

Abstract
Aldo-keto reductase 1C3 (AKR1C3), also known as type 5 17β-hydroxysteroid dehydrogenase, is a downstream steroidogenic enzyme and converts androgen precursors to the potent androgen receptor ligands: testosterone and 5α-dihydrotestosterone. Studies have shown that AKR1C3 is involved in the development of castration resistant prostate cancer (CRPC) and that it is a rational drug target for the treatment of CRPC. Baccharin, a component of Brazilian propolis, has been observed to exhibit a high inhibitory potency and selectivity for AKR1C3 over other AKR1C isoforms and is a promising lead compound for developing more potent and selective inhibitors. Here, we report the screening of fifteen baccharin analogs as selective inhibitors against AKR1C3 versus AKR1C2 (type 3 3α-hydroxysteroid dehydrogenase). Among these analogs, the inhibitory activity and selectivity of thirteen compounds were evaluated for the first time. The substitution of the 4-dihydrocinnamoyloxy group of baccharin by an acetate group displayed nanomolar inhibitory potency (IC50: 440 nM) and a 102-fold selectivity over AKR1C2. By contrast, when the cinnamic acid group of baccharin was esterified, there was a dramatic decrease in potency and selectivity for AKR1C3 in comparison to baccharin. Low or sub- micromolar inhibition was observed when the 3-prenyl group of baccharin was removed, and the selectivity over AKR1C2 was low. Although unsubstituted baccharin was still the most potent (IC50: 100 nM) and selective inhibitor for AKR1C3, these data provide structure-activity relationships required for the optimization of new baccharin analogs. They suggest that the carboxylate group on cinnamic acid, the prenyl group, and either retention of 4′-dihydrocinnamoyloxy group or acetate substituent on cinnamic acid are important to maintain the high potency and selectivity for AKR1C3.
Keywords: castration resistant prostate cancer, androgen, androgen receptor signaling
1. Introduction
Prostate cancer (CaP) is the most commonly diagnosed cancer and the second leading cause of cancer death in males in the United States [1]. The development of prostate cancer is androgen-dependent, and androgen deprivation therapy using surgical or chemical castration has been a main treatment for locally advanced or metastatic disease [2, 3]. However, within 2 to 3 years, the recurrence of CaP, also called castration resistant prostate cancer (CRPC), occurs despite castrate levels of circulating androgens (e.g. testosterone (T)), and has the potential to become more metastatic [4, 5]. Pathophysiological studies have shown that CRPC remains mainly androgen-driven, and can arise when the transcriptional activity of the androgen receptor (AR) is reactivated by the intratumoral conversion of weak adrenal androgens (e.g. dehydroepiandrosterone (DHEA) and 4-androstene-3,17-dione) into the potent AR ligands: T and 5α-dihydrotestosterone (DHT), and/or by androgen receptor amplification [5–7]. Thus, new therapeutics targeting AR signaling (either intratumoral androgen biosynthetic enzymes or AR) for the treatment of CRPC are being developed and tested in clinical trials [6, 7].
Recently, abiraterone acetate which inhibits the activities of cytochrome P450c 17 (17α-hydroxylase/17,20 lyase, Figure 1) blocks the formation of adrenal DHEA, improves overall survival in CRPC patients and has been approved by the FDA [8, 9]. However, P450c 17 catalyzes an early-step in steroidogenesis, and the inhibition of its hydroxylase activity leads to a loss in cortisol production and over stimulation of the gland by ACTH. This can lead to an increase in levels of mineralocorticoids which can cause serious adverse side effects such as hypertension. Consequently, patients taking abiraterone must be co-administered prednisone [10]. In addition, resistance to abiraterone has been reported due to an elevated expression level of CYP17A1 [11, 12]. Therefore, new molecular targets in the AR signaling pathway have been investigated to discover superior therapeutic agents [6, 13].
Figure 1.

Androgen metabolism in human prostate depicting the roles of AKR1C1, AKR1C2, and AKR1C3. AKR1C1: 20α-hydroxysteroid dehydrogenase; AKR1C2: type 3 3α-hydroxysteroid dehydrogenase; AKR1C3: type 5 17β-hydroxysteroid dehydrogenase; CYP 17A1: cytochrome P450 17α-hydroxylase/17,20 lyase; DHEA: dehydroepiandrosterone; HSD3B1: type 1 3β-hydroxysteroid dehydrogenase; SRD5A: 5α-reductase. Enzymes are identified by gene names.
Aldo-keto reductase 1C3 (AKR1C3) also known as type 5 17β-hydroxysteroid dehydrogenase in the prostate, converts 4-androstene-3,17-dione and 5α-androstane-3,17-dione to T and DHT respectively which are potent ligands for the AR (Figure 1) [14, 15]. AKR1C3 is overexpressed at both the mRNA and protein levels in prostate tumors from CRPC patients [16–19]. Reduction of AKR1C3 expression levels in CaP cells or inhibition of AKR1C3 activity significantly decreases the levels of T and DHT and androgen dependent gene expression e.g. prostate specific antigen (PSA). In vivo inhibition of AKR1C3 leads to a reduction in growth of xenograft models of CRPC [11, 19–21]. More recently, AKR1C3 was found to act as an AR coactivator which would provide an alternative mechanism by which it may promote the growth of prostate cancer cells and CRPC xenografts [21]. These findings have made AKR1C3 a promising therapeutic target for both androgen-dependent CaP and CRPC [13, 15, 19]. It has been proposed that inhibition of AKR1C3 might not be therapeutically efficacious based on insignificant changes in T levels in CaP cells after the treatment with the AKR1C3 inhibitor SN33638 [22]. However, data to support this notion may be circumspect because of the specificity of antibodies used in Western blot analysis, the reliance of ELISA measurements to quantitate androgens, and the maintenance of cancer cell lines in a fetal bovine serum (FBS) or fetal calf serum (FCS) media containing androgens which will suppress AKR1C3 expression [17, 18].
Significant efforts have been made by our group and others to discover and develop different classes of AKR1C3 inhibitors, including steroidal based compounds (e.g. medroxyprogesterone acetate) and repurposed nonsteroidal anti-inflammatory drugs (NSAIDs) which no longer inhibit COX-1 and COX-2 [15, 23–27]. One of the most important considerations in inhibitor development is to ensure that they do not inhibit other AKR1C isoforms (AKR1C1 and AKR1C2 in Figure 1). AKR1C1 and AKR1C2 share >86% sequence identity to AKR1C3 but inactivate DHT. Thus, screening for AKR1C3-selective inhibitors is imperative [6, 28, 29].
Baccharin (1, Table 1), a naturally occurring phenolic compound extracted from Brazilian propolis which is a resinous gum collected by bees from the plant exudate, has been commonly used in medicine and nutritional products and exhibits anti-tumor and anti-inflammatory activities [30–36]. Its ability to inhibit AKR1C3 with an IC50 value of 110 nM and without affecting other AKR1C isoforms has been reported [37]. In order to discover more potent and selective inhibitors and better understand the structure-activity relationship of how baccharin analogs inhibit AKR1C3, we tested the inhibitory activities of 15 analogs, thirteen of which represent new analogs for screening.
Table 1.
Inhibitory properties of group 1 analogs on AKR1C3 and AKR1C2.
| Compound | Structure | 1C3 IC50 (μM) | 1C2 IC50 (μM) | IC50 Ratio 1C2:1C3 |
|---|---|---|---|---|
| 1 (Baccharin) |
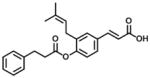
|
0.1 | 51 | 510 |
| 2 |
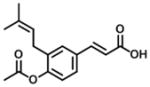
|
0.44 | 45 | 102 |
| Drupanin* |
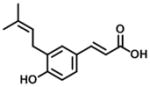
|
15 | 108 | 7 [37] |
data is from the reference [37].
2. Materials and methods
2.1. Reagents
Reagents were of ACS grade or higher and were purchased from Fisher Scientific (Pittsburgh, PA) and used without further purification. (S)-(+)-1,2,3,4-tetrahydro-1-naphthol (S-tetralol) was purchased from Sigma-Aldrich (St. Louis, MO). Nicotinamide adenine dinucleotide (NAD+) and nicotinamide adenine dinucleotide phosphate (NADP+) were purchased from Roche Diagnostics (Indianapolis, IN). Homogeneous recombinant enzymes AKR1C3 and AKR1C2 were prepared and purified as previously described [28]. The specific activities of AKR1C3 and AKR1C2 for the NAD+ dependent oxidation of S-tetralol are 2.0 and 1.5 μmol min−1 mg−1, respectively.
2.2. Chemistry
Baccharin (1) and compounds 2, 3, 4, 6, and 7 were synthesized using a modified literature procedure [38]. The general scheme for the synthetic procedure is shown in Scheme 1. Compounds 14 (p-coumaric acid), 15 (m-coumaric acid), and 16 (ferulic acid) were obtained from Sigma-Aldrich (Milwaukee, WI) and were used without further purification. Reaction progress was monitored by thin-layer chromatography (TLC) carried out on silica gel plates (2.5 cm × 7.5 cm, 200 μm thick, 60 F254) and visualized by using UV lamp (254 nm). Flash column chromatography was performed with silica gel (40–63 μm, 60 Å) using the mobile phase indicated in the following procedures. 1H NMR and 13C NMR spectra were recorded in the indicated solvent by a Bruker 400 MHz Advance III HD spectrometer at 400 and 100 MHz for 1H and 13C respectively with TMS as an internal standard. Multiplicities are indicated by s (single), d (doublet), dd (doublet of doublets) t (triplet), q (quartet), m (multiplet). Chemical shifts (δ) are given in parts per million (ppm), and coupling constants (J) are given in hertz (Hz).
Scheme 1.
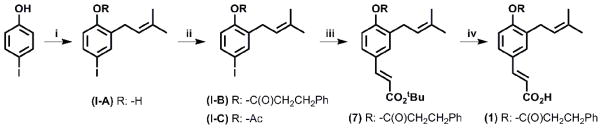
General synthetic procedure. Reagents i): NaH, PhMe, 3,3-dimethylallyl bromide; ii): RCl, NEt3, DMAP, DCM; iii): H2C=CHCO2tBu, Pd(OAc)2, (Ph)3P, NEt3, PhMe; iv): SiO2, PhMe.
4-iodo-2-(3-methylbut-2-en-1-yl)phenol (I-A)
To a solution of p-iodophenol (2 g, 9.08 mmol) in toluene (20 mL) was added NaH (60% dispersion in mineral oil, 540 mg, 13.62 mmol). When the effervescence ceased, 3,3-dimethylallyl bromide (1.1 mL, 9.5 mmol) was added and the reaction was stirred at room temperature overnight. The reaction mixture was acidified to pH 1 using AcOH, washed with H2O, extracted with DCM, dried (Na2SO4), filtered and concentrated. Purification by column chromatography (5–10% of Et2O in hexane, v:v) provided the title compound as a yellow oil (1.43 g, 4.96 mmol, 55%). Rf: 0.5 (4:1 Hexane:EtOAc, v:v).
1H NMR (400 MHz; CDCl3): δ 1.8 (6H, d, J = 8.0 Hz, CH3), 3.33 (2H, d, J = 6.88 Hz, CH2), 5.31 (1H, t, J = 6.6 Hz, CH), 5.46 (1H, s, -OH), 6.59 (1H, d, J = 8.2 Hz, Ar-H), 7.41 (2H, s, Ar-H). 13C NMR (100 MHz; CDCl3): δ 17.8, 25.8, 29.3, 82.7, 117.9, 120.7, 129.7, 135.4, 136.1. 138.3, 154.2.
4-iodo-2-(3-methylbut-2-en-1-yl)phenyl-3-phenylpropanoate (I-B)
To a solution of 4-iodo-2-(3-methylbut-2-en-1-yl)phenol (560 mg, 1.94 mmol) in DCM (4 mL) was added DMAP (25 mg, 0.2 mmol) followed by addition of 3-phenyl propionyl chloride (495 mg, 2.93 mmol) and NEt3 (0.4 mL, 2.91 mmol). The mixture was stirred overnight at room temperature. Saturated NaHCO3 was added and the layers were separated. The organic layer was washed with H2O, dried (Na2SO4), filtered and concentrated to provide the tile compound without further purification (490 mg, 1.16 mmol, 60 %).
1H NMR (400 MHz; CDCl3): δ 1.66 (3H, s, CH3), 1.76 (3H, s, CH3), 2.91 (2H, t, J = 7.5 Hz, CH2) 3.09 (4H, t, J = 6.0 Hz, CH2CH2), 5.16 (1H, t, J = 7.1 Hz, CH), 6.69 (1H, d, J = 8.3 Hz, ArCH), 7.21–7.36 (6H, m, ArCH), 7.53 (1H, s, ArCH). 13C NMR (100 MHz; CDCl3): δ 17.8, 25.9, 28.3, 30.2, 30.9, 35.7, 36.4, 90.5, 120.7, 124.4, 126.7, 128.4, 128.8, 133.9, 135.9, 136.3, 138.8, 140.0, 148.6, 171.0.
4-iodo-2-(3-methylbut-2-en-1-yl)phenyl acetate (I-C)
To a solution of 4-iodo-2-(3-methylbut-2-en-1-yl)phenol (332 mg, 1.15 mmol) in DCM (4 mL) was added DMAP (14 mg, 0.1 mmol) followed by addition of acetyl chloride (0.122 mL, 1.7 mmol) and NEt3 (0.236 mL, 1.7 mmol). The mixture was stirred overnight. Saturated NaHCO3 was added and the layers were separated. The organic layer was washed with H2O, dried (Na2SO4), filtered and evaporated in vacuo to provide the title compound (350 mg, 1.06 mmol, 92%).
1H NMR (400 MHz; CDCl3): δ 1.70 (3H, s, CH3), 1.76 (3H, s, CH3), 2.31 (3H, s, OCH3), 3.19 (2H, d, J = 7.1 Hz, CH2), 5.19 (1H, t, J = 7.1 Hz, CH), 6.79 (1H, d, J = 8.0 Hz, ArCH), 7.55 (2H, d, J = 9.4 Hz, ArCH). 13C NMR (100 MHz; CDCl3): δ 18.6, 20.9, 25.8, 28.4, 90.5, 120.6, 123.8, 124.3, 133.8, 135.9, 139.0, 148.8, 168.4.
(2E)-3-[3-(3-methylbut-2-en-1-yl)-4-[(3-phenylpropanoyl)oxy]phenyl]prop-2-enoic acid (1)
To a solution of tert-butyl(2E)-3-[3-(3-methylbut-2-en-1-yl)-4-[(3-phenylpropanoyl)-oxy]phenyl]prop-2-enoate (7) in toluene (10 mL) was added chromatography grade silica gel (3.3 g) and the mixture was refluxed with vigorous agitation overnight. Upon cooling the reaction mixture was diluted with 10% methanol in DCM and filtered over celite® pad using 10% methanol in DCM as the solvent. Purification by column chromatography with a stepwise elution (2:1 - 1:1 - 0:1 Hexane:EtOAc, v:v) provided the title compound (139.5 mg, 0.38 mmol, 58%). Rf: 0.11 (4:1 Hexane:EtOAc).
1H NMR (400 MHz, CDCl3): δ 1.69 (3H, s, CH3), 1.77 (3H, s, CH3), 2.94 (2H, t, J = 7.7 Hz, CH2), 3.11 (2H, t, J = 7.6 Hz, CH2), 3.17 (2H, d, J=7.4Hz, H-1″), 5.20 (1H, t, J = 7.3 Hz, CH), 6.40 (1H, d, J = 16.0 Hz, CH), 7.00 (1H, d, J = 8.3 Hz, ArCH) 7.24–7.42 (7H, m, ArCH), 7.75 (1H, d, J = 16.0 Hz, CH). 13C NMR (100 MHz, CDCl3): δ 17.8, 25.7, 28.4, 30.8, 35.8, 116.7, 120.8, 122.8, 126.5, 126.8, 128.6, 130.1, 131.9, 133.9, 134.3, 139.9, 146.3, 150.6, 170.6, 171.0. m/z (ESI): 363.1 [M−, 100%], 364.2(25%).
(2E)-3-[4-(acetyloxy)-3-(3-methylbut-2-en-1-yl)phenyl]prop-2-enoic acid (2)
4-iodo-2-(3-methylbut-2-en-1-yl)phenyl acetate (I-C) was exposed to the general method described for the synthesis of (1), and purification by column chromatography (4:1 Hexane:EtOAc) provided the title compound (86 mg, 99%). Rf: 0.11 (4:1 Hexane:EtOAc).
1H NMR (400 MHz, CDCl3): δ 1.73 (3H, s, CH3), 1.78 (3H, s, CH3), 2.34 (3H, s, CO2CH3), 3.27 (2H, d, J = 7.7 Hz, CH2), 5.24 (1H, t, J = 7.1 Hz, CH), 6.41 (1H, d, J = 15.9 Hz, CH), 7.09 (1H, d, J = 8.4 Hz, ArCH), 7.43 (2H, s, ArCH), 7.75 (1H, d, J = 15.8 Hz, CH). 13C NMR (100 MHz, CDCl3): δ 17.8, 20.8, 25.7, 28.6, 116.8, 120.8, 122.9, 126.9, 130.2, 132.0, 134.3, 146.1, 150.7, 169.0, 170.1, 206.9. m/z (ESI MS): 273.0 [M−] (100%).
(2E)-3-[4-(acetyloxy)-3-(3-methylbut-2-en-1-yl)phenyl]prop-2-enoate (3)
The general method described for the synthesis of (7) using 4-iodo-2-(3-methylbut-2-en-1-yl)phenyl acetate (350 mg, 1.06 mmol) as the starting material was followed, and purification by column chromatography (30:1 - 10:1 - 4:1 - 2:1 Hexane:EtOAc) provided the title compound (100 mg, 0.3 mmol, 28.5%). Rf: 0.9 (4:1 Hexane:EtOAc).
1H NMR (400 MHz, CDCl3): δ 1.55 (9H, s, CO2t-Bu), 1.72 (3H, s, CH3), 1.77 (3H, s, CH3), 2.33 (3H, s, CO2CH3), 3.25 (2H, d, J = 7.1 Hz, CH2), 5.23 (1H, t, J = 7 Hz, CH), 6.31 (1H, d, J = 15.9 Hz, CH), 7.05 (1H, d, J = 8.8 Hz, ArCH), 7.37(2H, s, ArCH), 7.56 (1H, d, J = 15.9 Hz, CH). 13C NMR (100 MHz, CDCl3): δ 17.8, 20.8, 25.7, 28.2, 28.7, 80.5, 120.1, 121.0, 122.0, 122.7, 126.5, 129.0, 129.7, 132.6, 133.7, 134.0, 142.8, 150.1, 166.2, 169.1. m/z (ESI MS): 215 (100%), 353.1 0 [M+Na]+ (50%).
(2E)-3-[4-hydroxy-3-(3-methylbut-2-en-1-yl)phenyl]prop-2-enoate (4)
To a solution 4-hydroxy methyl cinnamate (4.57 g, 25.81 mmol) in toluene (70 mL) was added NaH (1.132 g, 28.29 mmol) and the mixture was heated at 70°C for 3.5 hr. 3,3-dimethylallyl bromide (4.2 mL, 35.32 mmol) was added and the mixture was stirred at 80°C for 72 hr. Ice water (150 mL) was added, the solution acidified with AcOH, washed with NaHCO3, extracted with ether, dried (Na2SO4), filtered and concentrated. Purification by column chromatography (12:1 - 10:1 - 8:1 - 6:1 - 4:1 Hexane:EtOAc) provided the title compound (3.0 g, 12.19 mmol, 47%). Rf: 0.69 (4:1 Hexane:EtOAc).
1H NMR (400 MHz, CDCl3): δ 1.80 (6H, s, CH3), 3.38 (2H, d, J = 7.41 Hz, CH2), 3.81 (3H, s, CO2-CH3), 5.33 (1H, t, J = 7, CH), 5.37 (1H, s, -OH), 6.31 (1H, d, J = 15.91 Hz, CH), 6.82 (1H, d, J = 8.08 Hz, ArCH), 7.29–7.35 (2H, m, ArCH), 7.64 (1H, d, J = 15.92 Hz, CH). 13C NMR (100 MHz, CDCl3): δ 18.0, 25.8, 29.7, 51.6, 115.0, 116.2, 121.0, 127.2, 127.2, 127.9, 130.1, 135.7, 144.8, 156.5, 167.7. m/z (ESI MS): 247.1 [M+, 100%], 282.2 (35%), 248.2(15%).
(2E)-3-(3,3-dimethyl-3,4-dihydro-2H-1-benzopyran-6-yl)prop-2-enoate (5)
To a solution 4-hydroxy methyl cinnamate (720 mg, 4.06 mmol) in toluene (50 mL) was added NaH (292 mg, 6.10 mmol) and the mixture was heated at 70°C for 3.5 hr. 3,3-dimethylallyl bromide (0.70 mL, 6.10 mmol) was added and the mixture was stirred at 80°C for 72 hr. Ice water (150 mL) was added, acidified with AcOH, washed with NaHCO3 and extracted with ether, dried (Na2SO4), filtered, and concentrated. Purification by column chromatography (12:1 – 10:1 – 8:1 – 6:1 – 4:1 Hexane:EtOAc) provided the title compound (370 mg, 1.50 mmol, 41%).
1H NMR (400 MHz, CDCl3): δ 1.36 (6H, s, CH3), 1.84 (2H, t, J = 6.7 Hz, CH2), 2.80 (2H, t, J = 6.7 Hz, CH2), 3.80 (3H, s, CO2-CH3), 6.29 (1H, d, J = 15.99 Hz, CH), 6.79 (1H, d, J = 8.3 Hz, ArCH), 7.26–7.32 (2H, m, ArCH), 7.63 (1H, d, J = 15.9 Hz, CH). 13C NMR (100 MHz, CDCl3): δ 22.5, 27.0, 32.5, 51.5, 75.2, 114.5, 117.8, 121.6, 126.1, 127.3, 130.2, 145.3, 156.1, 168.1. m/z (ESI MS): 247.1 [M+, 100%], 282.2 (22.5%), 248.2(15%), 269.0(15%).
(2E)-3-[3-(3-methylbut-2-en-1-yl)-4-[(3-phenylpropanoyl)oxy]phenyl]prop-2-enoate (6)
The general method described for the synthesis of (7) using methyl acrylate was followed, and purification by column chromatography (30:1 – 20:1 Hexane:EtOAc) provided the title compound (100 mg, 0.26 mmol, 30%). Rf: 0.5 (4:1 Hexane:EtOAc).
1H NMR (400 MHz, CDCl3): δ 1.69 (3H, s, CH3), 1.78 (3H, s, CH3), 2.94 (2H, t, J = 7.6 Hz, CH2), 3.11 (2H, t, J = 7.6 Hz, CH2), 3.17 (2H, d, J = 7.1 Hz, CH2), 3.83 (3H, s, CO2CH3), 5.21 (1H, t, J = 7 Hz, CH), 6.40 (1H, d, J = 16 Hz, CH), 6.98 (1H, d, J = 8.7 Hz, ArCH) 7.24–7.40 (7H, m, ArCH), 7.67 (1H, d, J = 16 Hz, CH). 13C NMR (100 MHz, CDCl3): 17.8, 20.7, 22.6, 25.2, 25.7, 28.4, 30.9, 34.68, 117.6, 120.9, 122.7, 126.5, 126.5, 128.3, 128.6, 129.8, 132.3, 133.8, 134.2, 139.9, 144.1, 150.3, 167.3, 171.0. m/z (ESI MS): 401.1 [M+Na]+ (100%), 443.2(50%), 301.1(50%).
tert-butyl(2E)-3-[3-(3-methylbut-2-en-1-yl)-4-[(3-phenylpropanoyl)oxy]phenyl]prop-2-enoate (7)
To a solution of 4-iodo-2-(3-methylbut-2-en-1-yl)phenyl-3-phenylpropanoate (490 mg, 1.16 mmol) in anhydrous toluene (7 mL) was added PPh3 (46 mg, 0.175 mmol), Pd(OAc)2 (47 mg, 0.09 mmol) and NEt3 (0.245 ml, 1.75 mmol). The mixture was stirred at room temperature and t-Butyl acrylate (0.275 mL, 1.75 mmol) was added to the flask which was refluxed overnight. The reaction mixture was washed with a saturated solution of NH4Cl, water and extracted over DCM, dried (Na2SO4) and filtered. Purification by column chromatography (60:1 - 30:1 - 20:1 Hexane:EtOAc) provided the title compound (280 mg, 0.66 mmol, 57%). Rf: 0.6 (4:1 Hexane:EtOAc).
1H NMR (400 MHz, CDCl3): δ 1.55 (9H, s, CO2t-Bu), 1.69 (3H, s, CH3), 1.76 (3H, s, CH3), 2.93 (2H, t, J = 7.4 Hz, CH2), 3.10 (2H, t, J = 7.6 Hz, CH2), 3.16 (2H, d, J = 7 Hz, CH2), 5.20 (1H, t, J = 7 Hz, CH), 6.31 (1H, d, J = 15.9 Hz, CH), 6.96 (1H, d, J = 8.7 Hz, ArCH) 7.24–7.36 (7H, m, ArCH) 7.55(1H, d, J = 16 Hz, CH). 13C NMR (100 MHz, CDCl3): δ 17.8, 25.7, 28.2, 28.5, 29.7, 30.9, 35.8, 80.5, 120.0, 120.35, 121.0, 121.9, 122.6, 126.5, 128.3, 128.6, 129.0, 129.7, 132.6, 133.7, 134.0, 139.9, 142.3, 142.8, 150.0, 166.2, 171.0. m/z (ESI MS): 443.2 [M+Na]+ (100%), 444.3(35%).
(2E)-3-prop-2-enoic acid (8)
To a solution of p-coumaric acid (1.0 g, 6.09 mmol) in DCM (10 mL) was added 3-phenyl propionyl chloride (1.5 g, 9.14 mmol) and NEt3 (1.2 mL, 9.1 mmol) and the mixture was stirred at room temperature overnight. Saturated NaHCO3 was added and the layers were separated. The organic layer was washed with H2O, dried (Na2SO4), filtered and concentrated to provide the title compound as a white powder (400 mg, 2.43 mmol, 22%). Rf: 0.05 (4:1 Hexane:EtOAc).
1H NMR (400 MHz, CDCl3): δ 2.93 (2H, t, J = 7.6 Hz, CH2), 3.10 (2H, t, J = 7.6 Hz, CH2), 6.40 (1H, d, J = 16.2 Hz, CH), 7.31 (5H, m, ArCH), 7.08 (2H, d, J = 8.31 Hz, ArCH), 7.57 (2H, d, J = 8.41 Hz, ArCH), 7.77 (1H, d, J = 16 Hz, CH). 13C NMR (100 MHz, CDCl3): δ 30.9, 36.2, 117.2, 122.2, 122.3, 126.7, 128.3, 128.6, 129.4, 131.7, 131.8, 134.2, 139.8, 145.8, 152.5, 170.4, 171.0. m/z (ESI MS): 295.0 [M−, 100%], 296.1 [M, 22.5%], 230.9(10%), 152.7(10%).
(E)-3-(3-((3-phenylpropanoyl)oxy)phenyl)acrylic acid (9)
To a solution of m-coumaric acid (279 mg, 1.7 mmol) in DCM (50 mL) at 0 °C was added NEt3 (0.19 mL, 1.7 mmol) dropwise. A solution of 3-phenyl propionyl chloride (286 mg, 1.7 mmol) in DCM (20 mL) was added and the solution was warmed to room temperature and stirred overnight. Saturated NaHCO3 was added and the layers were seperated. The organic layer was washed with H2O, dried (Na2SO4), filtered and concentrated. Purification by column chromatography (4:1 petrol:EtOAc) provided the title compound as a white powder (49 mg, 0.16 mmol, 10%). Rf: 0.50 (4:1 Petrol:EtOAc).
1H NMR (400 MHz; CDCl3): δ 2.94 (2H, t, J = 7.6 Hz, CH2), 3.12 (2H, t, J = 7.6 Hz, CH2), 6.44 (1H, d, J = 15.9 Hz, CH), 7.09 (1H, m, ArCH), 7.19 (1H, m, ArCH), 7.28–7.31 (3H, m, ArCH), 7.35 (2H, d, J = 7.2 Hz, ArCH), 7.42 (2H, d, J = 5.0 Hz, ArCH), 7.75 (1H, d, J = 15.9 Hz, CH). 13C NMR (100 MHz; CDCl3): δ 30.7, 35.7, 118.2, 120.9, 123.7, 125.9, 126.5, 128.3, 128.8, 129.8, 135.5, 139.7, 145.8, 150.8, 171.2, 171.3.
(Z/E)-3-(4-((3-phenylpropanoyl)oxy)phenyl)acrylic acid (10)
To a solution of (Z/E)-tert-butyl 3-(4-(3-phenylpropanoyloxy)phenyl)acrylate (100 mg, 0.28 mmol) in toluene (10 mL) was added SiO2 (2.5 g) and the mixture was refluxed under nitrogen for 5 hours. The mixture was allowed to cool and diluted with 10% MeOH/DCM (v/v). Celite was added and the mixture was filtered through a pad of celite by washing with 10% MeOH/DCM. The solution was concentrated and excess toluene was removed by successive washing with EtOAc/Petrol. The yellow solid was purified by column chromatography (4:1 Petrol:EtOAc) to provide the title compound as a white powder (80 mg, 0.27 mmol) composed of a 7/3 mixture of E/Z isomers. Rf: 0.50 (4:1 Petrol:EtOAc).
1H NMR (400 MHz, CDCl3): δ 2.93 (2H, t, J = 7.7 Hz, CH2), 3.11 (2H, t, J = 7.6 Hz, CH2), 6.42 (1H, d, J = 15.9 Hz, CH), 7.07 (2H, d, J = 8.4 Hz, ArCH), 7.27 (3H, m, ArH), 7.34 (2H, d, J = 7.13 Hz, ArCH), 7.57 (2H, d, J = 8.53 Hz, ArCH), 7.77 (1H, d, J = 15.9 Hz, CH). Minor Z isomer: 5.99 (1H, d, J = 8.3 Hz, CH), 7.68 (1H, d, J = 8.3 Hz, CH). 13C NMR (100 MHz, CDCl3): δ 30.6, 35.9, 121.0, 121.9, 126.4, 128.4, 128.7, 129.4, 129.8, 131.7, 139.9, 145.8, 152.4, 171.0.
(2E)-3-[4-(benzoyloxy)phenyl]prop-2-enoic acid (11)
The general method described for the synthesis of (8) using benzoyl chloride (1.3 g, 9.14 mmol) was followed, and purification by column chromatography (4:1 Hexane:EtOAc) provided the title compound as a white solid (320 mg, 1.3 mmol, 20 %). Rf: 0.05 (4:1 Hexane:EtOAc).
1H NMR (400 MHz, CDCl3): δ 6.47 (1H, d, J = 16.1 Hz, CH2), 7.31 (2H, d, J = 8.4 Hz, ArCH), 7.5 (2H, t, J = 7.7 Hz, ArCH), 7.65 (3H, m, ArCH), 7.81 (1H, d, J = 15.9 Hz, CH), 8.23 (2H, d, J = 7.4 Hz, ArCH). 13C NMR (100 MHz, CD3OD): δ 106.8, 118.3, 119.6, 122.0, 128.4, 129.0, 129.1, 129.6, 132.3, 133.6, 143.6, 145.1, 152.4, 128.5, 129.1, 168.8. m/z (ESI MS): 266.9 [M−, 100%].
(2E)-3-[4-(pyridine-4-carbonyloxy)phenyl]prop-2-enoic acid (12)
The general method described for the synthesis of (8) using isonicotinoyl chloride (534 mg, 3.0 mmol) in THF (30 mL) was followed, and purification by column chromatography (4:1 Hexane:EtOAc) provided the title compound as a yellow solid (510 mg, 1.9 mmol, 99%). Rf: 0.1 (4:1 Hexane:EtOAc).
1H NMR (400 MHz, CD3OD,): δ 6.29 (1H, d, J = 15.9 Hz, CH), 6.82 (2H, d, J = 8.3Hz, ArCH), 7.46 (2H, d, J = 8.3Hz, ArCH), 7.61 (1H, d, J = 15.9 Hz, CH), 7.97 (2H, d, J = 4.9 Hz, ArCH), 8.74 (2H, d, J = 4.8Hz, ArCH). 13C NMR (100 MHz, CD3OD): δ 106.8, 114.1, 115.3, 123.3, 125.8, 129.6, 139.4, 145.2, 149.5, 159.7, 166.1, 169.5.
(Z/E)-tert-butyl 3-(4-(3-phenylpropanoyloxy)phenyl)acrylate (13)
To a solution of (E)-tert-butyl 3-(4-hydroxyphenyl)acrylate (500 mg, 2.27 mmol) in dry THF (25 mL) under N2 at 0 °C was added 3-phenylpropionic acid (0.33 mL, 2.27 mmol), followed by adding a solution of PPh3 (594 mg, 2.27 mmol) in dry THF (10 mL) and DEAD (1.12 mL of a 40% solution in toluene, 2.27 mmol). The solution was warmed to RT and stirred for 48 hrs. The solvent was removed in vacuo, and the residue was redissolved in EtOAc, washed with NaHCO3 and brine, dried (Na2SO4), filtered and concentrated. Purification by column chromatography (4:1 petrol:EtOAc) provided the title compound as a yellow oil (510 mg, 1.44 mmol) composed of a 7/3 mixture of E/Z isomers. Rf: 0.70 (4:1 Petrol:EtOAc).
1H NMR (300 MHz, CDCl3): δ 1.47 (9H, s, C(CH3)3), 2.80 (2H, t, J = 7.4 Hz, CH2), 2.91 (2H, t, J = 7.4 Hz, CH2), 6.23 (1H, d, J = 15.9 Hz, CH), 6.95 (1H, d, J = 8.5 Hz, ArCH), 7.12 – 7.30 (7H, m, ArCH), 7.44 (2H, d, J = 4.5 Hz, ArCH), 7.49 (1H, d, J = 15.9 Hz, CH). 13C NMR (100 MHz, CDCl3): δ 28.2, 30.9, 36.0, 80.6, 120.4, 121.1, 126.5, 128.4, 128.65, 129.1, 129.8, 130.9, 132.4, 140.0, 140.4, 142.4, 151.9, 166.2.
2.3. Assay of enzyme activity
2.3.1. AKR1C3 and AKR1C2
The specific activities of AKR1C3 and AKR1C2 were determined by measuring the formation of NADH at 340 nm using Beckman DU640 spectrophotometer. A typical assay solution contained 100 mM potassium phosphate pH 7.0, 2.3 mM NAD+, 3.0 mM (S)-(+)-1,2,3,4-tetrahydro-1-naphthol (S-tetralol), 4% acetonitrile (v/v). The mixtures were incubated at 37 °C for 3 min followed by adding a serial dilution of AKR1C3 or AKR1C2 solution to a final volume of 1 mL to initiate the reaction. After continuously monitoring for 5 min, the increase in UV absorption using different concentrations of enzyme were recorded to calculate the initial velocity and determine enzyme specific activity.
2.3.2. IC50 value determination
The inhibitory potency for each compound was represented by IC50 value and measured as described before [14, 26, 39]. The IC50 value of baccharin and baccharin analogs was determined by measuring their inhibition of the NADP+ dependent oxidation of S-tetralol catalyzed by AKR1C3 or AKR1C2. The concentration of S-tetralol used in this assay for AKR1C3 and AKR1C2 was 165 μM and 15 μM respectively, which was equal to the Km value for each enzyme isoform in order to make a direct comparison of IC50 values. The IC50 value of each compound was acquired from a single experiment with each inhibitor concentration run in quadruplicate and directly calculated by fitting the inhibition data to an equation using Grafit 5.0 software. In this equation, “range” is the fitted uninhibited value minus the “background”, and “S” is a slope factor. “I” is the concentration of inhibitor. The equation assumes that y falls with increasing “I”.
3. Results
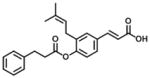
Baccharin (compound 1) and 15 baccharin analogs (compounds 2–16) were screened against recombinant human AKR1C3 and AKR1C2 (Table 1, 2, 3 and 4). The selectivity of AKR1C3 over AKR1C2 was determined based on the ratio of IC50 values between AKR1C2 and AKR1C3 [26]. As shown in Table 1, compound 1 inhibited AKR1C3 with an IC50 value of 100 nM and gave a 510-fold of selectivity for AKR1C3 over AKR1C2, which was comparable to the data reported by Endo et al [37]. The inhibitory potency and selectivity of 15 baccharin analogs were compared to compound 1. Based on the structure of 1 [3-prenyl-4-dihydrocinnamoyloxy) cinnamic acid], cinnamic acid is the core structure of baccharin (1) and the other 15 analogs were divided into 4 groups for comparison, which include removal of the 4-dihydrocinnamoyloxy group (compound 2 in Table 1), esterification of cinnamic acid (compounds 3–7 in Table 2), removal of 3-prenyl group (compounds 8–13 in Table 3) and cinnamic acid derivatives (compounds 14–16 in Table 4).
Table 2.
Inhibitory properties of group 2 analogs on AKR1C3 and AKR1C2.
| Compound | Structure | 1C3 IC50 (μM) | 1C2 IC50 (μM) | IC50 Ratio 1C2:1C3 |
|---|---|---|---|---|
| 1 (Baccharin) |
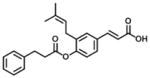
|
0.10 | 51 | 510 |
| 3 |
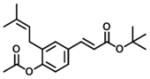
|
8.6 | 15 | 2 |
| 4 |
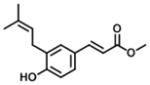
|
9.7 | 7.3 | 1 |
| 5 |
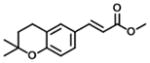
|
21 | 14 | 1 |
| 6 |
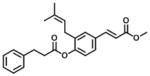
|
37% at 10 μM | 22 | N/A |
| 7 |
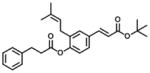
|
607 | 153 | 0.3 |
N/A: not available.
Table 3.
Inhibitory properties of group 3 analogs on AKR1C3 and AKR1C2.
| Compound | Structure | 1C3 IC50 (μM) | 1C2 IC50(μM) | IC50 Ratio 1C2:1C3 |
|---|---|---|---|---|
| 1 (Baccharin) |
|
0.10 | 51 | 510 |
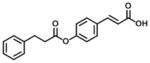
| 8 (100% E) |
|
4.7 | 88 | 19 |
| 9 (100% E) |
|
0.96 | 27 | 28 |
| 10 (7/3, E/Z) |
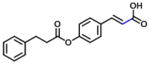
|
2.3 | 25 | 11 |
| 11 |
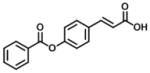
|
6.3 | 127 | 20 |
| 12 |
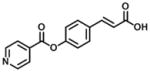
|
68 | 88 | 1 |
| 13 (7/3, E/Z) |
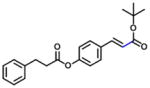
|
632 | 27 | 0.04 |
E/Z: E/Z geometric isomers (highlighted with blue bond).
Table 4.
Inhibitory properties of group 4 analogs on AKR1C3 and AKR1C2.
| Compound | Structure | 1C3 IC50 (μM) | 1C2 IC50 (μM) | IC50 Ratio 1C2:1C3 |
|---|---|---|---|---|
| 1 (Baccharin) |
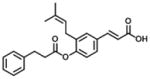
|
0.10 | 51 | 510 |
| 14 p-coumaric acid (100% E) |
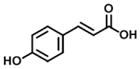
|
199 | 1297 | 7 |
| 15 m-coumaric acid (100% E) |
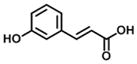
|
86 | 389 | 5 |
| 16 Ferulic acid (100% E) |
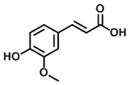
|
21 | 421 | 20 |
After the 4-dihydrocinnamoyloxy group was replaced by an acetate group (Table 1), it was found that compound 2 still displayed nanomolar inhibitory potency for AKR1C3 with an IC50 value of 440 nM and a 102-fold of selectivity for AKR1C3 versus AKR1C2 with no real change in inhibitory potency for AKR1C2.
Analogs in group 2 (Table 2) can also be separated into two subgroups. In the first group, the analogs are esterified and the 4-dihydrocinnamoyloxy group was removed (compounds 3–5). Esterification of the cinnamic acid on 2 to form the tertiary butyl ester (compound 3) resulted in 86-fold loss of inhibitory potency (IC50: 8.6 μM) for AKR1C3 and a slight increase of inhibition for AKR1C2, which resulted in a 255-fold decrease in selectivity (IC50 ratio: 2) in comparison to 1. Compound 4 was derived from drupanin (see Table 1) in which the dihydrocinnamoyloxy group of baccharin was replaced by a hydroxyl group, and the cinnamic acid portion was esterified to form the methyl ester. These substitutions gave a similar inhibitory potency (IC50: 9.7 μM) as compound 3 and drupanin [37], but the inhibitory potency for AKR1C2 was increased by 7-fold which made 4 lose inhibitory selectivity for AKR1C3 over AKR1C2 (IC50 ratio: 1). When the hydroxyl group on 4 reacted with the double bond of the 3-prenyl group to form a six-membered ring, the resultant compound 5 displayed a further 2-fold decrease of inhibitory potency for both AKR1C3 (IC50: 21 μM) and AKR1C2, and as a result had no inhibitory selectivity for AKR1C3 (IC50 ratio: 1). In the second group in Table 2, the methyl ester (compound 6) or the tertiary butyl ester (compound 7) of baccharin were found to cause a dramatic decrease in both the inhibitory potency and selectivity for AKR1C3. In addition, 6 did not inhibit AKR1C3 when the concentration was >10 μM.
In group 3 (Table 3), removal of the 3-prenyl group of baccharin resulted in compounds 8–11 which had low or sub- micromolar IC50 values for AKR1C3; however, the inhibitory selectivities of these compounds were decreased by 18 to 45-fold in comparison to 1. In comparison to compound 8 (IC50: 4.7 μM), the movement of the dihydrocinnamoyloxy group from the para to meta position (compound 9) increased inhibitory potency for both AKR1C3 (IC50: 960 nM) and AKR1C2, and displayed a 1.5-fold increase of the inhibitory selectivity (IC50 ratio: 28). Compound 10 which was composed of 70% (E)- and 30% (Z)-isomers also increased the inhibition for both AKR1C3 (IC50: 2.3 μM) and AKR1C2 (IC50: 25 μM) relative to compound 8, but the inhibitory selectivity was decreased by 1.7-fold in comparison to 8. Substitution of the dihydrocinnamoyloxy group in 8 with a benzoic acid group (compound 11) showed a similar inhibitory potency (IC50: 6.3 μM) and selectivity as 8 for AKR1C3. When the phenyl group of benzoic acid on 11 was replaced by a pyridine ring (compound 12), the inhibitory potency was decreased to yield an IC50 value of 68 μM and the selectivity was lost (IC50 ratio: 1). Esterification (compound 13) of cinnamic acid resulted in a large loss of inhibitory potency (IC50: 632 μM) and a reversal of selectivity (IC50 ratio: 0.04) for AKR1C3.
Compounds 14–16 in group 4 (Table 4) were derivatives of cinnamic acid, which are also found in propolis as natural products [33, 40]. The main characteristic in these compounds was the removal of the 3-prenyl group and the replacement of the 4-dihydrocinnamoyloxy group by a hydroxyl group. In comparison to 1, these natural products displayed much lower inhibition for both AKR1C3 and AKR1C2 and less selectivity for AKR1C3. However, in comparison to compounds 14 and 15, 16 displayed an increased inhibitory potency and selectivity for AKR1C3 due to the introduction of a small hydrophobic 5-methoxyl group.
4. Discussion
Our group and others have studied different classes of inhibitors for AKR1C3 in order to develop new therapeutics for the treatment of CRPC based on NSAID scaffolds [13, 14, 25, 26, 37, 39, 41–45]. Although inhibitors with nanomolar potency and high selectivity to AKR1C3 have been identified from these studies, the discovery of new compounds for preclinical development is still required. Among these compounds, baccharin, a natural component extracted from Brazilian propolis, displays a high potency and inhibitory selectivity against AKR1C3 [37]. This discovery has made baccharin a promising lead to develop a new series of potent and specific inhibitors against AKR1C3.
In this study, the inhibitory potency and selectivity of 15 baccharin analogs against AKR1C3 were investigated and compared to unmodified baccharin (compound 1). Thirteen baccharin analogs were studied for the first time. Compound 1 showed the most potent inhibition (IC50: 100 nM) and highest selectivity for AKR1C3 over AKR1C2 (IC50 ratio: 510) (Table 1, 2, 3 and 4). Compound 2 still displayed a good inhibitory potency (IC50: 440 nM) and selectivity (IC50 ratio: 102) for AKR1C3 and did not change the inhibitory potency to AKR1C2. This may result from the addition of the acetate group on the phenyl ring of cinnamic acid.
Analysis of crystal structures of AKR1C3.NADP+- inhibitor complexes have revealed that the ligand binding site can be dissected into five subsites: oxyanion site (consisting of catalytic residues Tyr55, His117 and cofactor NADP+), the steroid channel for the binding of steroid ligands, and three sub-pockets (SPs: SP1, SP2 and SP3 to accommodate other ligands) [13, 15]. The presence of either a carbonyl or carboxylate group is required to anchor many ligands to the oxyanion site, because it can form a strong hydrogen-bond (H-bond) with Tyr55 and His117 and bring the ligands into close proximity of the nicotinamide head group of the cofactor [15]. Based on the model of baccharin-docked into the AKR1C3-NADP+ complex structure (Figure 2) [37], the carbonyl group on the 4-dihydrocinnamoyloxy group of baccharin was found to be close to the oxyanion site and could form a H-bond interaction with Tyr55 and His117, which could contribute to the high inhibitory activity of baccharin. Thus, the carbonyl group on the acetate group in compound 2 could maintain the H-bond interaction in the oxyanion site to provide a good inhibitory potency. In comparison to compound 1, 2 displayed a 4-fold decrease of inhibitory potency and 5-fold decrease of inhibitory selectivity. This might be caused by the removal of the benzyl moiety to decrease the hydrophobic interactions in the SP3 pocket and the H-bond formation in the oxyanion site. The docked model predicted that the benzyl moiety of the 4-dihydrocinnamoyloxy group was located in the SP3 pocket and could form hydrophobic interactions with the side chain of Gln222 or Phe306 to provide the high binding affinity [37]. Thus, the loss of lipophilicity when the benzyl group is substituted by the acetyl group could affect the inhibitory potency. The same result was also observed in compounds 11 and 12, when the benzene ring was replaced by a pyridine ring. In addition, recent studies have shown that polar substitutions (e.g. hydroxyl and carboxylate group) on the phenyl ring of cinnamic acid or the phenyl ring of the 4-dihydroxycinnamoyloxy group decreased the inhibition for AKR1C3 [37, 46, 47]. For example, the displacement of 4-dihydrocinnamoyloxy group by a hydroxyl group to form drupanin (see Table 1) resulted in a loss of inhibitory potency (IC50: 15 μM) and only a 7-fold selectivity for AKR1C3 versus AKR1C2.
Figure 2.

Model of the AKR1C3.NADP+.Baccharin complex (left) and structure of baccharin in the orientation found in the model (right). AKR1C3 residues (light blue); NADP+ (pink); and baccharin (green). Dotted line: possible hydrogen bond; OX: oxyanion site; SP: subpocket. The docking model of the AKR1C3. NADP+.Baccharin complex was constructed by conducting calculations using the program Glide 5.0. The generated docked structure showing the highest score was selected for further structural analysis. The crystal structure of AKR1C3.NADP+ complex was chosen from RCSB protein data bank (PDB code: 1S2C) as a starting template. Reprinted with permission from (S. Endo et al., Selective inhibition of human type-5 17β-hydroxysteroid dehydrogenase (AKR1C3) by baccharin, a component of Brazilian propolis, Journal of Natural Products [37]). Copyright 2012, American Chemical Society.
Analogs in group 4 (Table 4) gave similar findings. In group 4, three naturally cinnamic acid derivatives (p-coumaric acid, m-coumaric acid and ferulic acid) in which a 4-hydroxyl group was introduced into the phenyl ring showed weak inhibition for AKR1C3; however, the introduction of 5-methoxyl group in ferulic acid (compound 16) increased the inhibitory potency and selectivity for AKR1C3 in comparison to p- and m-coumaric acid. We conclude it is important to either retain the 4′-dihydrocinnamolyoxy group or acetate substituent on cinnamic acid in baccharin analogs to maintain a high inhibitory potency towards AKR1C3 since none of the cinnamic acid analogs in Table 4 inhibited AKR1C3 with high potency.
In comparison to 1 and 2, esterification (compounds 3–7 shown in Table 2) of cinnamic acid group decreased inhibitory potency and selectivity to AKR1C3. In the AKR1C3.NADP+.Baccharin complex model [37], the carboxylate group on cinnamic acid was predicted to be bound in the SP1 pocket and could form hydrogen bonds with the side chain of Ser118 (Figure 2), which contributes to strong binding affinity for 1. In addition, several polar amino acids (e.g. Ser308 and Tyr319) also reside in the SP1 pocket of AKR1C3 which provide hydroxyl groups that can interact with the polar groups of these inhibitors. As such, these polar interactions also increase the selectivity of baccharin for AKR1C3 over the other AKR1C isoforms where the amino acids in the corresponding positions are Phe118, Leu308 and Phe319 in the SP1 pocket [15, 26]. Once the carboxylate group was esterified, the H-bond and polar interactions could be impaired or lost by the introduction of alkyl groups. Thus, both inhibitory potency and selectivity of 1 and 2 for AKR1C3 were greatly decreased and the inhibitory potency for AKR1C2 was slightly increased in compounds 3–6 (Table 2). A similar result was also observed when compound 13 was compared to compound 10 (Table 3). Notably, compound 7 in group 2 displayed much lower inhibitory potency for AKR1C3 and AKR1C2. This may be due to a steric effect caused by the introduction of the tertiary butyl group into baccharin, which may prevent it occupying the SP1 pocket. Therefore, the carboxylate group on cinnamic acid is essential to retain both inhibitory potency and selectivity of baccharin analogs and should not be blocked.
Although removal of 3-prenyl group in compounds 8–11 (group 3 analogs in Table 3) displayed a low or sub- micromolar inhibitory potency, the inhibitory selectivity was much lower than compound 1. The AKR1C3.NADP+.Baccharin docked structure predicted that the prenyl group also occupied the SP3 pocket [37]. The 3-prenyl group provides extra hydrophobic interactions between baccharin and the SP3 pocket and assists the orientation of the cinnamic acid and 4-dihydrocinnamoyloxy group in the SP1 and SP3 pockets, respectively. The loss of the 3-prenyl group may cause a change of the orientation of baccharin analogs and further decrease binding selectivity, which suggests that it is important to retain the 3-prenyl group on baccharin analogs to specifically bind to AKR1C3. Recently, Endo et al. reported that the replacement of the 3-prenyl group by either an aryl or alkyl ether yielded compounds that displayed less or similar inhibitory potency for AKR1C3. However, among these compounds, a more potent and selective inhibitor than baccharin was obtained when the 3-prenyl group was replaced by a 3-hydroxybenzyloxy group to yield a compound with an IC50 value of 20 nM for AKR1C3 and a 4–5 fold higher selectivity for AKR1C3 than baccharin [47]. Molecular docking showed that the benzyl ether group was close to Tyr24 in the SP3 pocket and that the addition of a hydroxy group on the phenyl ring produced a new H-bond with Tyr24, which improved the inhibitory potency and selectivity of baccharin [47]. These results suggest that the 3-prenly group should be retained in order to keep the inhibitory potency and selectivity of baccharin. Furthermore, inhibitory potency may be enhanced by the replacement of the 3-prenyl group by a rationally designed non-prenyl group. In addition, a comparison within the group 3 analogs (compounds 8–13) shows that compound 9 displayed the highest inhibitory potency and specificity to AKR1C3. A similar result was also observed in compounds 14 and 15 (Table 4), which suggests that the change in position of the dihydrocinnamoyloxy group from para- to meta- should be considered in the design of new baccharin analogs.
In conclusion, the current study focused on the screening of 15 baccharin analogs to discover new classes of inhibitors against AKR1C3. Although unsubstituted baccharin is still the most potent and selective inhibitor among these compounds, these data provide additional structural information for the future development of new baccharin analogs. The analyses of structure-activity relationship reveals that, in order to maintain a high inhibitory potency and selectivity, the carboxylate group on cinnamic acid, the prenyl group and either retention of 4′-dihydrocinnamoyloxy group or acetate substituent on cinnamic acid should be retained.
Highlights.
Inhibitory potencies of baccharin and fifteen analogs for AKR1C3 were evaluated.
Thirteen baccharin analogs were evaluated for the first time.
Unmodified baccharin still shows the highest inhibitory activity and selectivity.
Compound 2 shows a good inhibitory potency and high selectivity for AKR1C3.
Data provide structural information for the optimization of new baccharin analogs.
Acknowledgments
This work was supported by the NIH and NIEHS (R01-CA90744 and P30-ES013508 to T.M.P) and by Texas Tech University Health Sciences Center (P.C.T).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Miyamoto H, Messing EM, Chang C. Androgen deprivation therapy for prostate cancer: current status and future prospects. Prostate. 2004;61:332–353. doi: 10.1002/pros.20115. [DOI] [PubMed] [Google Scholar]
- 3.Perlmutter MA, Lepor H. Androgen deprivation therapy in the treatment of advanced prostate cancer. Rev Urol. 2007;9(Suppl 1):S3–8. [PMC free article] [PubMed] [Google Scholar]
- 4.Harris WP, Mostaghel EA, Nelson PS, Montgomery B. Androgen deprivation therapy: progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat Clin Pract Urol. 2009;6:76–85. doi: 10.1038/ncpuro1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, Ettinger SL, Gleave ME, Nelson CC. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008;68:6407–6415. doi: 10.1158/0008-5472.CAN-07-5997. [DOI] [PubMed] [Google Scholar]
- 6.Penning TM. Androgen biosynthesis in castration-resistant prostate cancer. Endocr Relat Cancer. 2014;21:T67–T78. doi: 10.1530/ERC-14-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bishr M, Saad F. Overview of the latest treatments for castration-resistant prostate cancer. Nat Rev Urol. 2013;10:522–528. doi: 10.1038/nrurol.2013.137. [DOI] [PubMed] [Google Scholar]
- 8.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB, Jr, Saad F, Staffurth JN, Mainwaring P, Harland S, Flaig TW, Hutson TE, Cheng T, Patterson H, Hainsworth JD, Ryan CJ, Sternberg CN, Ellard SL, Flechon A, Saleh M, Scholz M, Efstathiou E, Zivi A, Bianchini D, Loriot Y, Chieffo N, Kheoh T, Haqq CM, Scher HI C.-A.-. Investigators. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Logothetis CJ, Efstathiou E, Manuguid F, Kirkpatrick P. Abiraterone acetate. Nat Rev Drug Discov. 2011;10:573–574. doi: 10.1038/nrd3516. [DOI] [PubMed] [Google Scholar]
- 10.Rehman Y, Rosenberg JE. Abiraterone acetate: oral androgen biosynthesis inhibitor for treatment of castration-resistant prostate cancer. Drug Des Devel Ther. 2012;6:13–18. doi: 10.2147/DDDT.S15850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, Mostaghel EA, Marck B, Matsumoto AM, Simon NI, Wang H, Chen S, Balk SP. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011;71:6503–6513. doi: 10.1158/0008-5472.CAN-11-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, Matsumoto AM, Nelson PS, Montgomery RB. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011;17:5913–5925. doi: 10.1158/1078-0432.CCR-11-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adeniji AO, Chen M, Penning TM. AKR1C3 as a target in castrate resistant prostate cancer. J Steroid Biochem Mol Biol. 2013;137:136–149. doi: 10.1016/j.jsbmb.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adeniji AO, Twenter BM, Byrns MC, Jin Y, Winkler JD, Penning TM. Discovery of substituted 3-(phenylamino)benzoic acids as potent and selective inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3) Bioorg Med Chem Lett. 2011;21:1464–1468. doi: 10.1016/j.bmcl.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Byrns MC, Jin Y, Penning TM. Inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3): overview and structural insights. J Steroid Biochem Mol Biol. 2011;125:95–104. doi: 10.1016/j.jsbmb.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, Nelson PS. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447–4454. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hofland J, van Weerden WM, Dits NF, Steenbergen J, van Leenders GJ, Jenster G, Schroder FH, de Jong FH. Evidence of limited contributions for intratumoral steroidogenesis in prostate cancer. Cancer Res. 2010;70:1256–1264. doi: 10.1158/0008-5472.CAN-09-2092. [DOI] [PubMed] [Google Scholar]
- 18.Pfeiffer MJ, Smit FP, Sedelaar JP, Schalken JA. Steroidogenic enzymes and stem cell markers are upregulated during androgen deprivation in prostate cancer. Mol Med. 2011;17:657–664. doi: 10.2119/molmed.2010.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamid AR, Pfeiffer MJ, Verhaegh GW, Schaafsma E, Brandt A, Sweep FC, Sedelaar JP, Schalken JA. Aldo-keto reductase family 1 member C3 (AKR1C3) is a biomarker and therapeutic target for castration-resistant prostate cancer. Mol Med. 2012;18:1449–1455. doi: 10.2119/molmed.2012.00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Downs TM, Burton DW, Araiza FL, Hastings RH, Deftos LJ. PTHrP stimulates prostate cancer cell growth and upregulates aldo-keto reductase 1C3. Cancer Lett. 2011;306:52–59. doi: 10.1016/j.canlet.2011.02.027. [DOI] [PubMed] [Google Scholar]
- 21.Yepuru M, Wu Z, Kulkarni A, Yin F, Barrett CM, Kim J, Steiner MS, Miller DD, Dalton JT, Narayanan R. Steroidogenic enzyme AKR1C3 is a novel androgen receptor-selective coactivator that promotes prostate cancer growth. Clin Cancer Res. 2013;19:5613–5625. doi: 10.1158/1078-0432.CCR-13-1151. [DOI] [PubMed] [Google Scholar]
- 22.Yin YD, Fu M, Brooke DG, Heinrich DM, Denny WA, Jamieson SM. The activity of SN33638, an inhibitor of AKR1C3, on testosterone and 17β-estradiol production and function in castration-resistant prostate cancer and ER-positive breast cancer. Front Oncol. 2014;4:159. doi: 10.3389/fonc.2014.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bydal P, Luu-The V, Labrie F, Poirier D. Steroidal lactones as inhibitors of 17β-hydroxysteroid dehydrogenase type 5: chemical synthesis, enzyme inhibitory activity, and assessment of estrogenic and androgenic activities. Eur J Med Chem. 2009;44:632–644. doi: 10.1016/j.ejmech.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 24.Bauman DR, Rudnick SI, Szewczuk LM, Jin Y, Gopishetty S, Penning TM. Development of nonsteroidal anti-inflammatory drug analogs and steroid carboxylates selective for human aldo-keto reductase isoforms: potential antineoplastic agents that work independently of cyclooxygenase isozymes. Mol Pharmacol. 2005;67:60–68. doi: 10.1124/mol.104.006569. [DOI] [PubMed] [Google Scholar]
- 25.Byrns MC, Steckelbroeck S, Penning TM. An indomethacin analogue, N-(4-chlorobenzoyl)-melatonin, is a selective inhibitor of aldo-keto reductase 1C3 (type 2 3α-HSD, type 5 17β-HSD, and prostaglandin F synthase), a potential target for the treatment of hormone dependent and hormone independent malignancies. Biochem Pharmacol. 2008;75:484–493. doi: 10.1016/j.bcp.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adeniji AO, Twenter BM, Byrns MC, Jin Y, Chen M, Winkler JD, Penning TM. Development of potent and selective inhibitors of aldo-keto reductase 1C3 (type 5 17β-hydroxysteroid dehydrogenase) based on N-phenyl-aminobenzoates and their structure-activity relationships. J Med Chem. 2012;55:2311–2323. doi: 10.1021/jm201547v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Byrns MC, Penning TM. Type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase (AKR1C3): role in breast cancer and inhibition by non-steroidal anti-inflammatory drug analogs. Chem Biol Interact. 2009;178:221–227. doi: 10.1016/j.cbi.2008.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Penning TM, Burczynski ME, Jez JM, Hung CF, Lin HK, Ma H, Moore M, Palackal N, Ratnam K. Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem J. 2000;351:67–77. doi: 10.1042/0264-6021:3510067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steckelbroeck S, Jin Y, Gopishetty S, Oyesanmi B, Penning TM. Human cytosolic 3α-hydroxysteroid dehydrogenases of the aldo-keto reductase superfamily display significant 3β-hydroxysteroid dehydrogenase activity: implications for steroid hormone metabolism and action. J Biol Chem. 2004;279:10784–10795. doi: 10.1074/jbc.M313308200. [DOI] [PubMed] [Google Scholar]
- 30.Akao Y, Maruyama H, Matsumoto K, Ohguchi K, Nishizawa K, Sakamoto T, Araki Y, Mishima S, Nozawa Y. Cell growth inhibitory effect of cinnamic acid derivatives from propolis on human tumor cell lines. Biol Pharm Bull. 2003;26:1057–1059. doi: 10.1248/bpb.26.1057. [DOI] [PubMed] [Google Scholar]
- 31.Mishima S, Ono Y, Araki Y, Akao Y, Nozawa Y. Two related cinnamic acid derivatives from Brazilian honey bee propolis, baccharin and drupanin, induce growth inhibition in allografted sarcoma S-180 in mice. Biol Pharm Bull. 2005;28:1025–1030. doi: 10.1248/bpb.28.1025. [DOI] [PubMed] [Google Scholar]
- 32.Cestari SH, Bastos JK, Di Stasi LC. Intestinal anti-inflammatory activity of Baccharis dracunculifolia in the trinitrobenzenesulphonic acid model of rat colitis. Evid Based Complement Alternat Med. 2011;2011:524349. doi: 10.1093/ecam/nep081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teixeira EW, Message D, Negri G, Salatino A, Stringheta PC. Seasonal variation, chemical composition and antioxidant activity of Brazilian propolis samples. Evid Based Complement Alternat Med. 2010;7:307–315. doi: 10.1093/ecam/nem177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De P, Baltas M, Bedos-Belval F. Cinnamic acid derivatives as anticancer agents-a review. Curr Med Chem. 2011;18:1672–1703. doi: 10.2174/092986711795471347. [DOI] [PubMed] [Google Scholar]
- 35.Jitareanu A, Tataringa G, Zbancioc AM, Tuchilus C, Stanescu U. Antimicrobial activity of some cinnamic acid derivatives. Rev Med Chir Soc Med Nat Iasi. 2011;115:965–971. [PubMed] [Google Scholar]
- 36.Burdock GA. Review of the biological properties and toxicity of bee propolis (propolis) Food Chem Toxicol. 1998;36:347–363. doi: 10.1016/s0278-6915(97)00145-2. [DOI] [PubMed] [Google Scholar]
- 37.Endo S, Matsunaga T, Kanamori A, Otsuji Y, Nagai H, Sundaram K, El-Kabbani O, Toyooka N, Ohta S, Hara A. Selective inhibition of human type-5 17β-hydroxysteroid dehydrogenase (AKR1C3) by baccharin, a component of Brazilian propolis. J Nat Prod. 2012;75:716–721. doi: 10.1021/np201002x. [DOI] [PubMed] [Google Scholar]
- 38.Tani H, Hasumi K, Tatefuji T, Hashimoto K, Koshino H, Takahashi S. Inhibitory activity of Brazilian green propolis components and their derivatives on the release of cys-leukotrienes. Bioorg Med Chem. 2010;18:151–157. doi: 10.1016/j.bmc.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 39.Liedtke AJ, Adeniji AO, Chen M, Byrns MC, Jin Y, Christianson DW, Marnett LJ, Penning TM. Development of potent and selective indomethacin analogues for the inhibition of AKR1C3 (Type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase) in castrate-resistant prostate cancer. J Med Chem. 2013;56:2429–2446. doi: 10.1021/jm3017656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurek-Gorecka A, Rzepecka-Stojko A, Gorecki M, Stojko J, Sosada M, Swierczek-Zieba G. Structure and antioxidant activity of polyphenols derived from propolis. Molecules. 2013;19:78–101. doi: 10.3390/molecules19010078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gazvoda M, Beranic N, Turk S, Burja B, Kocevar M, Rizner TL, Gobec S, Polanc S. 2,3-Diarylpropenoic acids as selective non-steroidal inhibitors of type-5 17β-hydroxysteroid dehydrogenase (AKR1C3) Eur J Med Chem. 2013;62:89–97. doi: 10.1016/j.ejmech.2012.12.045. [DOI] [PubMed] [Google Scholar]
- 42.Heinrich DM, Flanagan JU, Jamieson SM, Silva S, Rigoreau LJ, Trivier E, Raynham T, Turnbull AP, Denny WA. Synthesis and structure-activity relationships for 1-(4-(piperidin-1-ylsulfonyl)phenyl)pyrrolidin-2-ones as novel non-carboxylate inhibitors of the aldo-keto reductase enzyme AKR1C3. Eur J Med Chem. 2013;62:738–744. doi: 10.1016/j.ejmech.2013.01.047. [DOI] [PubMed] [Google Scholar]
- 43.Brozic P, Turk S, Adeniji AO, Konc J, Janezic D, Penning TM, Lanisnik Rizner T, Gobec S. Selective inhibitors of aldo-keto reductases AKR1C1 and AKR1C3 discovered by virtual screening of a fragment library. J Med Chem. 2012;55:7417–7424. doi: 10.1021/jm300841n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jamieson SM, Brooke DG, Heinrich D, Atwell GJ, Silva S, Hamilton EJ, Turnbull AP, Rigoreau LJ, Trivier E, Soudy C, Samlal SS, Owen PJ, Schroeder E, Raynham T, Flanagan JU, Denny WA. 3-(3,4-Dihydroisoquinolin-2(1H)-ylsulfonyl)benzoic Acids: highly potent and selective inhibitors of the type 5 17β-hydroxysteroid dehydrogenase AKR1C3. J Med Chem. 2012;55:7746–7758. doi: 10.1021/jm3007867. [DOI] [PubMed] [Google Scholar]
- 45.Sinreih M, Sosic I, Beranic N, Turk S, Adeniji AO, Penning TM, Rizner TL, Gobec S. N-Benzoyl anthranilic acid derivatives as selective inhibitors of aldo-keto reductase AKR1C3. Bioorg Med Chem Lett. 2012;22:5948–5951. doi: 10.1016/j.bmcl.2012.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brozic P, Golob B, Gomboc N, Rizner TL, Gobec S. Cinnamic acids as new inhibitors of 17β-hydroxysteroid dehydrogenase type 5 (AKR1C3) Mol Cell Endocrinol. 2006;248:233–235. doi: 10.1016/j.mce.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 47.Endo S, Hu D, Matsunaga T, Otsuji Y, El-Kabbani O, Kandeel M, Ikari A, Hara A, Kitade Y, Toyooka N. Synthesis of non-prenyl analogues of baccharin as selective and potent inhibitors for aldo-keto reductase 1C3. Bioorg Med Chem. 2014 doi: 10.1016/j.bmc.2014.08.007. [DOI] [PubMed] [Google Scholar]