

Abstract
The human aldehyde dehydrogenase (ALDH) superfamily consists of at least 19 enzymes that metabolize endogenous and exogenous aldehydes. Currently, there are no commercially available inhibitors that target ALDH1A1 but have little to no effect on the structurally and functionally similar ALDH2. Here we present the first human ALDH1A1 structure, as the apoenzyme and in complex with its cofactor NADH to a resolution of 1.75 Å and 2.1 Å, respectfully. Structural comparisons of the cofactor binding sites in ALDH1A1 with other closely related ALDH enzymes illustrate a high degree of similarity. In order to minimize discovery of compounds that inhibit both isoenzymes by interfering with their conserved cofactor binding sites, this study reports the use of an in vitro, NAD+-independent, esterase-based high-throughput screen (HTS) of 64,000 compounds to discover novel, selective inhibitors of ALDH1A1. We describe 256 hits that alter the esterase activity of ALDH1A1. The effects on aldehyde oxidation of 67 compounds were further analyzed, with 30 selectively inhibiting ALDH1A1 compared to ALDH2 and ALDH3A1. One compound inhibited ALDH1A1 and ALDH2, while another inhibited ALDH1A1, ALDH2, and the more distantly related ALDH3A1. The results presented here indicate that this in vitro enzyme activity screening protocol successfully identified ALDH1A1 inhibitors with a high degree of isoenzyme selectivity. The compounds identified via this screen plus the screening methodology itself represent a starting point for the development of highly potent and selective inhibitors of ALDH1A1 that may be utilized to better understand the role of this enzyme in both normal and disease states.
Keywords: aldehyde dehydrogenase, retinaldehyde dehydrogenase, high-throughput screen, small molecule inhibitors, enzyme inhibition
1. Introduction
The aldehyde dehydrogenase (ALDH) superfamily of enzymes primarily catalyze the NAD(P)+-dependent oxidation of an aldehyde to its corresponding carboxylic acid[1]. The human genome has at least 19 ALDHs. A primary function of the ALDH1A subfamily (ALDH1A1, ALDH1A2, and ALDH1A3), whose members share over 70% protein sequence identity, is the oxidation of retinaldehyde to retinoic acid, a critical regulator in a number of cell growth and differentiation pathways. Other aldehydes also serve as substrates for ALDH1A1, including acetaldehyde during ethanol metabolism, 3,4-dihydroxyphenylacetaldehyde (DOPAL) in dopamine metabolism, and (±)-4-hydroxy-2E-nonenal (4-HNE), a toxic by-product of oxidative stress pathways. ALDH1A1 has been associated with a number of diseases. Down-regulation of ALDH1A1 has been reported in Parkinson’s disease, possibly due to the build-up of the neurotoxic aldehyde DOPAL in dopamine metabolism[2, 3]. ALDH1A1 knockout mice are able to resist diet-induced obesity[4], while rodents given the nonselective ALDH1A1 inhibitor citral also exhibit reduced weight gain[5], indicating that ALDH1A1 is playing a role in obesity and/or adipogenesis. Up-regulation of ALDH1A1 is a biomarker for both normal and cancer stem cells, but the role of ALDH1A1 in establishing and/or maintaining stem cells is not known[6–9]. ALDH1A1 and ALDH3A1 have long been linked to cancer drug resistance due to their roles in the metabolism of the anticancer agent cyclophosphamide[10]. It is evident that ALDH1A1 is involved in a number of biological processes, but its contributions to both normal and disease states, including retinoid-dependent processes, are not clearly understood. The development of selective activators and inhibitors of ALDH1A1 would provide chemical tools to help decipher the role of this enzyme. However, at this time there are no commercially available, ALDH1A1-selective modulators.
The development of compounds that selectively target ALDH1A1 has proven to be difficult as the ALDH superfamily of enzymes shares many common structural and mechanistic features. These members generally function as homodimers or homotetramers, with each subunit containing three structural domains, a catalytic domain, a cofactor binding domain, and an oligomerization domain. The NAD(P)+ binding domain is a Rossmann-fold, a nucleotide binding site that consists of two sets of parallel beta sheets and alpha helices. The Rossmann-fold structure motif is found in the NAD+ binding domains of multiple dehydrogenase families, including ALDHs, lactate dehydrogenases, alcohol dehydrogenases, and glyceraldehyde-3-phosphate dehydrogenase[11–13]. There are differences in the Rossmann fold between ALDH and other oxidoreductases that could possibly be exploited for the development of small molecule modulators of various ALDH isoenzymes compared to other NAD+-binding enzyme families[14]. However, there exists much structural similarity in the NAD+- binding site within the ALDH family and the development of selective modulators that target this site may present difficulties.
A number of ALDH’s, including ALDH1A1 also possess esterase activity. Based on the ALDH2 sequence, site-directed mutagenesis has shown that Cys-302 is the essential nucleophile for both the esterase and dehydrogenase reaction, with Glu-268 acting as the general base to activate Cys-302[15, 16]. The proposed catalytic steps for both the dehydrogenase and esterase reactions have been recently reviewed[17], although minor details still need to be resolved including the roles of second sphere residues in assisting proton transfer to solvent[18, 19]. The use of common active site residues for the two reactions makes it likely that modulators of the esterase reaction would also modulate aldehyde oxidation activity. In support of this hypothesis, the ALDH2 activator Alda-1 activates both the esterase and dehydrogenase activity of the enzyme and daidzen inhibits both reactions[13, 20, 21]. An additional advantage of the esterase reaction is that it does not require the cofactor NAD+ to be present, and so allows the screen to be less influenced by compounds binding to this site.
The human ALDH1 family, which shares over 60% protein sequence identity, is a particularly difficult challenge for inhibitor development since it contains the highest number of orthologs in the genome at seven (ALDH1A1, ALDH1A2, ALDH1A3, ALDH1B1, ALDH1L1, ALDH1L2, and ALDH2). Compounds such as diethylaminobenzaldehyde (DEAB) and disulfiram are potent inhibitors of ALDH1A1, with IC50’s in the nM range, but both also inhibit ALDH2[17, 22]. DEAB is also a relatively potent inhibitor for a number of other ALDH1 family members, although not ALDH1L1 [22, 23]. An in vitro high throughput screen (HTS) is one method of discovering novel, small molecule modulators for a particular enzyme. Typically, the rate of aldehyde oxidation by ALDHs is studied by monitoring the formation of NADH at 340 nm on a spectrophotometer (molar extinction coefficient of 6220 M−1 cm−1) (Figure 1A). However, this approach is not ideal for the screening assay as it is common for compounds in the libraries to absorb light in the same wavelength range as NADH and leads to interference in this analytical approach. Therefore, another assay design is needed for an ALDH1A1 HTS. One approach is to couple aldehyde oxidation to a second reaction that can be monitored by either fluorescence or UV/Vis spectrophotometry. For example, the dehydrogenase activity of ALDH2 was coupled to the NADH-dependent reduction of resazurin to resorufin to discover the ALDH2 activator Alda-1[20]. However, a second approach would be to use the inherent esterase activity of ALDH1A1 to identify modulators. The ALDH1A1 ester substrate para-nitrophenylacetate (pNPA) is hydrolyzed to p-nitrophenol, which absorbs light at 405 nm and can be monitored spectrophotometrically, with minimal interference from library compounds (Figure 1B).
Figure 1.
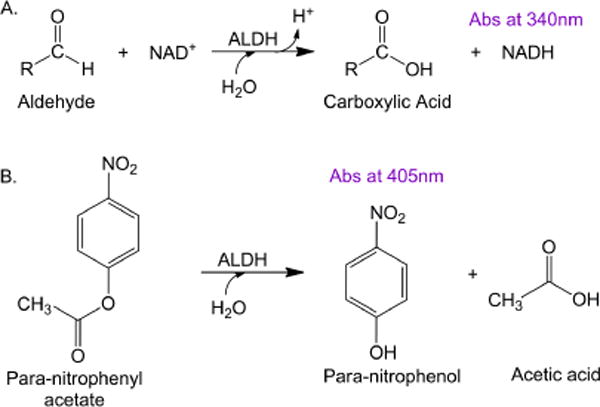
Reactions used to discover ALDH1A1 modulators. A. NAD+-dependent aldehyde oxidation reaction monitored formation of NADH at 340 nm. B. HTS used an NAD+-independent esterase reaction that monitored the formation of p-nitrophenol at 405 nm.
In this paper, we used an in vitro esterase assay to identify compounds that modulate ALDH1A1 activity but have little to no effect on either ALDH2, an isoenzyme that has approximately 70% protein sequence identity with ALDH1A1, or ALDH3A1, a more distantly related isoenzyme with 30% protein sequence identity. Comparison of the cofactor binding sites of human ALDH1A1 and ALDH2 points to a high degree of similarity, suggesting that development of selective modulators that bind at this location would be challenging. Use of the esterase assay allowed us to minimize two potential problems: 1) identification of compounds that bind to the highly conserved cofactor site, and 2) monitor activity at a wavelength with minimal spectral overlap to that of the library compounds. Of the 64,000 compounds screened, 256 were identified as modulators of ALDH1A1 esterase activity. We examined the dehydrogenase activity and selectivity of 67 hits and nearly half selectively inhibited ALDH1A1 dehydrogenase activity. These results indicate that this simple esterase-based in vitro HTS was successful in identifying novel, selective inhibitors of ALDH1A1.
2. Materials and Methods
2.1 Materials
All chemicals and reagents including para-nitrophenylacetate, propionaldehyde, NAD+, and buffers were purchased from Sigma Aldrich unless where noted otherwise.
2.2 Expression and Purification of ALDH Proteins
ALDH1A1, ALDH2, and ALDH3A1 were prepared as described elsewhere[24–26]. Protein used for kinetics was flash frozen in liquid nitrogen and stored at −80°C. ALDH1A1 protein used for X-ray crystallography was stored at −20°C in a 50% (v/v) solution with glycerol and dialyzed against 10 mM Na+-ACES pH 6.6 and 1 mM dithiothreitol at 4°C. The ALDH1A1 protein used for the screen was produced from a cDNA obtained from Dr. Henry Weiner containing a known A-to-G SNP at position 72928972 on chromosome 9 (NCBI rs1049981), resulting in an Asn-to-Ser missense mutation at protein position 121[27]. This SNP has been found in a small percentage of the HapMap-CEU population representing Utah residents with Northern and Western European ancestry, but there is no known clinical significance to the mutation. The NCBI reference sequence for ALDH1A1 (wild-type) was constructed using the forward primer 5′- CTC TAT TCC AAT GCA TAT CTG AAT GAT TTA GCA GGC TGC ATC -3′ and its complement, using the QuikChange site-directed mutagenesis protocol. Unless where noted otherwise, ALDH1A1 WT protein was used for all aldehyde oxidation assays and the X-ray crystallography of the ALDH1A1-NADH structure. ALDH1A1-N121S was used for the HTS and the apo-enzyme structure. For the kinetic assays, although the enzymes have more activity at a higher pH, a more physiologically relevant pH of 7.5 was used for both the HTS and dehydrogenase assays. This also kept the spontaneous hydrolysis of the ester substrate to a minimum and allowed direct comparison between the esterase and dehydrogenase assays.
2.3 Structural determination of human ALDH1A1
For the apo-enzyme structure, crystals of ALDH1A1 N121S at 3–5 mg/mL concentration were equilibrated against a crystallization solution of 100 mM sodium BisTris, pH 6.2–7.0, 8–12% PEG3350 (Hampton Research), 200 mM NaCl, and 5–10 mM YbCl3 at 25°C. Freezing of the crystals occurred in crystallization solution plus 20% (v/v) ethylene glycol. For the ALDH1A1-NADH structure, apo-enzyme crystals (WT) were prepared in the same manner as ALDH1A1 N121S crystals and were soaked for 2 hours with crystallization solution containing 1 mM NAD+. Freezing of the crystals occurred in crystallization solution with NAD+ plus 20% (v/v) ethylene glycol. Diffraction data was collected at Beamline 19-ID operated by the Structural Biology Consortium at the Advanced Photon Source, Argonne National Laboratory. Diffraction data were indexed, integrated, and scaled using either the HKL2000 or HKL3000 program suites[28]. The CCP4 program suite[29] was used for molecular replacement and refinement, using the sheep ALDH1 structure (PDB Code 1BXS) as a model for the apo-ALDH1A1 structure. The Coot molecular graphics application[30] was used for model building and the TLSMD (Translation/Libration/Screw Motion Determination) server was used to determine dynamic properties of the protein[31, 32].
2.4 Esterase based high throughput screen on ALDH1A1
The high throughput screen was performed in 384-well, clear-bottomed plates, monitoring the change in absorbance of p-nitrophenol at 405 nm wavelength (molar extinction coefficient of 18000 M−1 · cm−1) on a Spectramax plate reader. The chemical library consisted of 64,000 compounds from ChemDiv Corp (San Diego, CA) at a final concentration of 10 μM. The 50 μL assay contained 730 nM ALDH1A1, 800 μM substrate para-nitrophenylacetate (pNPA), 10 μM compound, and 2% DMSO in 25 mM Na+-HEPES, pH 7.5 at 25°C. The non-selective ALDH1A1 inhibitor Aldi-1[33] at 25 μM final concentration was used as a positive control of ALDH1A1 esterase inhibition in each plate. Following a 2 minute incubation of enzyme and compound, the reaction was initiated by adding the substrate pNPA and monitored for 7 minutes. A Z-factor for the HTS was calculated by comparing the values of ALDH1A1 plus/minus Aldi-1 under the conditions of the HTS assay, each at n = 384 to determine the quality of the HTS conditions. A second control using no enzyme was also performed to determine whether our control inhibitor concentration had nearly 100% inhibition. An activator was defined as having 2-fold or higher esterase activity compared to control, while an inhibitor had 50% or less activity. After one round of screening, compounds identified as activators and inhibitors were rescreened using the same protocol and cutoffs to confirm the initial readings.
2.5 ALDH1A1, ALDH2, and ALDH3A1 aldehyde oxidation activity assays
Hits from the HTS were ordered from ChemDiv to determine if they had any effect on aldehyde oxidation and whether they were selective for ALDH1A1 compared to ALDH2 and ALDH3A1. Dehydrogenase activity of the three isoenzymes were assayed by monitoring the production of NADH at 340 nm (molar extinction coefficient of 6220 M−1 · cm−1) on a Beckman DU-640 or Cary 300 Bio UV-Vis spectrophotometer for 2 to 3 minutes. For ALDH1A1 and ALDH2, the reaction contained 100–200 nM enzyme, 200 μM NAD+, 100 μM propionaldehyde, and 1% DMSO in 50 mM Na+ BES, pH 7.5 at room temperature. For ALDH3A1, the reaction contained 20 nM enzyme, 200 μM NAD+, 300 μM benzaldehyde, and 1% DMSO in either 100 mM sodium phosphate or 50 mM Na+ BES at pH 7.5 at room temperature. For most compounds, 20 μM concentration was used for the selectivity assays. However, due to solubility issues for CM307, 10 μM of compound was used. Following a 2 minute incubation of enzyme, compound, and NAD+, the reaction was initiated by adding substrate. For compounds with over 60% inhibition at 20 μM, IC50 values for propionaldehyde oxidation were calculated by varying the concentration of the compounds from 0–200 μM under the same conditions as the selectivity assays. Data were fit to the four parameter EC50 equation using SigmaPlot (StatSys v12.3).
3. Results
3.1 Structure of human ALDH1A1
X-ray crystallography was used to compare the structure of human ALDH1A1 with other members of the ALDH enzyme superfamily. The structure of human ALDH1A1 had not been previously reported (Figure 2 and Table 1, PDB Code 4WJ9). As expected, it is highly similar to both the human ALDH2 enzyme (PDB code 3N80), with which ALDH1A1 shares about 70% sequence identity, and the sheep ALDH1A1 (PDB code 1BXS), with over 90% sequence identity. The structure of ALDH1A1 with NADH was determined to a resolution of 2.1 Å (Figure 3 and Table 1, PDB Code 4WB9). A comparison of the respective alpha-carbons in the structure of the N121S apo-enzyme and those of the wild-type ALDH1A1 structure complexed with NADH, generated an RMSD of 0.2 Å, consistent with a high degree of functional and structural similarity. The side chains of Ser and Asn both form similar hydrogen bonding interactions with Tyr297. Although wild-type apo-crystals were soaked with NAD+, the cofactor is bound in the hydrolysis position, characteristic of NADH binding, in the structure which suggests it could have been reduced via oxidation of PEG aldehydes. The hydrolysis conformation observed here is similar to that seen in ALDH2 (PDB Code 1O02)[25] and the sheep ALDH1A1 (PDB Code 1BXS)[34] with cofactor, with the exception of the interaction of Glu-349 with a ytterbium cation bound to the pyrophosphate of NADH. Comparison of the structure of ALDH1A1, ALDH2, and ALDH3A1 (PDB Code 4L2O) with cofactor illustrates the difficulty of developing selective inhibitors for ALDH1A1 that target this site. There is a high degree of similarity between the cofactor binding sites of ALDH1A1 and ALDH2 (Figure 4), supporting our hypothesis to select an assay independent of the cofactor binding site in order to develop selective inhibitors for the ALDH1/2 class of enzymes. ALDH3A1 is the least similar both by structural topology and sequence identity, and as expected based on its ability to utilize both NAD+ and NADP+, these differences are most obvious near the adenosine ribose and pyrophosphate binding site.
Figure 2.
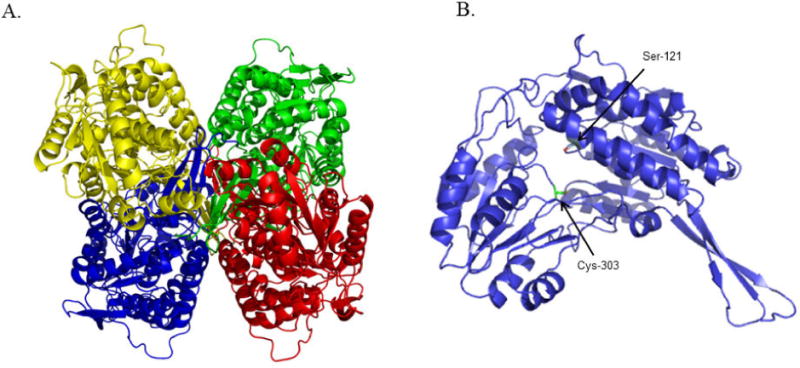
Structure of human ALDH1A1 (N121S) apo-enzyme. (A) Ribbon representation of the structure of the homotetrameric ALDH1A1 with each monomer colored separately. (B) Ribbon representation of an ALDH1A1 monomer showing the location of cysteine 303 in the active site plus the location of ALDH1A1 N121S used for the HTS (PDB Code 4WJ9).
Table 1.
Data collection and refinement statistics of ALDH1A1-NADH.
| Data Collection | Apo-ALDH1A1 N121S (PDB 4WJ9) |
ALDH1A1-NADH Wild-type (PDB 4WB9) |
|---|---|---|
| Space Group | P422 | P422 |
| Cell Dimensions | ||
| a,b,c [Å] | 109, 109, 83 | 109, 109, 83 |
| α,β,γ [°] | 90, 90, 90 | 90, 90, 90 |
| Resolution [Å] | 50 – 1.75 | 50 – 2.1 |
| Rmerge | 0.056 (0.59) | 0.09 (0.52) |
| I/σi | 31.5 (3.9) | 17.8 (4.9) |
| Completeness [%] | 99 (97) | 99 (100) |
| Redundancy | 9.6 (8.5) | 8.3 (7.9) |
| Refinement | ||
| No. of Reflections | 48862 | 29814 |
| Rwork/Rfree | 0.19/0.21 | 0.18/0.23 |
| No. of Atoms | ||
| Protein | 3839 | 3837 |
| Ligand/Ions | 2 | 50 |
| Water | 246 | 215 |
| R.M.S. Deviations | ||
| Bond Lengths [Å] | 0.010 | 0.017 |
| Bond Angles [°] | 1.21 | 1.8 |
Numbers in parenthesis represent value of highest resolution shell.
Figure 3.
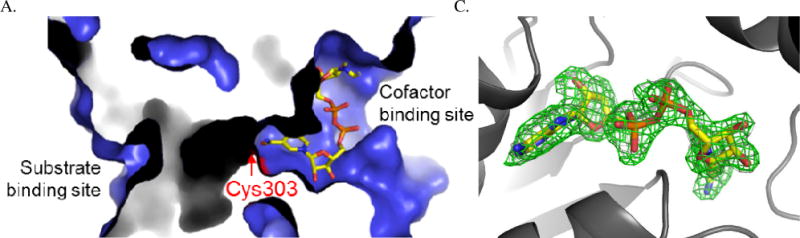
Structure of ALDH1A1 with reduced cofactor NADH. The location of the active site Cys-303 is shown in red. A. Surface rendition of NADH near the active site Cys-303. B Electron density maps of NADH with the original Fo – Fc in green contoured at 2.5 standard deviations and the final 2Fov – Fc map in grey contoured at 1.0 standard deviations.
Figure 4.
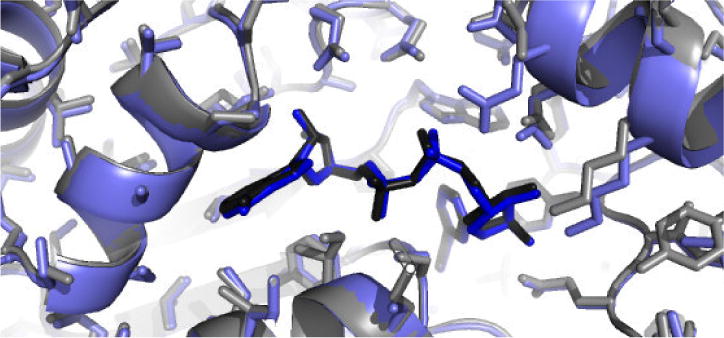
Overlap of the structure of ALDH1 A1 + NADH, in blue, with the structure of ALDH2 + NADH, in grey.
3.2 High throughput screen to identify modulators of ALDH1A1 esterase activity
The Z-factor for the HTS comparing ALDH1A1 plus/minus inhibitor (Aldi-1) under screening conditions was 0.67 (n = 384), indicating the screen is capable of identifying inhibitors from single assays. As shown in Figure 5, there is a clear separation between the control reaction containing enzyme and substrate (ES Control) represented in blue, and the inhibitor control reaction containing enzyme, substrate plus an ALDH1A1 inhibitor (ESI control) represented in red. Also, the average value for ALDH1A1 with control inhibitor was similar to the no enzyme, blank control (mean rate of change of 0.70 vs 0.60), indicating our inhibition control (25 μM Aldi-1, IC50 = 2.2 μM[33]) strongly inhibited ALDH1A1. For the HTS, we used an ALDH1A1 protein with a known SNP at residue 121. This N121S “mutant” is the open reading frame cloned by the Weiner group [26] and utilized for all their published work on ALDH1A1. The enzyme is active and behaved similarly to ALDH1A1 WT (Km of 12 μM vs 15 μM, respectfully, with identical kcat/Km values at 2.7 min−1 · μM−1 for the substrate propionaldehyde). The screen used a saturating amount of the esterase substrate pNPA (Km = 5μM[35]). Each plate contained a control column with enzyme and substrate (ES control) and the average (n = 16) of this intraplate control served as the basis to determine whether a compound modified esterase activity. An activator was defined as having 2-fold or higher esterase activity compared to this control, while an inhibitor had 50% or less activity. Each plate also contained a positive control for inhibition (ESI control) using the inhibitor Aldi-1. The initial round of the in vitro es terase-based screen of 64000 compounds yielded 631 compounds that activated ALDH1A1 and 278 compounds that inhibited ALDH1A1. A sample plate from the first round of screening is shown in Figure 6, illustrating 3 activators and 1 inhibitor out of 352 compounds tested. Following rescreening of the 909 compounds identified in the first round under identical conditions, nearly 75% did not meet these same selection criteria during the second, validation assay set. After two rounds, the esterase screen identified 241 activators and 15 inhibitors of ALDH1A1 esterase activity (Supplementary Tables 1 and 2).
Figure 5.
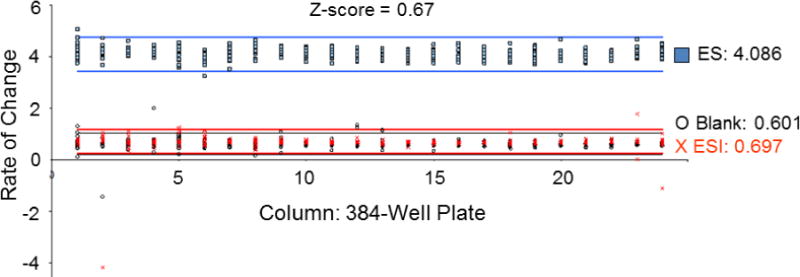
Z-factor determination for esterase screen. Each point represents the rate of change in absorbance at 405 nm of a reaction. The x-axis is the column (1 – 24) on the 384-well plate of the reaction. The blue data points represent the enzyme + substrate (ES) control, with an average value of 4.086; the red is enzyme + substrate + inhibitor, with an average value of 0.697; the open circles are the no enzyme control (blank). The lines represent 3× standard deviation from ES control (blue lines), ESI control (red lines), and blank (black lines). Each condition (ES, ESI, blank) performed on a separate plate with n = 384.
Figure 6.
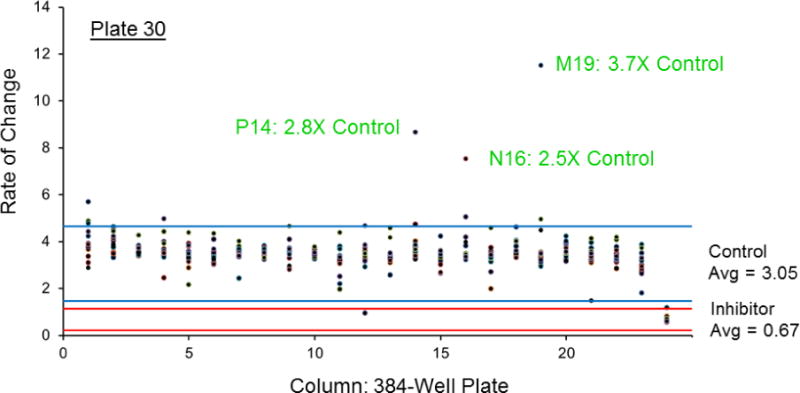
Representative plate from esterase HTS. Each point represents one well, with the x-axis the column (1–24) on the plate and the y-axis, the rate of change measured at wavelength 405 nm. Column 23 is the ES control, with an average value of 3.05 (n=16). Column 24 is the inhibition (ESI) control containing 25 μM Aldi-1. For this plate, an activator had a value ≥ 6.1 while an inhibitor had a value ≤ 1.22. Lines are 3× standard deviation, blue for ES and red for ESI. On this plate, we identified 3 activators (P14, N16, M19) and 1 inhibitor (D12) out of 352 compounds, with labeling based on their row and column on 384-well plate.
3.3 ALDH1A1, ALDH2, and ALDH3A1 aldehyde oxidation activity assays
The 256 compounds were grouped based on structural similarities. From this set of compounds, we selected 57 esterase activators and 10 esterase inhibitors and tested their ability to alter aldehyde oxidation (Figure 7). Of the 15 esterase inhibitors identified by HTS, only eight were commercially available. However, close analogs of three were purchased and analyzed, hit 3343–2924 was substituted by 2188–3302 (CM310), hit C699-0615 was substituted by C699-0244 (CM306), and hit K788-2754 was substituted by K938-0803 (CM307). The effects the hits have on aldehyde oxidation were tested using the standard assays performed in our laboratory to study these three ALDH isoenzymes. For ALDH1A1 and ALDH2, 100 μM propionaldehyde is near to saturation (ALDH1A1 Km ~15 μM and ALDH2 Km < 1 μM). For ALDH3A1, the concentration of benzaldehyde used was set at its Km. None of the 67 compounds tested activated aldehyde oxidation by ALDH1A1, ALDH2, or ALDH3A1 by more than 20%. However, of the 57 esterase activators examined at 20 μM concentration, 28 inhibited ALDH1A1 propionaldehyde oxidation at least 50%. Of the 10 esterase inhibitors tested at 20 μM concentration, four inhibited ALDH1A1 propionaldehyde oxidation at least 50%, but two inhibitors (CM302 and CM303) also exhibited at least 50% inhibition of ALDH2 and therefore were not selective for ALDH1A1. To a limited degree, CM302 also inhibited ALDH3A1 but none of the remaining 66 hits altered ALDH3A1 benzaldehyde oxidation more than 20% from control. Based on the selectivity assays of 67 esterase hits, 30 compounds selectivity inhibited ALDH1A1 compared to ALDH2 and ALDH3A1, while two compounds inhibited both ALDH1A1 and ALDH2 at least 50% but not ALDH3A1.
Figure 7.
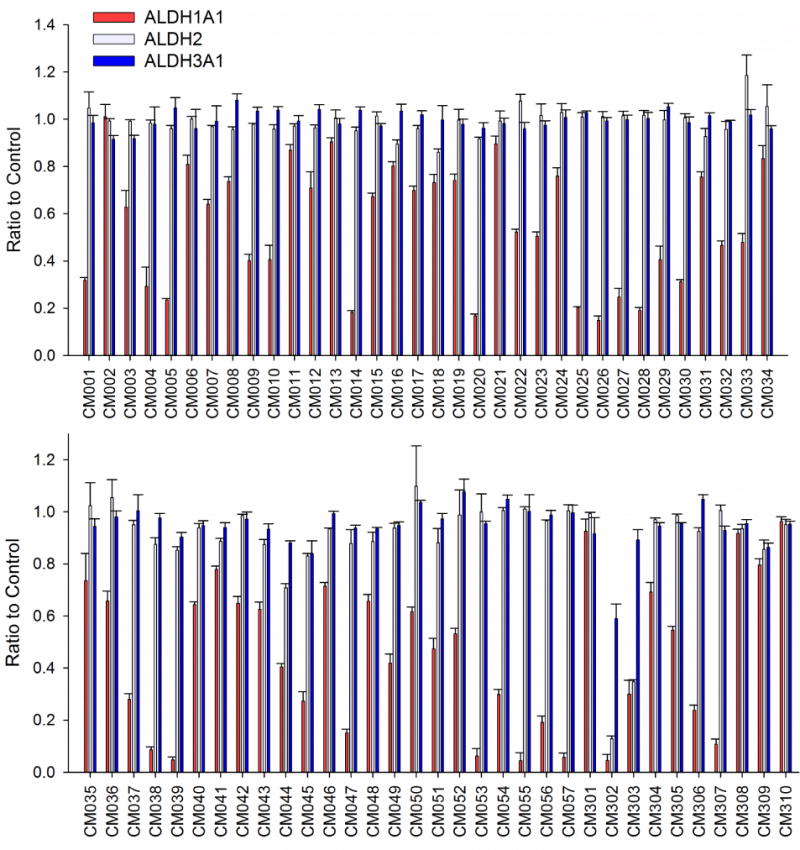
The effect on dehydrogenase activity of 67 compounds identified via an esterase HTS on three ALDH isoenzymes. The reactions used 20 μM compound and each bar represents mean/SEM (n = 3). Only one compound for ALDH3A1 and two compounds for ALDH2 altered the respective activity of these enzymes more that 20%, while nearly half inhibited ALDH1A1 at least 50%.
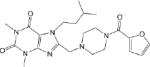
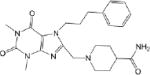
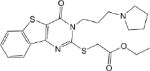
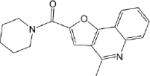
IC50 values were determined for compounds that inhibited propionaldehyde oxidation at least 60% at 20 μM concentration, with the most potent inhibitors and their IC50 values shown in Table 2. Of the 57 esterase activators, 17 were structurally similar (CM022-031, CM051-057) with all but one (CM024) inhibiting ALDH1A1 at 20 μM compound concentration. Based on IC50 values, the most potent inhibitors selective for ALDH1A1 were CM038 and two structural analogs, CM053 and CM055, with all three hits having IC50 values less than 300 nM. CM0302 was a potent inhibitor of both ALDH1A1 and ALDH2, with IC50 values of 1.0 ± 0.1 μM and 2.2 ± 0.3 μM, respectfully. To a limited extent, CM302 also inhibited ALDH3A1, but with an IC50 value greater than 10-fold higher compared to ALDH1A1 and ALDH2. In comparison, the nonselective inhibitor Aldi-1, which was used as a control during the esterase HTS, has an IC50 value of 2.2 μM[33]. DEAB is a nonselective ALDH1 inhibitor used as a control for the ALDEFLUOR Assay (Stemcell Technologies, Vancouver, Canada), a flow cytometry assay commonly used to identify stem cells based on ALDH activity. DEAB has an IC50 value of approximately 60 nM under these same conditions, but is also a potent inhibitor of other ALDH isoenzymes[23].
Table 2.
IC50 values with ALDH1A1 for compounds that inhibit dehydrogenase activity.
| Compound IC50 [μM] |
Structure |
|---|---|
| CM001 1.1 ± 0.1 * |
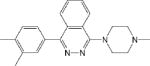
|
| CM009 5.3 ± 0.3 * |
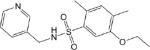
|
| CM010 1.3 ± 0.1 * |
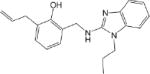
|
| CM020 0.45 ± 0.10 |
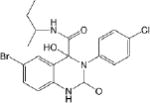
|
| CM025 2.1 ± 0.7 |
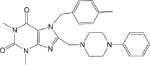
|
| CM026 0.80 ± 0.06 |
|
| CM028 2.0 ± 0.1 |
|
| CM037 4.6 ± 0.8 |
|
| CM038 0.26 ± 0.01 |
|
| CM039 0.41 ± 0.01 |
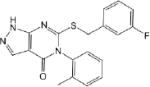
|
| CM045 2.5 ± 0.5 * |
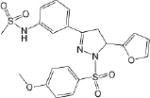
|
| CM047 0.31 ± 0.03 * |
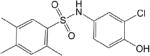
|
| CM053 0.21 ± 0.04 |
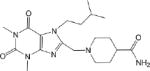
|
| CM055 0.24 ± 0.04 |
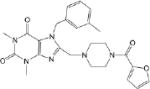
|
| CM056 5.4 ± 0.8 |
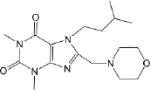
|
| CM057 0.92 ± 0.2 |
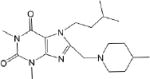
|
| CM302 1.1 ± 0.1 |
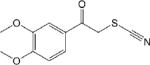
|
| CM306 3.5 ± 0.6 |
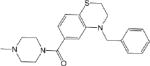
|
| CM307 0.57 ± 0.09 |
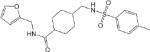
|
Each value represents mean/SEM for three independent assays, each n = 3. Values calculated using 100 μM Propionaldehyde and 200 μM NAD+.
Maximum inhibition < 70%.
Discussion
Comparison of the structures of human ALDH1A1, ALDH2, and ALDH3A1 indicate they exhibit a high degree of structural similarity, but demonstrate distinct differences within their substrate binding sites. In contrast, their respective coenzyme binding sites are less dissimilar, especially between ALDH1A1 and ALDH2 (Figure 4). This supports our screening approach to avoid identifying compounds that interact at this location, as they are less likely to be selective for ALDH1/2 class members. However, it might be possible to utilize this approach for inter-class selectivity (Figure 8).
Figure 8.
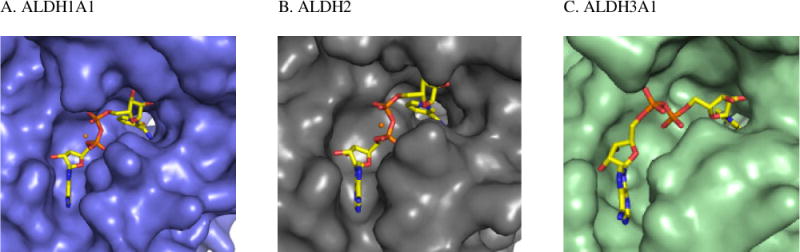
Surface topography of the cofactor binding site for ALDH1A1, ALDH2 (PDB 1O02), and ALDH3A1 (PDB 4L2O). The orange sphere in ALDH1A1 and ALDH2 represent cations, Yb for ALDH1A1 and Mg for ALDH2, that are present during crystallization.
The esterase screen used in this study was modeled after a previously reported screen for ALDH3A1 inhibitors that successfully identified two classes of selective ALDH3A1 inhibitors capable of increasing mafosfamide sensitivity in cancer cells[26, 36, 37]. By adapting this assay to ALDH1A1, we screened a 64,000 compound library and following one round of screen, identified over 900 compounds. Rescreening of these compounds under identical conditions resulted in 256 confirmed hits that modified ALDH1A1 esterase activity. Therefore, the effect on esterase activity of <30% of the identified activators/inhibitors identified in round one were successfully repeated in round two. Although these replicability results may seem low, HTS are inherently noisy to begin with, producing many false positives that can be eliminated in the second round. As shown in Figure 5, simply calculating the Z-factor produced outliers despite identical conditions within one plate. Some reasons for poor replicability include inaccuracies in compound concentration, spectral interference from the compounds, errors in robot pipetting, and debris or bubbles in the well that interfered with the reading. The second round of screening is designed to remove these false positives from consideration, conserving both time and resources. Since the HTS identified 256 compounds, the large number of false positives from round one was not a concern. We examined the effect on dehydrogenase activity of 67 of these compounds and found that 30 selectively inhibited ALDH1A1 compared to ALDH2 and ALDH3A1, while 2 inhibited both ALDH1A1 and ALDH2. Therefore, nearly 50% of the esterase modifiers identified also altered aldehyde oxidation and almost all of the compounds did so selectively for ALDH1A1 compared to two other ALDH’s.
Of the 57 esterase activators tested, none activated the dehydrogenation reaction of ALDH1A1, but nearly half inhibited it. The esterase reaction is independent of NAD+, but the presence of either NAD+ or NADH will increase the rate of ester hydrolysis, depending on assay conditions[35, 38]. The substrate and cofactor binding sites are linked to the active site by a tunnel through the enzyme. For ester hydrolysis, the substrate can likely enter the active site via either end of this tunnel. To activate esterase activity, it is proposed that cofactor binding slows transit of the ester substrate out of the tunnel, increasing the number of productive encounters with the active site nucleophile and also possibly by directly activating the nucleophile (Figure 2B) [13]. Compounds that function as esterase activators but dehydrogenase inhibitors likely bind to the substrate-binding end of this tunnel. In a manner similar to activation via cofactor binding, compound binding slows the transit of pNPA out of the active site tunnel and increases the likelihood of a productive encounter with the active site cysteine. However, the effect these esterase activators have on the NAD+-dependent aldehyde oxidation reaction is the opposite. Binding of the compound along with cofactor binding alters access to the active site at both ends and therefore depending on the structure of the compound could inhibit dehydrogenase activity. However, as seen with the ALDH2 activator Alda-1, a compound that binds at the substrate binding end of the active site tunnel could also result in a dehydrogenase activator, depending on binding position, location relative to the active site residues and substrate size[13]. It is possible that a number of our esterase activators that had no effect on aldehyde oxidation acted at the cofactor binding site, activating the esterase reaction like NAD+/NADH. However, the levels of NAD+ used in the assays (approximately 4 × KM) might minimize their effect on aldehyde oxidation. If a compound did bind at the cofactor site, only an extremely potent or covalent modulator would be identified under these conditions.
Of the 241 esterase activators identified, 78 were structural analogs with a common xanthine ring core structure. Of these 78 compounds, 17 were tested (CM022-031, CM051-057) and 16 selectively inhibited dehydrogenase activity of ALDH1A1 with no effect on either ALDH2 or ALDH3A1. The esterase HTS also produced 8 other structural groups containing between 7 and 20 analogs each. As a consequence, 65% of the esterase hits could be classified into 9 structural groups (Table 3). There were an additional 8 structural classes containing between 2 – 6 analogs and 22 structurally unique compounds.
Table 3.
Structural classes of hit compounds
| Structure | HTS Hits | Dehydrogenase Activity | |
|---|---|---|---|
| Tested | Results | ||
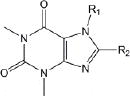
|
78 | 17 | 16 Inhibitors |
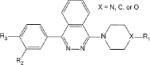
|
20 | 3 | CM001 |
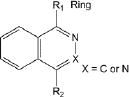
|
10 | 1 | No effect |
|
9 | 0 | |
|
13 | 1 | No effect |
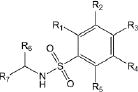
|
11 | 4 | 3 Inhibitors |
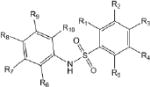
|
8 | 3 | CM047 |
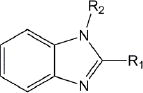
|
7 | 4 | CM010 |
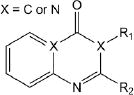
|
7 | 0 | |
Results of nine structural classes of esterase modulators representing 65% of the compounds identified from the esterase HTS.
CM037 was one of the structurally unique esterase activators that was found to be a potent and selective inhibitor of ALDH1A1 (IC50 = 4.6 ± 0.8 μM). A recent publication has shown that this compound, published as A37, is capable of disrupting spheroid formation in an ovarian cancer cell model by targeting ALDH1A1 activity[39]. These results show that this esterase-based HTS identified a novel compound selective for ALDH1A1 compared to ALDH1A2, ALDH1A3, ALDH1B1, ALDH2, and ALDH3A1 and that this compound could enter a cell and alter a cancer phenotype by inhibiting ALDH1A1[39]. Further studies of CM037 as well as the other compounds identified in this screen are needed.
Conclusion
Aldehyde dehydrogenases are critical enzymes involved in the metabolism of a variety of aldehyde substrates. ALDH1A1 has been identified as a marker for both normal and cancer stem cells and has been linked to such diseases as obesity, Parkinson’s disease, and cancer. Small molecule probes are urgently needed to elicit the role of this enzyme in both normal and disease states. However at this time there are no commercially available small molecules that selectively modulate ALDH1A1 activity compared to other ALDHs due to the high degree of structural and functional similarity, particularly within the ALDH1 family. In this paper, we report the first structure of the human ALDH1A1 protein, complexed to NADH. As anticipated, ALDH1A1 is structurally similar to other ALDH proteins, particularly other ALDH1 family members, illustrating the difficulties in discovering selective modulators of this enzyme family. Also, we present an in vitro, esterase-based HTS that identified 256 compounds capable of modulating ALDH1A1 esterase activity. Of these 256 hits, we examined the effect on aldehyde oxidation of 67 compounds and nearly 50% (32 compounds) also modified aldehyde oxidation. These results indicate that the esterase activity of ALDH1A1 can be used to reliably identity small molecule modulators of the enzyme’s dehydrogenase activity. As presented, the HTS should identify both activators and inhibitors of aldehyde oxidation. However, of the 57 esterase activators tested, none activated and 32 inhibited aldehyde oxidation. It is possible that an ALDH1A1 activator is present in the remaining compounds whose effect on aldehyde oxidation has yet to be tested. The screen was successful in identifying potent and selective inhibitors of ALDH1A1, with CM037 already characterized as a potent, selective inhibitor of ALDH1A1 capable of altering spheroid formation in an ovarian cancer cell model. Future work is needed on the remaining compounds identified in order to develop small molecule activators and inhibitors of ALDH1A1.
Supplementary Material
Highlights.
Development of in vitro esterase-based HTS for discovery of modulators of ALDH1A1
Identified 30 hits that selectively inhibited ALDH1A1 compared ALDH2 and ALDH3A1
Determined first structure of human apo-ALDH1A1 and with its cofactor NADH
Acknowledgments
The authors would like to thank Indiana University Chemical Genomics Core Facility (especially Lan Chen, Ph.D. and Andrea Gunawan) for assistance with the high-throughput screen, including access to the chemical library and use of their facility. Special thanks to Lanmin Zhai and Bibek Parajuli for assistance making ALDH proteins. The coordinates and structure factors for human ALDH1A1 and its complex with NADH have been deposited with the RCSB under codes 4WJ9 and 4WB9. This research was supported by NIH R01 AA018123 to TDH. CAM was supported by NIH R13-AA023149 to present this work at the 17th Enzymology and Molecular Biology of Carbonyl Metabolism Meeting held at Skytop Lodge, Poconos, PA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
Thomas D. Hurley holds significant financial equity in SAJE Pharma, LLC. However, none of the work described in this study is related to, based on or supported by the company.
References
- 1.Black W, Vasiliou V. The aldehyde dehydrogenase gene superfamily resource center. Hum Genomics. 2009;4(2):136–42. doi: 10.1186/1479-7364-4-2-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Durrenberger PF, et al. Inflammatory Pathways in Parkinson’s Disease; A BNE Microarray Study. Parkinsons Dis. 2012;2012:214714. doi: 10.1155/2012/214714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaiter D, et al. ALDH1 mRNA: presence in human dopamine neurons and decreases in substantia nigra in Parkinson’s disease and in the ventral tegmental area in schizophrenia. Neurobiol Dis. 2003;14(3):637–47. doi: 10.1016/j.nbd.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 4.Ziouzenkova O, et al. Retinaldehyde represses adipogenesis and diet-induced obesity. Nat Med. 2007;13(6):695–702. doi: 10.1038/nm1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ress NB, et al. Toxicology and carcinogenesis studies of microencapsulated citral in rats and mice. Toxicol Sci. 2003;71(2):198–206. doi: 10.1093/toxsci/71.2.198. [DOI] [PubMed] [Google Scholar]
- 6.Marcato P, et al. Aldehyde dehydrogenase: its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle. 2011;10(9):1378–84. doi: 10.4161/cc.10.9.15486. [DOI] [PubMed] [Google Scholar]
- 7.Kastan MB, et al. Direct demonstration of elevated aldehyde dehydrogenase in human hematopoietic progenitor cells. Blood. 1990;75(10):1947–50. [PubMed] [Google Scholar]
- 8.Jones RJ, et al. Assessment of aldehyde dehydrogenase in viable cells. Blood. 1995;85(10):2742–6. [PubMed] [Google Scholar]
- 9.Storms RW, et al. Isolation of primitive human hematopoietic progenitors on the basis of aldehyde dehydrogenase activity. Proc Natl Acad Sci USA. 1999;96(16):9118–23. doi: 10.1073/pnas.96.16.9118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emadi A, Jones RJ, Brodsky RA. Cyclophosphamide and cancer: golden anniversary. Nat Rev Clin Oncol. 2009;6(11):638–47. doi: 10.1038/nrclinonc.2009.146. [DOI] [PubMed] [Google Scholar]
- 11.Dixon M, Webb EC. Enzymes. 3. New York: Academic Press; 1979. p. xxiv, 1116 p. [Google Scholar]
- 12.Rossmann MG, et al. Evolutionary and Structural Relationships among Dehydrogneases. In: Boyer PD, editor. The Enzymes. Academic Press; New York City: 1975. pp. 61–102. [Google Scholar]
- 13.Perez-Miller S, et al. Alda-1 is an agonist and chemical chaperone for the common human aldehyde dehydrogenase 2 variant. Nat Struct Mol Biol. 2010;17(2):159–64. doi: 10.1038/nsmb.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu ZJ, et al. The first structure of an aldehyde dehydrogenase reveals novel interactions between NAD and the Rossmann fold. Nat Struct Biol. 1997;4(4):317–26. doi: 10.1038/nsb0497-317. [DOI] [PubMed] [Google Scholar]
- 15.Farres J, et al. Investigation of the active site cysteine residue of rat liver mitochondrial aldehyde dehydrogenase by site-directed mutagenesis. Biochemistry. 1995;34(8):2592–8. doi: 10.1021/bi00008a025. [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Weiner H. Involvement of glutamate 268 in the active site of human liver mitochondrial (class 2) aldehyde dehydrogenase as probed by site-directed mutagenesis. Biochemistry. 1995;34(1):237–43. doi: 10.1021/bi00001a028. [DOI] [PubMed] [Google Scholar]
- 17.Koppaka V, et al. Aldehyde dehydrogenase inhibitors: a comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol Rev. 2012;64(3):520–39. doi: 10.1124/pr.111.005538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gonzalez-Segura L, et al. The crystal structure of a ternary complex of betaine aldehyde dehydrogenase from Pseudomonas aeruginosa Provides new insight into the reaction mechanism and shows a novel binding mode of the 2′-phosphate of NADP+ and a novel cation binding site. J Mol Biol. 2009;385(2):542–57. doi: 10.1016/j.jmb.2008.10.082. [DOI] [PubMed] [Google Scholar]
- 19.Tsybovsky Y, et al. Crystal structures of the carboxyl terminal domain of rat 10-formyltetrahydrofolate dehydrogenase: implications for the catalytic mechanism of aldehyde dehydrogenases. Biochemistry. 2007;46(11):2917–29. doi: 10.1021/bi0619573. [DOI] [PubMed] [Google Scholar]
- 20.Chen CH, et al. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321(5895):1493–5. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keung WM, Vallee BL. Daidzin: a potent, selective inhibitor of human mitochondrial aldehyde dehydrogenase. Proc Natl Acad Sci USA. 1993;90(4):1247–51. doi: 10.1073/pnas.90.4.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moreb JS, et al. The enzymatic activity of human aldehyde dehydrogenases 1A2 and 2 (ALDH1A2 and ALDH2) is detected by Aldefluor, inhibited by diethylaminobenzaldehyde and has significant effects on cell proliferation and drug resistance. Chem Biol Interact. 2012;195(1):52–60. doi: 10.1016/j.cbi.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morgan CA, et al. N,N-diethylaminobenzaldehyde (DEAB) as a substrate and mechanism-based inhibitor for human ALDH isoenzymes. Chemico-Biological Interactions. 2014 doi: 10.1016/j.cbi.2014.12.008. This issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hammen PK, et al. Multiple conformations of NAD and NADH when bound to human cytosolic and mitochondrial aldehyde dehydrogenase. Biochemistry. 2002;41(22):7156–68. doi: 10.1021/bi012197t. [DOI] [PubMed] [Google Scholar]
- 25.Perez-Miller SJ, Hurley TD. Coenzyme isomerization is integral to catalysis in aldehyde dehydrogenase. Biochemistry. 2003;42(23):7100–9. doi: 10.1021/bi034182w. [DOI] [PubMed] [Google Scholar]
- 26.Parajuli B, et al. Discovery of novel regulators of aldehyde dehydrogenase isoenzymes. Chem Biol Interact. 2011;191(1–3):153–8. doi: 10.1016/j.cbi.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng CF, Wang TT, Weiner H. Cloning and expression of the full-length cDNAS encoding human liver class 1 and class 2 aldehyde dehydrogenase. Alcohol Clin Exp Res. 1993;17(4):828–31. doi: 10.1111/j.1530-0277.1993.tb00849.x. [DOI] [PubMed] [Google Scholar]
- 28.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 29.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(Pt 5):760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 30.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 31.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 4):439–50. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 32.Painter J, Merritt EA. A molecular viewer for the analysis of TLS rigid-body motion in macromolecules. Acta Crystallogr D Biol Crystallogr. 2005;61(Pt 4):465–71. doi: 10.1107/S0907444905001897. [DOI] [PubMed] [Google Scholar]
- 33.Khanna M, et al. Discovery of a novel class of covalent inhibitor for aldehyde dehydrogenases. J Biol Chem. 2011;286(50):43486–94. doi: 10.1074/jbc.M111.293597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore SA, et al. Sheep liver cytosolic aldehyde dehydrogenase: the structure reveals the basis for the retinal specificity of class 1 aldehyde dehydrogenases. Structure. 1998;6(12):1541–51. doi: 10.1016/s0969-2126(98)00152-x. [DOI] [PubMed] [Google Scholar]
- 35.MacGibbon AK, et al. Kinetic studies on the esterase activity of cytoplasmic sheep liver aldehyde dehydrogenase. Biochem J. 1978;171(3):533–8. doi: 10.1042/bj1710533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parajuli B, Fishel ML, Hurley TD. Selective ALDH3A1 inhibition by benzimidazole analogues increase mafosfamide sensitivity in cancer cells. J Med Chem. 2014;57(2):449–61. doi: 10.1021/jm401508p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parajuli B, et al. Development of Selective Inhibitors for Human Aldehyde Dehydrogenase 3A1 (ALDH3Al) for the Enhancement of Cyclophosphamide Cytotoxicity. Chembiochem. 2014 doi: 10.1002/cbic.201300625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ho KK, Hurley TD, Weiner H. Selective alteration of the rate-limiting step in cytosolic aldehyde dehydrogenase through random mutagenesis. Biochemistry. 2006;45(31):9445–53. doi: 10.1021/bi060718c. [DOI] [PubMed] [Google Scholar]
- 39.Condello S, et al. beta-Catenin-regulated ALDH1A1 is a target in ovarian cancer spheroids. Oncogene. 2014 doi: 10.1038/onc.2014.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.