [PubMed] [Google Scholar]
- 80.Kinzig KP, Honors MA, Hargrave SL. Insulin sensitivity and glucose tolerance are altered by maintenance on a ketogenic diet. Endocrinology. 2010;151:3105–14. doi: 10.1210/en.2010-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Garbow JR, Doherty JM, Schugar RC, Travers S, Weber ML, Wentz AE, et al. Hepatic steatosis, inflammation, and ER stress in mice maintained long term on a very low-carbohydrate ketogenic diet. Am J Physiol Gastrointest Liver Physiol. 2011;300:G956–67. doi: 10.1152/ajpgi.00539.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kennedy AR, Pissios P, Otu H, Roberson R, Xue B, Asakura K, et al. A high-fat, ketogenic diet induces a unique metabolic state in mice. Am J Physiol Endocrinol Metab. 2007;292:E1724–39. doi: 10.1152/ajpendo.00717.2006. [DOI] [PubMed] [Google Scholar]
- 83.Badman MK, Kennedy AR, Adams AC, Pissios P, Maratos-Flier E. A very low carbohydrate ketogenic diet improves glucose tolerance in ob/ob mice independently of weight loss. Am J Physiol Endocrinol Metab. 2009;297:E1197–204. doi: 10.1152/ajpendo.00357.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell. 2011;44:177–90. doi: 10.1016/j.molcel.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jornayvaz FR, Jurczak MJ, Lee HY, Birkenfeld AL, Frederick DW, Zhang D, et al. A high-fat, ketogenic diet causes hepatic insulin resistance in mice, despite increasing energy expenditure and preventing weight gain. Am J Physiol Endocrinol Metab. 2010;299:E808–15. doi: 10.1152/ajpendo.00361.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Srivastava S, Baxa U, Niu G, Chen X, Veech RL. A ketogenic diet increases brown adipose tissue mitochondrial proteins and UCP1 levels in mice. IUBMB Life. 2013;65:58–66. doi: 10.1002/iub.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Freedland SJ, Mavropoulos J, Wang A, Darshan M, Demark-Wahnefried W, Aronson WJ, et al. Carbohydrate restriction, prostate cancer growth, and the insulin-like growth factor axis. Prostate. 2008;68:11–9. doi: 10.1002/pros.20683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mavropoulos JC, Buschemeyer WC, 3rd, Tewari AK, Rokhfeld D, Pollak M, Zhao Y, et al. The effects of varying dietary carbohydrate and fat content on survival in a murine LNCaP prostate cancer xenograft model. Cancer Prev Res (Phila) 2009;2:557–65. doi: 10.1158/1940-6207.CAPR-08-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McDaniel SS, Rensing NR, Thio LL, Yamada KA, Wong M. The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia. 2011;52:e7–11. doi: 10.1111/j.1528-1167.2011.02981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nordmann AJ, Nordmann A, Briel M, Keller U, Yancy WS, Jr, Brehm BJ, et al. Effects of low-carbohydrate vs low-fat diets on weight loss and cardiovascular risk factors: a meta-analysis of randomized controlled trials. Arch Intern Med. 2006;166:285–93. doi: 10.1001/archinte.166.3.285. [DOI] [PubMed] [Google Scholar]
- 91.Paoli A, Rubini A, Volek JS, Grimaldi KA. Beyond weight loss: a review of the therapeutic uses of very-low-carbohydrate (ketogenic) diets. European journal of clinical nutrition. 2013;67:789–96. doi: 10.1038/ejcn.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ajala O, English P, Pinkney J. Systematic review and meta-analysis of different dietary approaches to the management of type 2 diabetes. Am J Clin Nutr. 2013;97:505–16. doi: 10.3945/ajcn.112.042457. [DOI] [PubMed] [Google Scholar]
- 93.Harvie M, Wright C, Pegington M, McMullan D, Mitchell E, Martin B, et al. The effect of intermittent energy and carbohydrate restriction v. daily energy restriction on weight loss and metabolic disease risk markers in overweight women. The British journal of nutrition. 2013:1–14. doi: 10.1017/S0007114513000792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Westman EC, Yancy WS, Jr, Mavropoulos JC, Marquart M, McDuffie JR. The effect of a low-carbohydrate, ketogenic diet versus a low-glycemic index diet on glycemic control in type 2 diabetes mellitus. Nutr Metab (Lond) 2008;5:36. doi: 10.1186/1743-7075-5-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Frigolet ME, Ramos Barragan VE, Tamez Gonzalez M. Low-carbohydrate diets: a matter of love or hate. Annals of nutrition & metabolism. 2011;58:320–34. doi: 10.1159/000331994. [DOI] [PubMed] [Google Scholar]