

Abstract
Doxorubicin is highly effective at inducing DNA double-strand breaks in rapidly dividing cells, which has led to it being a widely used cancer chemotherapeutic. However, clinical administration of doxorubicin is limited by off-target cardiotoxicity, which is thought to be mediated by doxorubicinol, the primary alcohol metabolite of doxorubicin. Carbonyl reductase 1 (CBR1), a well-characterized monomeric enzyme present at high basal levels in the liver, is known to exhibit activity toward doxorubicin. Little is known about a closely related enzyme, carbonyl reductase 3 (CBR3), which is present in the liver at low basal levels but is highly inducible by the transcription factor Nrf2. Genetic polymorphisms in CBR3, but not CBR1, are associated with differential cardiac outcomes in doxorubicin treated pediatric patients. Cbr3 mRNA and CBR3 protein are highly expressed in the livers of Gclm −/− mice (a mouse model of glutathione deficiency) relative to wild type mice. In the present study, we first investigated the ability of CBR3 to metabolize doxorubicin. Incubations of doxorubicin and purified recombinant murine CBR3 (mCBR3) were analyzed for doxorubicinol formation using HPLC, revealing for the first time that doxorubicin is a substrate of mCBR3. Moreover, hepatocytes from Gclm −/− mice produced more doxorubicinol than Gclm +/+ hepatocytes. In addition, differentiated rat myoblasts (C2C12 cells) co-cultured with primary Gclm −/− murine hepatocytes were more sensitive to doxorubicin-induced cytostasis/cytotoxicity than incubations with Gclm +/+ hepatocytes. Our results indicate a potentially important role for CBR3 in doxorubicin-induced cardiotoxicity. Because there is likely to be variability in hepatic CBR3 activity in humans (due to either genetic or epigenetic influences on its expression), these data also suggest that inhibition of CBR3 may provide protection from doxorubicinol cardiotoxicity.
Keywords: Doxorubicin, glutathione deficiency, pharmacogenetics, cardiotoxicity, carbonyl reductase
Introduction
1.1 The anthracycline doxorubicin (also known as Adriamycin) is indicated for a broad range of malignant neoplasms, including blood cancers, carcinomas, and sarcomas. Its central role in hematological cancer regimens also makes it one of the most widely prescribed chemotherapeutics in children [1,2]. Despite the general effectiveness of doxorubicin in destroying cancer cells, its use is dose-limited by off-target complications, namely cardiomyopathies [1,3]. A dose-dependent increase in cardiomyopathy risk is observed in patients; approximately 50–60% of patients given high doses of doxorubicin develop cardiomyopathies [1,4]. The risk of developing cardiomyopathy resulting from administration represents a major impediment to the continued use of doxorubicin in chemotherapy regimens. Elimination of doxorubicin-associated cardiotoxicity would thus represent a major advance in the clinical setting, especially in the context of chronic doxorubicin administration.
Doxorubicinol, an alcohol metabolite of doxorubicin, has been implicated in the cardiotoxicity observed in doxorubicin-treated patients, largely due to doxorubicinol targeting Na+/K+ channels present in cardiac sarcolemma [5,6]. The NADPH-dependent two-electron reduction of doxorubicin to doxorubicinol is shown in Figure 1. Carbonyl reductase 1 (CBR1) has been shown to catalyze the reduction of doxorubicin to doxorubicinol [7]. However, another carbonyl reductase, CBR3, has been less well characterized [8]. Interestingly, in childhood cancer patients treated with doxorubicin, a relatively common polymorphism in the CBR3 gene (present in ~30% of Caucasians) encodes for a non-synonymous amino acid change (V244M), which is associated with decreased risk of developing cardiomyopathy, while a polymorphism in the gene encoding CBR1 (1096 G>A) is not associated with differential cardiomyopathy risk [3].
Figure 1.
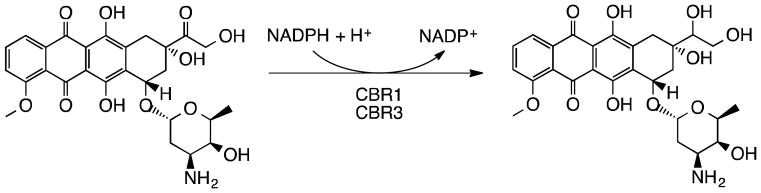
Two-electron reduction of doxorubicin to the putative cardiotoxic alcohol metabolite, doxorubicinol, at the 13th carbon. NADPH-dependent monomeric carbonyl reductase CBR1 is known to mediate this reaction. Here, we demonstrate that this reaction is also carried out by a protein that shares high sequence identity with Cbr1, carbonyl reductase 3.
Furthermore, another CBR3 variant (11 G>A) has been shown to influence the relative expression of CBR3—and subsequent doxorubicinol formation—in a cohort of Southeast Asian breast cancer patients [9]. Though the importance of specific CBR3 variants remains controversial, currently available data taken as a whole suggest an important role for this protein in doxorubicin-induced cardiotoxicity [3,9,10,11]; factors such as tissue-specific expression, polymorphisms present in other genes, patient age, duration of treatment, dosage, and co- therapies, among others, likely influence the relative role of CBR3.
While CBR1 and CBR3 share high amino acid identity (~78%) and are both NADPH-dependent, the endogenous substrate(s) and function(s) of these enzyme appear distinct, and the endogenous role of CBR3 remains unknown. We were initially drawn to CBR3 due to our lab’s interest in the tripeptide glutathione (GSH), an abundant low-molecular weight antioxidant thiol within cells. GSH synthesis is rate-limited by the conjugation of glutamate to cysteine by glutamate cysteine ligase (GCL), which is composed of catalytic (GCLC) and modifier (GLCM) subunits. The level of GSH synthesized in the livers of mice lacking two copies of Gclm (Gclm −/−) is only ~10% relative to that of wild-type mice [12]. To compensate for low GSH levels, Gclm −/− mice have up-regulated a number of genes, especially those involved in antioxidant defense. Cbr3 mRNA is the most highly up-regulated gene in the livers of Gclm −/− mice. On average, primary transcripts of Cbr3 are increased approximately 10-fold relative to Gclm +/+ mice, a trend mirrored in another model of thiol insufficiency—conditional hepatic knockout of Txnrd1 [13,14]. This is especially relevant in the context of doxorubicin metabolism, given the liver’s critical role in xenobiotic biotransformation and detoxification.
While we are currently working to identify the endogenous substrate(s) of CBR3, which currently remain unknown, we present here evidence that doxorubicin is an exogenous substrate of mouse CBR3, a previously undocumented finding. We demonstrate a significantly higher rate of doxorubicinol formation in doxorubicin-treated Gclm −/− mouse hepatocytes relative to Gclm +/+ mouse hepatocytes, and also show that differentiated rat myoblasts (C2C12 cells) co-cultured with primary Gclm −/− mouse hepatocytes are more sensitive to doxorubicin-induced changes in cell growth and/or viability relative to those co-cultured with Gclm +/+mouse hepatocytes. Our findings are important for the ongoing elucidation of mechanisms of doxorubicin-induced cardiotoxicity, and, given the central role of doxorubicin use in chemotherapy regimens, our results may prove clinically relevant upon further investigation.
Materials and Methods
2.1 Chemicals
All reagents for cell culture and biochemical analyses were purchased from Life Technologies (Carlsbad, CA) and/or Sigma-Aldrich (Saint Louis, MO), unless otherwise noted.
2.2 Purification of recombinant murine CBR3
The Cbr3 expression plasmid was made in two steps. First, the complete open reading frame (ORF) was amplified by polymerase chain reaction (PCR) using a RIKEN cDNA clone as template and primers that placed a Nde1 site overlapping the AUG start codon. The PCR product was blunt end-ligated into Sma1-cut pBluescipt II KS (Stratagene California, La Jolla, CA). The Nde1 site and a downstream Hind3 site in the polylinker were then used to move the ORF into a similarly cleaved pET28a expression vector (EMD Millipore, Billerica, MA). The plasmid was shuttled to BL21, and a 500 ml culture was induced by addition of 1 mM isopropyl thiogalactopyranoside when it reached an A600 of 0.6. Six hours later, cells were collected by centrifugation, and disrupted by sonication (five one-minute bursts, on ice) in 20 ml of extraction buffer (300 mM NaCl, 50 mM Na2HPO4, pH 7), containing 20 mM imidazole and protease inhibitors (1 mM PMSF, 1μg/ml leupeptin, 1μg/ml pepstatin). Lysate was clarified by centrifugation (15 min at 15,000 rpm, SS-34 rotor) and mixed on a rotating wheel with a 1 ml slurry of Talon resin (Clontech Laboratories, Inc., Mountain View, CA). After collecting the resin by centrifugation and washing twice with 50 ml extraction buffer containing 20 mM imidazole, the resin was transferred in small volume of washing buffer to a nickel mini-column, and protein was eluted in extraction buffer containing 150 mM imidazole. The first milliliter of eluate contained the highest concentration of recombinant protein and was used for gel and enzymatic analyses.
2.3 Animals
All procedures for animal use were in accordance with the National Institutes of Health Guide for the Use and Care of Laboratory Animals and were approved by the University of Washington Institutional Animal Care and Use Committee (IACUC). Female Gclm −/− mice were derived by homologous recombination techniques in mouse embryonic stem (ES) cells, as previously documented [12]. Mice were anesthetized with 0.02 ml/g of a mixture of ketamine and xylazine prior to liver perfusion and/or liver excision.
2.4 Liver excision and homogenization
Livers were excised from anesthetized mice and placed in TES/SB buffer (20 mM Tris, 1 mM ethylenediaminetetraacetic acid, 250 mM sucrose, 20 mM sodium borate, 2 mM serine) with protease inhibitor cocktail (Roche Molecular Biochemicals, Indianapolis, IN) on ice, and homogenized. Homogenates were then centrifuged at 9000 x g for 20 minutes at 4°C; the resulting supernatant was extracted and prepared for dialysis.
2.5 Primary murine hepatocyte isolation
Mouse livers were perfused and hepatocytes isolated by cannulating the hepatic portal vein and perfusing with Hanks’ Balanced Salt Solution (HBSS) containing 0.5 mM ethylene glycol tetraacetic acid (EGTA) for 4 minutes, after which livers were perfused with 15 mg TH Research Grade Liberase (Roche, Madison, WI) per 100 ml perfusion medium (William’s E medium supplemented with 2 mM L-glutamine, 10 mM HEPES (pH 7.55), and 5 ug/ml ITS+ Premix (BD Biosciences, Bedford, MA)) for 6 minutes. Digested liver lobes were immediately placed in William’s E Medium (supplemented with 2 mM L-glutamine, 5 ug/ml ITS+ Premix, 100 U penicillin and 100 ug/ml streptomycin, and 25 mM dexamethasone; WECM), on ice and then centrifuged at 130 x g for 5 minutes at 4 °C. Following centrifugation, enriched cell pellets were resuspended in Hepatozyme medium (Life Technologies) supplemented with 2 mM L-glutamine,100 U penicillin and 100 ug/ml streptomycin. Hepatocyte yields were assessed by trypan blue exclusion using a hemacytometer. A typical perfusion yielded approximately 20–30 million hepatocytes per liver.
2.6 Cell culture
All cells were cultured in 5% CO2/95% air in a humidified incubator at 37 °C .
2.6.1 Primary hepatocytes
Murine hepatocytes were seeded on collagen (25 ug/well)-coated six well plates (Corning Inc., Corning, NY) at a density of ~500,000 cells/well. After 4 hours incubation at 37 °C to allow for attachment, fresh WECM was added to each well
2.6.2 C2C12 culture and differentiation
Undifferentiated mouse C2C12 myoblasts were cultured in 75 cm2 flasks at 37 °C for 24 hours using Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum and 100 IU penicillin plus 100 ug/ml streptomycin . C2C12 cells were then trypsinized and reseeded onto 6-well Transwell Permeable Supports (0.4 μm polyester membrane; Corning Inc., Corning, NY), at a density of 75,000 cells/well, at which time medium was switched to DMEM supplemented with 2% horse serum and 100 U penicillin plus 100 μg/ml streptomycin (differentiation medium) to allow for differentiation. Differentiation medium was replaced every 24 hours for 3 days.
2.7 Treatment and co-culture of primary mouse hepatocytes with C2C12 cells
After four hours incubation at 37 °C in Hepatozyme to allow for attachment, primary mouse hepatocytes were given 2 ml of fresh WECM, and treated with 0 (vehicle control), 2, 5, or 10 μM doxorubicin hydrochloride (dissolved in dimethyl sulfoxide (DMSO) vehicle; volume of DMSO did not exceed 0.2% for any treatment). Concomitantly, transwell inserts containing differentiated C2C12 cells were placed in wells containing hepatocytes cultured on the bottom of the wells. Medium within each insert was replaced with fresh differentiation medium (1.5 ml/insert). Underlying doxorubicin-treated hepatocytes and overlying C2C12 cells were co-cultured for 18 hours following treatment, after which time metabolic and toxicological endpoints were assessed.
2.8 High pressure liquid chromatography
HPLC analyses for hepatocyte lysates, culture media, and liver homogenate incubations were performed using a Shimadzu LC6A reverse-phase HPLC, with an RFIOAxI fluorescence detector and a C-R5A integrator (Kyoto, Japan). Samples were loaded in 50 mM monobasic potassium phosphate buffer (pH 2.5) and eluted under isocratic conditions, with the mobile phase containing 27% acetonitrile, 73% 50 mM monobasic potassium phosphate (pH 2.5) and a flow rate of 1 ml/minute through a 150-mm Synergi 5u Hydro – RP 80A column (Phenomenex, Torrance, CA). HPLC analysis of purified mCBR3 incubations was performed on a Waters 2690 Separations Module, with a Waters 474 Scanning Fluorescence Detector (Waters Corporation, Milford, MA). A slightly modified gradient to more effectively separate doxorubicin and doxorubicinol peaks was employed—22% Acetonitrile, 78% in 50 mM monobasic potassium phosphate (pH 2.5) and a flow rate of 1 ml/minute through a 150-mm Synergi 5u Hydro – RP 80A column (Phenomenex, Torrance, CA). Excitation and emission wavelengths were 470 nm and 590 nm, respectively, and Waters Millenium Software V2.0 was used for data processing (Waters Corporation, Milford, MA).
2.8.1 Detection of doxorubicinol in media and primary mouse hepatocytes
Following 18 hours doxorubicin treatment, 5% 5-sulfosalicylic acid (SSA) was added to culture media in microfuge tubes, on ice, for 10 min and then centrifuged at 14,000 x g for 2 minutes at room temperature. Resulting supernatants were analyzed by HPLC. For hepatocytes, cells were washed with phosphate-buffered saline and trypsinized for 15 min at 37 °C using 0.25% trypsin/2.21 mM EDTA in HBSS (Cellgro, Corning Inc., Corning, NY). They were then removed from the dishes by scraping, triturated 3–5 times and transferred to microfuge tubes. Next, hepatocytes were centrifuged for five minutes at 130 x g, 4°C, resuspended in TES/SB and sonicated on ice. Cell lysates were centrifuged for 2 minutes at 14,000 x g (4°C) and the resulting supernatant was added to 5% SSA, incubated on ice for 15 minutes, and centrifuged again at 14,000 x g (room temperature) for 2 minutes. Resulting supernatants (pH=2.5) were used for HPLC analyses.
2.8.2 Detection of doxorubicinol formation in liver homogenates
Aliquots of liver cytosolic fractions prepared by centrifugation of liver homogenates at 9000 x g (S9 fraction) from female Gclm +/+ and Gclm −/− mice were placed in 3.0 mL dialysis cassettes (Thermo Scientific, Waltham, MA) overnight in a solution of 100 mM monobasic potassium phosphate, pH 7.4. The following day, 20 μl of the dialyzed liver S9 fraction (approximately 50–70 mg/ml concentration) were transferred to microfuge tubes containing 15 μl of 10 mM nicotinamide adenine dinucleotide phosphate (NADPH) in 100 mM monobasic potassium phosphate (pH 7.4), 1.5 μl of 1 mg/ml doxorubicin hydrochloride (in DMSO), and 113.5 μl of 100 mM monobasic potassium phosphate buffer (pH 7.4). Samples were briefly vortexed, then placed on a rocker in the dark at 37 °C, and allowed to incubate for one hour. Next, 50 μl of 5% SSA was added to each sample, on ice, for 15 minutes, and samples were centrifuged at 14,000 x g for 2 minutes. Resulting supernatants were used for HPLC analysis.
2.8.3 Detection of doxorubicinol formation using purified recombinant murine CBR3
Incubations with purified recombinant mouse CBR3 (mCBR3), followed the same protocol as incubations with liver homogenates. Briefly, 1 μl of 55 mM mCbr3, 1 μl of 1 mg/ml doxorubicin, 15 μl of 10 mM NADPH, and 133 μl 100 mM monobasic potassium phosphate buffer (pH 7.4) were briefly vortexed, then placed on a rocker in the dark at 37 °C, and allowed to incubate for one hour. Next, 50 μl of 5% SSA was added to each sample, on ice, for 15 min, and samples were centrifuged at 14,000 X g for 2 minutes. Resulting supernatants were used for HPLC analysis. For control experiments, the same protocol was followed, with the additional step of boiling mCBR3 for ten minutes prior to incubation with doxorubicin and NADPH.
2.9 Liquid Chromatography/Mass Spectrometry (LC/MS)
Gclm WT and KO murine liver homogenates were incubated with doxorubicin (1 μg/150 μl total incubation volume) for 60 minutes at 37 °C in the dark, as described in section 2.8.2 of Methods. Following incubation and derivitization, samples were analyzed using LC/MS to confirm the presence of the doxorubicinol metabolite. Samples were run on an Agilent Technologies (Santa Clara, CA) MS QQQ Mass Spectrometer using a Poroshell SB-C18 (3.0mm x 50mm x 2.7 μ) column at 0.4 ml/minute flow rate (0.1% formic acid buffer). An internal standard of epirubicin (4’-epimer of doxorubicin) was used to differentiate between doxorubicinol and doxorubicin. The mass to charge (m/z) ratio for the precursor ions of doxorubicin and epirubicin were 544.2, and 546.1 for doxorubicinol (determined using standards of each compound). The m/z of the product ions were 361.1 and 397.1 for epirubicin and doxorubicin, respectively, and 363.1 and 399.2 for doxorubicinol. Analyses were performed in triplicate for each Gclm genotype.
2.10 Assessment of C2C12 cell viability
Cellular cytostasis and/or cytotoxicity of C2C12 cells following 18 hour co-culture with doxorubicin-treated primary mouse hepatocytes was determined spectrophotometrically by measuring the reduction of the tetrazolium salt 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to formazan as previously described [15].
2.11 Protein isolation
Total protein concentration in cell lysates from primary mouse hepatocytes and S9 liver homogenate fractions was determined using the Bio-Rad Protein Assay Dye Reagent (Bio-Rad Laboratories Inc., Hercules, CA), in 96-well microtiter plates, following the manufacturer’s protocol.
2.12 Western Blotting
Western blotting was performed using standard techniques. Briefly, ~15 ug protein per sample were separated by electrophoresis using 12% Tris-Glycine mini-gels in a Novex XCell SureLock Mini-Cell (Life Technologies, Carlsbad, CA). Following electrophoresis, proteins were transferred to polyvinyldiene difluoride (PVDF) membranes (Immobilon-P, Millipore, Billerica, MA). Membranes were blocked with 5% milk in tris-buffered saline with Tween 20 (TBS-T) and incubated with primary antibodies raised against mouse Cbr3 (1:1000 dilution; rabbit polyclonal antisera; GFM, unpublished), β-actin (1:1000 dilution; Cell Signaling, Beverly, MA), and Gclm (1:50000; rabbit polyclonal antisera, as described previously [12]), followed by incubation with horseradish peroxidase-conjugated secondary rat anti-rabbit antibody (Millipore, Billerica, MA), diluted 1:10,000 in 5% milk TBS-T.
2.13 Statistical Analyses
Data are expressed as the mean and SEM, unless otherwise noted. Data were analyzed by one- or two-way ANOVA, followed by a Dunnett’s post-hoc test (for cytotoxicity experiments), or a Student’s t-test (for pair-wise comparisons) using a statistical software package (Prism, GraphPad, Inc., San Diego, CA). Differences yielding a p value of less than 0.05 were considered statistically significant.
Results
3.1 Carbonyl reductase 3 levels in the livers of Gclm −/− and Gclm +/+ mice
Western blotting of primary hepatocyte lysates and dialyzed liver homogenates (S9 fraction) confirmed that CBR3 protein is highly up-regulated in the livers of Gclm −/− mice relative to Gclm +/+ mice (Figure 2).
Figure 2.
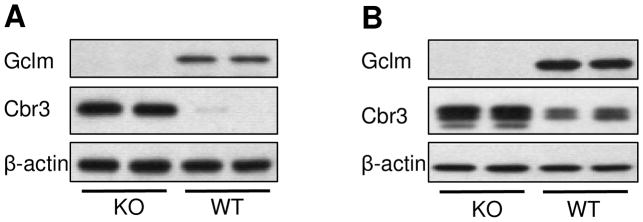
Representative Western blot analyses of GCLM, CBR3, and β-actin (loading control) proteins in (A) primary murine hepatocytes and (B) dialyzed liver homogenates from Gclm −/− and Gclm +/+ mice.
3.2 Assessment of the ability of mCBR3 to metabolize doxorubicin to doxorubicinol
We report here that doxorubicin is a substrate of murine CBR3. Incubation of purified mCBR3 with doxorubicin and NADPH, revealed the formation of doxorubicinol with HPLC analysis, at the expected elution time of approximately 10 minutes (Figure 3).
Figure 3.

HPLC chromatograms showing doxorubicinol formation with (A) catalytically inactive and (B) catalytically active purified mCBR3. Doxorubicinol eluted at ~10 minutes, while doxorubicin eluted at ~15 minutes.
3.3 Doxorubicinol formation in primary mouse hepatocytes and liver homogenates
Primary Gclm −/− mouse hepatocytes treated for 18 hours with 2, 5, or 10 uM doxorubicin had significantly higher levels (~2.5- to 3.5-fold) of both cellular and extracellular doxorubicinol than Gclm +/+ mouse hepatocytes, as measured by HPLC (Figure 4; n=12, p < 0.001 for all doses). A graded dose-response of doxorubicinol formation was observed with increasing doxorubicin treatment (Figure 4). Additionally, dialyzed liver S9 homogenates (cytosolic + microsomal fraction) from Gclm −/− mice produced approximately 2.5-fold more doxorubicinol than S9 fractions from Gclm +/+ homogenates (Figure 5; n=4, p < 0.05).
Figure 4.

Doxorubicinol formation in Gclm −/− and Gclm +/+ hepatocytes. (A) Representative HPLC chromatograms of doxorubicinol formation in Gclm −/− (top panel) and Gclm +/+ (bottom panel) hepatocytes. (B) Quantification by HPLC of doxorubicinol formation (area units, AU) in hepatocytes and (C) media taken from doxorubicin treatments; n=12 for each treatment. *** indicates p < 0.001 for Student’s t-test relative to wild-type for each dose.
Figure 5.

Doxorubicinol formation by Gclm −/− and Gclm +/+ liver homogenates. (A) Representative HPLC chromatograms of doxorubicinol formation in Gclm −/− (top panel) and Gclm +/+ (bottom panel) dialyzed liver homogenates. (B) Quantification by HPLC of doxorubicinol formation (area units, AU) in dialyzed liver homogenates following incubation with 50 mM doxorubicin; n=4 per genotype. *indicates p < 0.05 for Student’s t-test relative to wild-type.
3.4 LC/MS Analysis of doxorubicinol formation in liver homogenates
Analysis of doxorubicinol formation using mass spectrometry confirmed the identity of the putative doxorubicinol peak observed in HPLC analyses. Additionally, following the trend observed in HPLC analyses, 2- to 3-fold more doxorubicinol was formed in incubations containing Gclm −/− liver homogenate relative to Gclm +/+ liver homogenate (Figure 6).
Figure 6.


LC/MS analysis of liver homogenate-doxorubicin incubations. Confirmation of doxorubicinol formation in (A) Gclm +/+ and (B) Gclm −/− liver homogenates. Precursor ions for doxorubicin and epirubicin had a m/z of 544.2, while the precursor ion for doxorubicinol had a m/z of 546.1. The mass-to-charge ratio of the product ions for doxorubicin and epirubicin were 397.1 and 361.1, respectively, while doxorubicinol had two product ions with m/z ratios of 399.2 and 363.1.
3.5 Co-culture of differentiated mouse myoblasts with doxorubicin-treated primary murine hepatocytes
C2C12 cells differentiated on transwell inserts were placed on top of primary Gclm −/− or Gclm +/+ mouse hepatocytes and then treated with doxorubicin to assess the effect of hepatocyte genotype on the production of compound(s) affecting myocyte viability, an in vitro proxy of liver-mediated metabolism resulting in cardiotoxicity. Significantly decreased C2C12 cell viability and/or cell growth (as measured by the MTT assay) was observed in C2C12 cells co-cultured with primary Gclm −/− hepatocytes relative to those co-cultured with primary Gclm +/+ hepatocytes (Figure 7; n=6 per dose and genotype, p < 0.001 by two-way ANOVA and Dunnet’s post-hoc analysis).
Figure 7.

Cytotoxicity in C2C12 cells following co-culturing with either Gclm −/− or Gclm +/+ primary hepatocytes. Two-way ANOVA analysis, *** indicates p < 0.001 relative to controls, # indicates p < 0.001 relative to wild-type at each; representative of two independent experiments, n=3 per dose and genotype per experiment; n.s. - not significant.
Discussion
4. 1 In this study, we investigated the potential role of a poorly characterized enzyme, mouse CBR3, in doxorubicin metabolism. Preliminary epidemiological data implicate human CBR3 polymorphisms as being important in predicting doxorubicinol formation [3,9,16,17]. Because doxorubicinol is believed to contribute significantly to cardiotoxicity in cancer patients given doxorubicin as part of chemotherapy regimens, further investigation into the role of CBR3 in doxorubicin metabolism was warranted [6].
We first tested the ability of purified recombinant mCBR3 to metabolize doxorubicin. Our results indicate that murine CBR3 is indeed able to convert doxorubicin to doxorubicinol, a previously undocumented finding. The Nrf2-regulated CBR3 enzyme [18] is highly up-regulated in Gclm −/− mice (a model of glutathione deficiency) relative to Gclm +/+ mice. Accordingly, we evaluated the ability of primary mouse hepatocytes and liver homogenates from Gclm −/− and Gclm +/+ mice to produce doxorubicinol. Gclm −/− hepatocytes produced significantly higher levels of doxorubicinol following 18 hours of doxorubicin treatment in a dose-dependent fashion, relative to Gclm +/+ hepatocytes. Additionally, dialyzed liver homogenates from Gclm −/− mice produced significantly higher amounts of doxorubicinol following incubation with doxorubicin.
Following these observations, we expanded our focus to a physiologically-relevant in vitro system to model cardiotoxicity induced by doxorubicin treatment. We used differentiated C2C12 cells (a murine myoblast cell line) as a surrogate for cardiomyocytes, and co-cultured them with primary murine hepatocytes from Gclm −/− and Gclm +/+ mice. As anticipated, a greater impact on C2C12 cell viability/numbers was observed in co-cultures with doxorubicin-treated Gclm −/− mouse hepatocytes relative to cultures with Gclm +/+ mouse hepatocytes. Moreover, there was an inverse relationship between doxorubicinol concentration and cell numbers and/or viability in these in vitro cell culture studies.
Because of their enhanced CBR3 expression, Gclm −/− mice are likely to exhibit increased sensitivity to doxorubicin-induced cardiomyopathy, relative to Gclm +/+ mice, a hypothesis that we are currently in the process of investigating with in vivo studies..
Ultimately, the overarching goal of this research is to reduce doxorubicin-induced cardiomyopathy to a level at which doxorubicin administration is not dose-limited by off-target cardiotoxicity. Doxorubicin continues to be and will likely remain a widely used chemotherapeutic central to numerous chemotherapy regimens [1]. Despite the discovery and use of targeted, less toxic therapies for specific cancer types (e.g., Trastuzumab), little progress has been made in reducing off-target cardiotoxicity induced by anthracyclines since their clinical introduction nearly 40 years ago [1,19]. Our findings not only implicate CBR3 in doxorubicin metabolism, but underscore the potential importance of CBR3 inhibition in the context of doxorubicin-containing therapies. Finally, if validated in appropriate pre-clinical models, such inhibition would represent a novel advancement in the treatment of a wide variety of cancers.
Highlights.
Doxorubicin is a substrate of murine Cbr3
Gclm −/− hepatocytes express higher levels of Cbr3 than Gclm +/+ hepatocytes
More doxorubicinol is made by Gclm −/− hepatocytes than Gclm +/+ hepatocytes
Gclm −/− hepatocytes sensitize co-cultured C2C12 cells to doxorubicin toxicity
Acknowledgments
Funding Information
5. 1 This work was supported by National Institutes of Health grants P30ES007033 and T32AG000057, a grant to TJK from the University of Washington Department of Environmental and Occupational Health Sciences, and a grant to GFM from the Medical Research Foundation of Oregon.
6. 1 We wish to thank members of the Kavanagh laboratory, and Drs. Chad Weldy, Trevor Penning, Edmund Maser, and Udo Oppermann, for their encouragement and helpful suggestions, and Brian Philips and the laboratory of Dr. Danny Shen for assisting with the LC/MS analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lipshultz SE, Cochran TR, Franco VI, Miller TL. Treatment-related cardiotoxicity in survivors of childhood cancer. Nat Rev Clin Oncol. 2013;10(12):697–710. doi: 10.1038/nrclinonc.2013.195. [DOI] [PubMed] [Google Scholar]
- 2.Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339(13):900–905. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- 3.Blanco JG, Sun CL, Landier W, Chen L, Esparza-Duran D, Leisenring W, Mays A, Friedman DL, Ginsberg JP, Hudson MM, Neglia JP, Oeffinger KC, Ritchey AK, Villaluna D, Relling MV, Bhatia S. Anthracycline-related cardiomyopathy after childhood cancer: role of polymorphisms in carbonyl reductase genes—A report from the Children’s Oncology Group. J Clin Oncol. 2012;30(13):1415–1421. doi: 10.1200/JCO.2011.34.8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kremer LCM, Caron H. Anthracycline cardiotoxicity in children. N Engl J Med. 2004;351:120–121. doi: 10.1056/NEJMp048113. [DOI] [PubMed] [Google Scholar]
- 5.Boucek RJ, Olson RD, Brenner DE, Ogunbunmi EM, Inui M, Fleischer S. The major metabolite of doxorubicin is a potent inhibitor of membrane-associated ion pumps. A correlative study of cardiac muscle with isolated membrane fractions. J Biol Chem. 1987;262(33):15851–15856. [PubMed] [Google Scholar]
- 6.Olson RD, Mushlin PS, Brenner DE, Fleischer S, Cusack BJ, Chang BK, Boucek RJ. Doxorubicin cardiotoxicity may be caused by its metabolite, doxorubicinol. Proc Natl Acad Sci USA. 1988;85(10):3585–3589. doi: 10.1073/pnas.85.10.3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kassner N, Huse K, Martin HJ, Gödtel-Armbrust U, Metzger A, Meineke I, Brockmöller J, Klein K, Zanger UM, Maser E, Wojnowski L. Carbonyl reductase 1 is a predominant doxorubicin reductase in the human liver. Drug Metab Dispos. 2005;36(10):2113–2120. doi: 10.1124/dmd.108.022251. [DOI] [PubMed] [Google Scholar]
- 8.Oppermann U. Carbonyl reductases: the complex relationships of mammalian carbonyl- and quinone-reducing enzymes and their role in physiology. Annu Rev Pharmacol Toxicol. 2007;47:293–322. doi: 10.1146/annurev.pharmtox.47.120505.105316. [DOI] [PubMed] [Google Scholar]
- 9.Fan L, Goh BC, Wong CI, Sukri N, Lim SE, Tan SH, Guo JY, Lim R, Yap HL, Khoo YM, Iay F, Lee HS, Lee SC. Genotype of human carbonyl reductase CBR3 correlates with doxorubicin disposition and toxicity. Pharmacogenetics and Genomics. 2008;18(7):623–631. doi: 10.1097/FPC.0b013e328301a869. [DOI] [PubMed] [Google Scholar]
- 10.Lubieniecka JM, Liu J, Heffner D, Graham J, Reid R, Hogge D, Grigliatti TA, Riggs WK. Single-nucleotide polymorphisms in aldo-keto and carbonyl reductase genes are not associated with acute cardiotoxicity after daunorubicin chemotherapy. Cancer Epidemiol Biomarkers Prev. 2012;21(11):2118–2120. doi: 10.1158/1055-9965.EPI-12-1037. [DOI] [PubMed] [Google Scholar]
- 11.Bains OS, Karkling MJ, Lubieniecka JM, Grigliatti TA, Reid RE, Riggs KW. Naturally occurring variants of human CBR3 alter anthracycline in vitro metabolism. J Pharmacol Exp Ther. 2010;332(3):755–763. doi: 10.1124/jpet.109.160614. [DOI] [PubMed] [Google Scholar]
- 12.McConnachie LA, Mohar I, Hudson FN, Ware CB, Ladiges WC, Fernandez C, Chatterton-Kirchmeier S, White CC, Pierce RH, Kavanagh TJ. Glutamate cysteine ligase modifier subunit deficiency and gender as determinants of acetaminophen-induced hepatotoxicity in mice. Toxicol Sci. 2007;99(2):628–636. doi: 10.1093/toxsci/kfm165. [DOI] [PubMed] [Google Scholar]
- 13.Haque JA, McMahan RS, Campbell JS, Shimizu-Albergine M, Wilson AM, Botta D, Bammler TK, Beyer RP, Montine TJ, Yeh MM, Kavanagh TJ, Fausto N. Attenuated progression of diet-induced steatohepatitis in glutathione-deficient mice. Lab Invest. 2010;90(12):1704–1717. doi: 10.1038/labinvest.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bondareva AA, Capecchi MR, Iverson SV, Li Y, Lopez NI, Lucas O, Merrill GF, Prigge JR, Siders AM, Wakamiya M, Wallin SL, Schmidt EE. Effects of thioredoxin reductase-1 deletion on embryogenesis and transcriptome. Free Radic Biol Med. 2007;43(6):911–23. doi: 10.1016/j.freeradbiomed.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carmichael J, Degraff WG, Gazdar AF, Minna JD, Mitchell JB. Evaluation of a Tetrazolium-based Semiautomated Colorimetric Assay: Assessment of Chemosensitivity Testing. Cancer Res. 1987;47:936. [PubMed] [Google Scholar]
- 16.Blanco JG, Leisenring WM, Gonzalez-Covarrubias VM, Kawashima TI, Davies SM, Relling MV, Robison LL, Sklar CA, Stovall M, Bhatia S. Genetic polymorphisms in the carbonyl reductase 3 gene CBR3 and the NAD(P)H: quinone oxidoreductase 1 gene NQO1 in patients who developed anthracycline-related congestive heart failure after childhood cancer. Cancer. 2008;112(12):2789–2795. doi: 10.1002/cncr.23534. [DOI] [PubMed] [Google Scholar]
- 17.Jamieson D, Boddy AV. Pharmacogenetics of genes across the doxorubicin pathway. Expert Opin Drug Metab Toxicol. 2011;7(10):1201–1210. doi: 10.1517/17425255.2011.610180. [DOI] [PubMed] [Google Scholar]
- 18.Ebert B, Kisiela M, Malátková P, El-Hawari Y, Maser E. Regulation of human carbonyl reductase 3 (CBR3; SDR21C2) expression by Nrf2 in cultured cancer cells. Biochemistry. 2010;49(39):8499–8511. doi: 10.1021/bi100814d. [DOI] [PubMed] [Google Scholar]
- 19.Hudis CA. Trastuzumab--mechanism of action and use in clinical practice. N Engl J Med. 2007;357(1):39–51. doi: 10.1056/NEJMra043186. [DOI] [PubMed] [Google Scholar]