

Abstract
Mutation of human chromosome 15q13.3 increases the risk for autism and schizophrenia. One of the noteworthy genes in 15q13.3 is CHRNA7, which encodes the nicotinic acetylcholine receptor alpha 7 subunit (α7nAChR) associated with schizophrenia in clinical studies and rodent models. This study investigates the role of α7nAChR in maternal immune activation (MIA) mice model, a murine model of environmental risk factor for autism and schizophrenia. We provided choline, a selective α7nAChR agonist among its several developmental roles, in the diet of C57BL/6N wild-type dams throughout the gestation and lactation period and induced MIA at mid-gestation. The adult offspring behavior and gene expression profile in the maternal spleen-placenta-fetal brain axis at mid-gestation were investigated. We found that choline supplementation prevented several MIA-induced behavioral abnormalities in the wild-type offspring. Pro-inflammatory cytokine interleukin-6 (IL-6) and Chrna7 gene expression in the wild-type fetal brain were elevated by poly(I:C) injection and were suppressed by gestational choline supplementation. We further investigated the gene expression level of IL-6 in Chrna7 mutant mice. We found that the basal level of IL-6 was higher in Chrna7 mutant fetal brain, which suggests that α7nAChR may serve an anti-inflammatory role in the fetal brain during development. Lastly, we induced MIA in Chrna7+/− offspring. The Chrna7+/− offspring were more vulnerable to MIA, with increased behavioral abnormalities. Our study shows that α7nAChR modulates inflammatory response affecting the fetal brain and demonstrates its effects on offspring behavior development after MIA.
Keywords: maternal immune activation (MIA), maternal infection, poly(I:C), nicotinic acetylcholine receptor alpha 7 subunit (α7AChR; CHRNA7; Chrna7), choline supplementation, interleukin-6 (IL-6), schizophrenia, autism
1. Introduction
Autism and schizophrenia are neurodevelopmental disorders that stem from genetic inheritance and environmental influences (Hallmayer et al., 2011; Persico and Bourgeron, 2006; Tsuang, 2000). Epidemiology studies indicate that viral or bacterial infections of pregnant mothers increase the risk for schizophrenia and autism in the offspring (Patterson, 2009). A promising explanation for the link between maternal infection and the occurrence of autism and schizophrenia is MIA (Boksa, 2010; Brown and Patterson, 2011; Meyer and Feldon, 2010; Meyer et al., 2005; Patterson, 2002, 2009, 2011a, b). Briefly, MIA represents a state of activated immune system in infected, pregnant women that leads to immunological or physiological changes in the mothers, eventually perturbing fetal neurodevelopment (Garbett et al., 2012; Meyer et al., 2008a; Soumiya et al., 2011; Stolp et al., 2011).
The hypothesis that MIA increases the risk for autism and schizophrenia in the offspring has been tested by injecting a viral mimic compound, poly(I:C), into pregnant mice. Injection of poly(I:C) alters the cytokine levels in both mother and fetuses (Abazyan et al., 2010; Connor et al., 2012; Garay et al., 2013; Harvey and Boksa, 2012; Hsiao and Patterson, 2011; Meyer et al., 2006; Missault et al., 2014; O’Leary et al., 2014; Pratt et al., 2013; Vuillermot et al., 2012). In particular, pro-inflammatory cytokine IL-6 has been shown to play a critical role in MIA associated autistic- and schizophrenia-like behaviors (Hsiao and Patterson, 2011; Smith et al., 2007). IL-6 was found to be elevated in maternal serum, placenta, and fetal brain after poly(I:C) injection into pregnant mice (Connor et al., 2012; Harvey and Boksa, 2012; Hsiao and Patterson, 2011; O’Leary et al., 2014; Pratt et al., 2013; Vuillermot et al., 2012). Blockade of IL-6 with antibody prevents the MIA offspring from displaying autistic- and schizophrenia-like behaviors (Smith et al., 2007). Furthermore, injecting recombinant IL-6 into pregnant mice led to behavioral abnormalities in the offspring similar to those found in MIA offspring (Hsiao and Patterson, 2011; Smith et al., 2007). This evidence points to IL-6 as a key mediator in the development of behavioral abnormalities caused by MIA.
Molecular mechanisms underlying MIA have been investigated by induction of MIA in genetically altered mice and testing for their physiological or behavioral changes. Autism and schizophrenia susceptibility genes are considered as identifying possible mechanisms involved in MIA. To date, induction of MIA in the mice with disrupted-in-schizophrenia 1 (Disc1), neuregulin 1 (Nrg1), nuclear receptor related 1 protein (Nurr1), tuberous sclerosis 2 (Tsc2), or interleukin 10 (Il10) mutation produced exacerbated effect in offspring behaviors and brain functionalities (Abazyan et al., 2010; Ehninger et al., 2012; Ibi et al., 2010; Lipina et al., 2013; Meyer et al., 2008b; O’Leary et al., 2014; Vuillermot et al., 2012). Dysfunction of the cholinergic system has been associated with the etiology of autism and schizophrenia (Adams and Stevens, 2007; Karvat and Kimchi, 2014; McTighe et al., 2013; Miwa et al., 2011; Pratt et al., 2013; Ross et al., 2010), a process that, in the MIA system, may act through α7nAChR agonism. Supporting this hypothesis, mutations or splice variations of the nicotinic acetylcholine receptor alpha 7 subunit (Protein: α7nAChR, Human: CHRNA7, Mouse: Chrna7) have been associated with autistic and schizophrenic patients (Freedman et al., 1997; Leonard et al., 2002; Ross et al., 2010; Yasui et al., 2011). α7nAChR-expressing neurons emerge at an early stage of development and serve different functions, many of which are involved in inhibition in the hippocampus and regulation of sensory gating (Adams et al., 2002; Miwa et al., 2011; Ross et al., 2010). Perinatal choline dietary supplementation increases hippocampal α7nAChR levels and improves hippocampal sensory gating in DBA/2 mice, which have sensory gating deficit similar to that found in schizophrenia and associated with Chrna7 and hippocampal α7nAChRs (Stevens et al., 2008). Although choline has many roles in fetal development, its role includes acting as a specific agonist of α7nAChR (Alkondon et al., 1997). Its specific activity at the α7nAChR to promote the development of sensory inhibition was confirmed by the demonstration that DBA/2 Chrna7 null mutants do not demonstrate the ameliorating effect of perinatal choline supplementation found in the parental strain (Stevens et al., 2014). We hypothesized that α7nAChR agonism may be involved in MIA-induced behavioral abnormalities.
In this study, we investigate how α7nAChRs interact with the environmental factor, MIA, to alter behavior in the offspring. We provided mid-gestational poly(I:C)-injected wild-type dams with choline supplementation in the daily diet through the period of gestation and lactation and tested for offspring behavior. To understand how α7nAChRs agonism alters MIA fetal brain IL-6 production, we analyzed gene expression in the maternal spleen-placenta-fetal brain axis at the early stage of MIA with perinatal choline supplementation. To further characterize the role of α7nAChR in MIA-induced cytokine changes in fetus and behavioral abnormalities in the adult offspring, we examined cytokine changes in Chrna7 mutant fetal brain and adult behavior alteration in Chrna7 mutant offspring.
2. Materials and Methods
2.1. Mice
Wild-type C57BL/6N mice were obtained through Caltech’s Braun animal facility (originally from Charles River, Wilmington, MA, USA). Chrna7−/− mice were obtained from Jackson Laboratory (Bar Harbor, ME, USA) and then crossed with the wild-type mice to yield heterozygous mice. The colony was maintained by crossing heterozygous breeding pairs. Genotyping of Chrna7 mutant mice followed instructions from Jackson Laboratory. Mice were transferred and maintained at Caltech’s Broad animal facility. All mice were group housed (2–5 per cage) with a 13 hours light/11 hours dark cycle (lights on at 06:00) at 21–23°C and 45% relative humidity within a range of 30–70% in ventilated cages (Super Mouse 750™, Lab Products Inc, Seaford, DE, USA). We fed the mice which were not involved in maternal choline supplementation experiment with the Irradiated PicoLab Rodent Diet (5053, Lad Diet, St. Louis, MO, USA). For the pregnant and lactating mice, we fed the mice with a mix of half 5053 PicoLab Rodent Diet and half 5058 PicoLab Rodent Diet (5053, Lad Diet, St. Louis, MO, USA). Total number of mice we used in this study is listed in supplementary Tables 1 and 2. All experiments were performed under the approval of the California Institute of Technology Institutional Animal Care and Use Committee.
2.2. Timed-mating for Chrna7 mutant and C57BL/6N wild-type mice
Timed-mating pairs were set up in the late phase of the light period. Vaginal plugs were checked the next morning. The day of vaginal plug presence was termed E0.5. Two independent mouse lines were used in this study-C57BL/6N wild-type line and Chrna7 mutant line. For the maternal choline supplementation experiments, both genders were wild-type C57BL/6N mice (Originally from Charles River, Wilmington, MA, USA). For testing the MIA-induced autistic and schizophrenia behavior in Chrna7 mutant mice, our goal was to compare the wild-type and heterozygous mice, instead of knockout mice. The timed-mating pair we used was male Chrna7+/− and female wild-type mice. For the cytokine-related gene expression analysis in Chrna7 fetal brain, we included all three genotypes. The timed-mating pair we used was male Chrna7+/− and female Chrna7+/− mice.
2.3. Maternal immune activation (MIA)
Potassium salt poly(I:C) (P9582; Sigma, St. Louis, MO, USA) was used to induce MIA. 20mg/kg Poly(I:C) was injected i.p. into pregnant female mice at E12.5. We injected saline into pregnant female mice as control. All female mice were 8–16 weeks of age, and this was their first pregnancy.
2.4. Maternal choline supplementation
5 g/kg choline supplemented (#110184) and 1.1g/kg control choline (#110098) diets were purchased from DYETS INC (Bethlehem, PA, USA). The dose of choline supplementation was chosen because of the previous report of the effect of this dose of choline supplementation on the DBA/2 sensory gating deficit (Stevens et al., 2008; Stevens et al., 2014). After the timed-mating, plugged female mice were single housed at E0.5 and randomly assigned to either choline supplemented or control diet. Poly(I:C) or saline injection was randomly given to both groups at E12.5. Choline supplemented and control diets were provided through the entire pregnancy until weaning. After weaning, all offspring were given regular mouse chow (#5053; LabDiet; St. Louis, MO, USA).
2.5. Tissue sampling from E12.5 pregnant mice after MIA
Three hours after poly(I:C) injection, mice were deeply anesthetized by Euthasol (Virbac Animal Health, Fort Worth, TX, USA) and tissues harvested. This time point was based on prior work in MIA on the placenta (Hsiao and Patterson, 2011). Spleen, placenta and fetal brain were harvested from poly(I:C)- and saline-injected pregnant mice. Fetal brains were dissected under a stereomicroscope (M5A, Wild Heerbrugg, Switzerland). For gene expression analysis, tissues were stored in RNAlater (Qiagen, Gaithersburg, MD, USA) at −80°C. For immunohistochemistry, fetal brains were postfixed in 4% paraformaldehyde at 4°C for 30 min and then dehydrated in 30% sucrose at room temperature overnight. After dehydration, brains were embedded in OCT (Tissue-Tek, Torrance, CA, USA) and stored at −80 °C.
2.6. Splenic macrophage culture
The isolation of splenic macrophages was based on published methods with modifications (Zhu et al., 1995). Mice were deeply anesthetized by Euthasol and spleens immediately dissected and thoroughly washed in cold Dulbecco’s Modified Eagle Medium (DMEM), high glucose, GlutaMAX medium (Life Technologies, Carlsbad, CA, USA). Spleen pieces were ground between sterilized microscope slides to make a single cell suspension. The cell suspension was centrifuged at 1000 rpm for 15min. The pellet was re-suspended in 3 ml medium with 10% fetal bovine serum (FBS) and then seeded into 6 well cell culture plates (22.6 mm diameter, Corning Inc, Acton, MA, USA). After seeding, 1 ml DMEM with 10% FBS was added to each well. The plates were then incubated at 37 °C in a 5% CO2 for 2 hr, after which non-adherent cells were removed by washing with DMEM. At this stage, the majority of the viable cells adherent to the plate appeared to be macrophages, based on morphology. For experimentation, the macrophages were collected by mechanically washing the plate with DMEM.
2.7. RNA extraction and quantitative reverse transcription (qRT-PCR)
The RNA extraction of spleen, macrophages, placenta, and fetal brain was based on the manufacturer’s protocol (Trizol; Life Technologies, Grand Island, NY, USA). The RNA concentration and quality were measured by NanoDrop (Thermo Scientific, Wilmington, DE, USA). Before the reverse transcription, RNA was treated with DNase I (Promega, San Luis Obispo, CA, USA) to eliminate DNA contamination. 1μg RNA from each sample was reverse transcribed by using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA). mRNA expression was measured using FastStart Universal SYBR Green master mix with ROX (Roche, El Cerrito, CA, USA) on the ABI 7300 real-time PCR system (Life Technologies, Carlsbad, CA, USA). Gene expression was normalized to either β-actin or GAPDH mRNA. Data are presented as fold-change in gene expression in each group relative to that in the maternal saline control group. The primer sequences were adapted from PrimerBank database (Spandidos et al., 2010).
2.8. Immunohistochemistry
For fetal brain immunostaining, whole embryos were cut in 16 μm thickness in sagittal direction and the sections were adhered to Superfrost Plus microscope slide (Fisher Scientific, Tustin, CA, USA). After adherence, a barrier was drawn on the edge of microscope slide by using ImmEdge Hydrophobic Barrier Pen (Vector Laboratories Inc, Burlingame, CA, USA) for the convenience of staining.
For staining, the slides were incubated with blocking solution (10% horse serum, 0.1% triton X-100, and 0.02% sodium azide in PBS) for 1 hr at room temperature. After blocking, slides were incubated in primary antibody overnight at 4 °C. The next day, the slides were incubated in fluorescence-conjugated secondary antibody for 1.5 hr at room temperature, and then washed in PBS. PBST (PBS with 0.1% triton X-100) served as the first wash to increase the penetration of the antibody and PBS served as rest of the washing solutions. The primary antibodies and their dilutions were goat anti-α7nAChR (1:100; Santa Cruz, CA, USA), rabbit anti-IL-6R (1:100; Santa Cruz, CA, USA), mouse anti-NeuN (1:500; Millipore, Billerica, MA, USA), mouse anti-MAP2 (1:100; Sigma, St. Louis, MO, USA), rabbit anti-DCX (1:100; Cell Signaling, Danvers, MA, USA), mouse anti-Nestin (1:100; Millipore, Billerica, MA, USA). The secondary antibodies were fluorescence-conjugated donkey anti-goat (1:1000), donkey anti-rabbit (1:1000), and donkey anti-mouse (1:1000) (all from Molecular Probes, Life Technologies, Carlsbad, CA, USA). After staining, ProLong gold, anti-fade mounting medium (Molecular Probe, Life Technologies, Carlsbad, CA, USA) was applied to the slide before coverslip mounting. For imaging, the fluorescence images were taken by fluorescence microscope (Nikon DIAPHOT 300, Nikon, Tokyo, Japan) with SPOT software (V4.6, Sterling Heights, MI, USA). For confocal imaging, TCS SP confocal microscope (Leica, Exton, PA, USA) equipped with argon and krypton was used to image the sections. Quantification of α7nAChR optical density was analyzed by ImageJ software (NIH, Bethesda, MD, USA).
2.9. α-bungarotoxin binding
2.9.1. [125I]-α-bungarotoxin (BTX) binding
Fresh-frozen total and nonspecific tissue sections (12 μM) were collected through the dorsal hippocampus, and preincubated in a Tris/saline (TBS) buffer containing bovine serum albumin (BSA) (TBS/BSA) (50 mM Tris-HCl, 120 mM NaCl, 2 mg/ml bovine serum albumin, pH 7.4, Sigma Aldrich, St. Louis, MO) for 30 minutes at room temperature (RT), with nonspecific binding defined by blocking with 50 nM unlabeled α-BTX (Tocris Bioscience, Bristol, UK). Sections were incubated in TBS/BSA buffer containing [125I]-α-BTX (5 nM, specific activity 2000 Ci/mmol, Perkin-Elmer, Waltham, MA) at 37°C for 3 hours followed by rinsing with TBS/BSA buffer for 5 minutes, with TBS without BSA for 15 minutes and with PBS for 5 minutes, all at 37°C. Nonspecific binding was again defined by blocking with 50 nM unlabeled α-BTX. The slides were dipped briefly in ice cold, distilled water, dried quickly under a stream of cool air, dried overnight and placed on radiation-sensitive Hyperfilm (Amersham, Piscataway, NJ) for 72 hours with [14C] standards (Amersham, Piscataway, NJ) of known radioactivity to generate autoradiograms for quantitative analysis.
2.9.2 Quantitative autoradiography
Film images of tissue sections labeled with [125I]-α-BTX were captured using an LED light box (Schott North America, Inc., Elmsford, NY) and Retiga CCD camera (Qimaging, Surrey, BC, Canada) using Simple PCI software (Hamamatsu Corp., Sewickley, PA). Calibration curves were generated from [14C] standards to generate equations for converting gray values into tissue equivalent values (nCi/g tissue, ImageJ, NIH). Gray values were measured in all major subdivisions of the hippocampal formation for total and nonspecific binding. The gray values were converted to tissue equivalents and the nonspecific tissue binding values were subtracted from the total tissue equivalent values, resulting in specific tissue equivalent values, which were statistically analyzed.
2.10. Choline measurements
Choline/acetylcholine quantification kit (MAK056; Sigma, St. Louis, MO, USA) was used to determine choline level. The measurement was based on manufacturer’s protocol. Briefly, 50 μg fetal brain was homogenized in 200 μl of choline assay buffer respectively and centrifuged at 13,000 g for 10 min. The supernatants were stored at −20°C until used. 50μl of each sample was added to a 96 well plate (Corning Inc, Acton, MA, USA). Reaction mix cocktail was added to each sample and was mixed by gentle pipetting. The plate was then covered by foil and place on horizontal shaker for 30 min. Each plate contained choline standards of 0–5 nmole/well. The concentration in each sample was detected by microplate reader (SpectraMax 190, Molecular Devices, Sunnyvale, CA, USA) using absorbance at 570 nm (A570). All samples were measured in duplicate. Total protein of each sample was measured by BCA assay (Thermo Scientific, Rockford, IL, USA) according to the manual.
2.11. Behavior
Mice were labeled by ear punch and tail snips obtained for genotyping immediately after weaning. Both genders were used for the behavior assay. Unless specified, prepulse inhibition (PPI) was performed at 6 weeks of age. Open-field testing was performed at 7 weeks of age. Marble burying was also performed at 8 weeks of age. All the apparatuses were cleaned with 70% ethanol and then tap water between subjects. Cage bedding was not changed 3 days prior to behavioral testing. Mice were acclimated to the testing room at least 30 min before each behavior test.
2.11.1. Prepulse inhibition (PPI)
PPI is a typical test for detecting sensorimotor gating deficits. The procedure has been previously described (Smith et al., 2007). SR-LAB apparatus (San Diego Instruments, San Diego, CA, USA) was used to measure the PPI of mice. Mice were restrained in a plexiglass cylinder that has a piezo-electric sensor underneath it. Mice were acclimated to chamber for 5 min and then exposed to six trials of 120 dB white noise (Startle). After white noise exposure, mice were exposed to randomized mixtures of 14 trials of background noise, 14 trials of Startle, 14 trials of prepulse 5 dB (5 dB higher than background noise) + Startle (PPI5), and 14 trials of prepulse 15 dB (15 dB higher than background noise + Startle (PPI15). Each value was yielded from the average of 14 values. PPI was defined as (Startle - PPI5 or PPI15)/Startle.
2.11.2. Open field test
Open-field test is a task for testing anxiety level and general locomotion. The procedure has been previously described (Hsiao and Patterson, 2011). The open-field apparatus is a square open arena (50 × 50 cm) bordered by opaque plastic walls. Each mouse was placed in the arena, along a wall, and behavior was recorded for 10 min. The center zone (17 × 17 cm) was defined as the middle of the open-field chamber. The behavior in the open field was recorded by a video camera mounted over the arena. Ethovision (Noldus Information Technology, Leesburg, VA, USA) was used to analyze the number of entries and the duration in the center zone.
2.11.3. Marble burying test
Marble burying is a test for repetitive behavior. The procedure has been previously described (Malkova et al., 2012) with modification. The mouse was first acclimated to a test cage with compressed, 5-cm deep, clean Aspen pine bedding. After this habituation the mouse was returned to its home cage. 20 navy blue glass marbles (15 mm diameter) were gently placed on the bedding of the test cage (4 × 5 arrangement). The mouse was then returned to this test cage and, after 10 min, the number of buried marbles was counted. The criteria for a buried marble was over 50% of the marble covered by bedding.
2.12. Statistical analysis
All behavior results were analyzed by 2-way ANOVA with Fisher’s LSD post-hoc test. qPCR and immunohistochemistry results were compared by Student’s t test. Data were analyzed by a 3-way ANOVA assessing differences in [125I]-α-BTX binding in hippocampal across gender, choline treatment and saline/poly(I:C) treatment, with Holm-Sidak post-hoc test. All Pairwise Multiple Comparison Procedures a posterior analyses were performed where appropriate.
3. Results
3.1. Maternal choline supplementation prevented C57BL/6N wild-type offspring autistic- and schizophrenia-like behaviors induced by MIA
As a rodent model of autism and schizophrenia, MIA had previously been observed to decrease PPI, decrease center zone entering in open field test, and increase marble burying behavior (Hsiao and Patterson, 2011; Malkova et al., 2012; Smith et al., 2007). Choline treatment had a range of effects on these observations (Fig. 1). The clearest example of an alteration of the effect of MIA by choline treatment was in open field entries, where there was no change in behavior of the offspring who did not receive MIA, regardless of choline status, but for offspring who received MIA, choline treatment prevented the decrement in center entries normally observed with MIA (Saline v.s. Poly(I:C), p = 0.0641; Poly(I:C) v.s. Saline+Choline, p < 0.01; Poly(I:C) v.s. Poly(I:C)+Choline, p < 0.01) (Fig. 1A). MIA offspring showed decrement of time in center zone. There were no changes by choline treatment in the time spent in center zone for the offspring who received MIA (Saline v.s. Poly(I:C), p = 0.0689; Saline+Choline v.s. Poly(I:C)+Choline, p = 0.0570; Poly(I:C) v.s. Saline+Choline, p < 0.01) (Fig. 1B). An alteration of the effect of MIA by choline treatment was found in marble burying behavior. For the offspring who received MIA, choline treatment prevented the increment in marble burying behavior (Poly(I:C) v.s. Poly(I:C)+Choline, p < 0.05), which is normally observed with MIA (Saline v.s. Poly(I:C), p < 0.01). However, choline treatment also changed marble burying behavior for the offspring who did not receive MIA (Saline v.s. Saline+Choline, p < 0.05)(Fig. 1C). In PPI, MIA offspring showed reduction of inhibition in PPI 5db (Saline v.s. Poly(I:C), p < 0.01; Saline+Choline v.s. Poly(I:C)+Choline, p < 0.01) and PPI 15db (Saline v.s. Poly(I:C), p < 0.05; Saline+Choline v.s. Poly(I:C)+Choline, p = 0.0593), regardless of choline treatment (Fig. 1D–E).
Fig. 1.

Maternal choline supplementation ameliorated autistic- and schizophrenia-like behaviors in MIA wild-type offspring. (A–B) Maternal choline supplementation prevented the MIA-induced open field anxiety phenotype as assayed by center entries (A) but not center duration (B). (C) Maternal choline supplementation prevented repetitive/compulsive behavior in the marble burying test that was induced by MIA. MIA offspring given the control diet buried significantly more marbles than saline offspring. The MIA-induced marble burying behavior was reduced by maternal choline supplementation. (D–E) Maternal choline supplementation did not prevent the MIA-induced PPI deficit. Saline-Control diet: n = 6 litters; Poly(I:C)-Control diet: n = 7 litters; Saline-Choline diet: n = 7 litters; Poly(I:C)-Choline diet: n = 8 litters. Data are presented as Mean ± SEM. Significant difference between groups is labeled as * p < 0.05, ** p < 0.01, *** p < 0.001. n.s. : not significant.
3.2. α7nAChR level in adult hippocampus of MIA offspring
Hippocampal α7nAChR has been reported as a critical mediator for sensory gating (Stevens et al., 2008). Since some of the MIA offspring behavioral abnormalities can be ameliorated by maternal choline supplementation, changes in hippocampal α7nAChR expression levels is one of the mechanisms to explain the effect of maternal choline supplementation on the MIA wild-type offspring. We examined the α7nAChR level in hippocampus by qPCR of Chrna7 mRNA level and the binding of the α7nAChR ligand α-BTX. We found that the Chrna7 mRNA expression was significantly higher in MIA wild-type offspring hippocampus (p < 0.05) (Fig. 2A). Maternal choline supplementation also showed higher Chrna7 mRNA expression than saline wild-type offspring (p < 0.01)(Fig. 2A). Next, we further examined α7nAChR in MIA wild-type hippocampus by 125Iα-BTX binding (Fig. 2B–E). However, neither MIA nor maternal choline supplementation resulted in a difference of 125Iα-BTX binding at the hippocampal CA3, CA1 and dentate gyrus (DG) regions (Fig. 2D–E). This result indicates that hippocampal expression of α7nAChRs in adult animals may not directly contribute to the behavioral abnormalities induced by MIA.
Fig. 2.

α7nAChR expression in the hippocampus of adult MIA offspring. (A) Compared to control offspring, hippocampal Chrna7 mRNA expression was higher in MIA offspring. The elevation of Chrna7 mRNA expression was not affected by maternal choline supplementation diet, however. Chrna7 mRNA expression was normalized by Gapdh. Saline n = 4; Poly(I:C) n = 12, Poly(I:C)+Choline n = 8. (B) Film images of 125Iα-bungarotoxin binding to transverse sections through the hippocampus of saline and MIA offspring given a control or choline supplementation diet. The dashed lines illustrate how the subdivisions of the hippocampus were delineated. (C–E) The density of 125Iα–bungarotoxin (α–BTX) binding to sections of hippocampal formation from saline and MIA offspring given control or choline supplementation diets. Generally, no difference was detected among the groups. Male, Saline-Control diet: n = 3 litters; Poly(I:C)-Control diet: n = 3 litters; Saline-Choline diet: n = 3 litters; Poly(I:C)-Choline diet: n = 4 litters. Female. Saline-Control diet: n = 2 litters; Poly(I:C)-Control diet: n = 4 litters; Saline-Choline diet: n = 5 litters; Poly(I:C)-Choline diet: n = 4 litters. MIA and maternal choline supplementation did not affect the density of α–BTX binding in (C) CA3, (D) CA1 and (E) dentate gyrus (DG). Chrna7: nicotinic acetylcholine receptor alpha 7 subunit. Data are presented as Mean ± SEM. Significant difference between groups is labeled as * p < 0.05, ** p < 0.01.
3.3. Changes in cytokines and cholinergic gene expression after MIA in C57BL/6N wild-type dam spleen, splenic macrophages, and placenta
Studies show that α7nAChR is involved in the anti-inflammatory reflex of the CNS and periphery (Rosas-Ballina et al., 2011; Tracey, 2009). MIA had been shown to induce an acute inflammatory response in maternal-placental-fetal axis (Boksa, 2010; Patterson, 2011a). Therefore, we examined the MIA-induced acute inflammatory reaction in dam spleen, placenta and fetal brain. In the dam spleen and splenic macrophages, the increase in interleukin-6 (Il6) and tumor necrosis factor (Tnf) mRNA normally observed after poly(I:C) injection was moderately blunted by choline administration (Spleen-Saline v.s. Poly(I:C): Il6, p < 0.01; Tnf, p < 0.001; Saline v.s. Poly(I:C)+Choline: Il6, p < 0.01)(Splenic macrophage-Saline v.s. Poly(I:C): Il6, p < 0.05; Tnf, p = 0.0596;)(Fig 3A–B). The decrease in acetylcholinesterase (Ache) and beta-2 adrenergic receptor (Adrb2) mRNA seen in the dam spleen after MIA was not blunted by choline administration (Saline v.s. poly(I:C) and Saline v.s. Poly(I:C)+Choline, Ache, p < 0.01; Adrb2, p < 0.001) (Fig 3A). In the placenta, the increasing trend in Il6 mRNA was not blunted by choline treatment (Fig 4). Choline caused a fall in both Chrna7 (Saline vs. Poly(I:C)+Choline, p < 0.05; Poly(I:C) v.s. Poly(I:C)+Choline, p = 0.0555) and Ache mRNA expression (Poly(I:C) v.s. Poly(I:C)+Choline, p < 0.01)(Fig 4).
Fig. 3.

Cytokines and cholinergic signaling genes were altered in maternal spleen and splenic macrophage by maternal poly(I:C) injection, but not by maternal choline supplementation. (A) Il6 and Tnf mRNA were increased while Ache and Adrb2 were decreased in the dam’s spleen 3 hr after maternal poly(I:C) injection. Choline supplementation did not affect these changes in gene expression. Saline n = 3 dams; Poly(I:C) n = 4 dams, Poly(I:C)+Choline n = 4 dams. (B) Consistent with the results from the spleen, Il6 and Tnf mRNA were increased in macrophages after maternal poly(I:C) injection, and choline supplementation had little effect. Gene expression was normalized by β-actin. Saline n = 3; Poly(I:C) n = 4, Poly(I:C)+Choline n = 4. Ache: acetylcholinesterase, Adrb2: beta-2 adrenergic receptor, Chrna7: nicotinic acetylcholine receptor alpha 7 subunit, Il6: interleukin-6, Tnf: tumor necrosis factor alpha. Data are presented as Mean ± SEM. Significant difference between groups is labeled as * p < 0.05, ** p < 0.01, *** p < 0.001.
Fig. 4.

Cytokines and cholinergic signaling genes were altered in placenta by maternal poly(I:C) injection and maternal choline supplementation. Maternal poly(I:C) injection increased Il6 mRNA in the placenta, and maternal choline supplementation did not prevent this induction. Maternal choline supplementation lowered Chrna7 and Ache mRNA in placenta after poly(I:C) injection. Gene expression was normalized by β-actin. Saline n = 3 litters; Poly(I:C) n = 4 litters, Poly(I:C)+Choline n = 3 litters. Ache: acetylcholinesterase, Chrna7: nicotinic acetylcholine receptor alpha 7 subunit, Il6: interleukin-6. Data are presented as Mean ± SEM. Significant difference between groups is labeled as * p < 0.05, ** p < 0.01.
3.4. Maternal choline supplementation prevented Il6 and Chrna7 elevation in the C57BL/6N wild-type MIA fetal brain
IL-6 and cholinergic signaling mRNA levels were next analyzed in the wild-type fetal brain 3 hours after maternal poly(I:C) injection. Il6 mRNA was significantly increased in the wild-type fetal brain after MIA (p < 0.05)(Fig. 5A). Choline treatment significantly reduced the Il6 elevation in the fetal brains who received MIA (p < 0.05)(Fig. 5A). Several genes involved in the cholinergic system of the wild-type fetal brain were examined after poly(I:C) injection. Similar to the fluctuation of Il6, Chrna7 mRNA level was elevated in the wild-type fetal brain after MIA (p < 0.05). The elevation of Chrna7 mRNA in the MIA fetal brain was reduced with choline administration (p < 0.05)(Fig. 5A). No change was found in Ache and Slc5a7 (Choline transporter) mRNA expression (Fig. 5A). The choline content was next analyzed in the wild-type fetal brain who received MIA with maternal choline supplementation. The choline level was only increased in the offspring when the dams were given the choline supplementation (Saline v.s. Poly(I:C)+Choline, p < 0.05; Poly(I:C) v.s. Poly(I:C)+Choline, p < 0.05) (Fig. 5B).
Fig. 5.

Cytokines and cholinergic signaling genes were altered in fetal brain by maternal poly(I:C) injection and maternal choline supplementation. (A) Maternal poly(I:C) injection increased Il6 and Chrna7 mRNA in fetal brain, and maternal choline supplementation prevented these increases. No changes were found in Ache and Slc5a7 among groups. Gene expression was normalized by β-actin. Saline n = 3 litters; Poly(I:C) n = 3–4 litters, Poly(I:C)+Choline n = 3–4 litters. (B) Maternal choline supplementation was associated with increased trend of choline levels in MIA fetal brain. Each n = 3 litters. Ache: acetylcholinesterase, Chrna7: nicotinic acetylcholine receptor alpha 7 subunit, Il6: interleukin-6, Slc5a7: choline transporter 1. Data are presented as Mean ± SEM. Significant difference between groups is labeled as * p < 0.05.
3.5. MIA increased α7nAChR in mature neurons in the wild-type fetal brain
With immunohistochemistry staining for α7nAChR in the wild-type fetal brain, we observed an increase in the number of α7nAChR positive cells in the fetal brain 3 hours post maternal poly(I:C) injection. α7nAChR staining was expressed in several fetal brain regions. The most notable elevation of α7nAChR is in the fetal hindbrain region. More specifically, the α7nAChR positive staining was increased in prepontine hindbrain (PPH) and pontine hindbrain (PH) of fetal brain (Fig. 6A–B). The optic density of α7nAChR was significantly higher in MIA group than in saline group in PPH and PH of fetal hindbrain (p < 0.05)(Fig. 6C). By co-staining α7nAChR with neuron makers, we could identify the cell types of the α7nAChR positive cells in the MIA fetal hindbrain (Fig. 6D). α7nAChR was expressed with MAP2 and DCX, but not with nestin (Fig. 6D). This indicated that the α7nAChR positive cells we found in MIA fetal hindbrain are differentiated and migrated neurons. The increase of α7nAChR in MIA fetal brain reflects the alteration of neuron development by immune activation.
Fig. 6.

MIA increased α7nAChR in fetal brain neurons. MIA E12.5 fetal hindbrain displayed more α7nAChR immunostaining than control hindbrain 3 hours following maternal poly(I:C) injection. In hindbrain subregions (A) PPH and (B) PH, α7nAChR was increased in MIA offspring. (C) Optical density quantitation revealed the increased α7nAChR immunostaining in MIA fetal hindbrain. PPH: Saline: n = 3 litters, Poly(I:C): n = 3 litters; PH: Saline: n = 3 litters, Poly(I:C): n = 4 litters. (D) Confocal images demonstrated α7nAChR-positive cells were double-labeled with mature neuronal marker MAP2 (left panel) and the immature neuronal marker DCX (middle panel), but not with the neural stem cell marker nestin (right panel). White arrows indicate the colocalization between α7nAChR and the other markers. PPH: prepontine hindbrain, PH: pontine hindbrain, MAP2: microtubule-Associated Protein 2, DCX: doublecortin. Data are presented as mean ± SEM. Significant difference between groups is labeled as * p < 0.05.
3.6. Il6 gene expression increased in the Chrna7 mutant fetal brain
To further explore the role of α7nAChR in fetal brain during maternal infection, we injected poly(I:C) into Chrna7+/− dams and compared the acute inflammatory response in the wild-type (+/+), heterozygous (+/−), and knockout (−/−) fetal brain. Chrna7 mRNA expression was changed in genotype-dependent manner; there was no expression in −/− fetal brain and intermediate levels of expression in +/− fetal brain (Among genotypes: p < 0.0001. Except Saline +/+ v.s. Saline +/−, p < 0.001; Saline +/+ v.s. Poly(I:C) +/−, p < 0.01)(Fig. 7A). The increase in Chrna7 mRNA and Il6 mRNA by poly(I:C) injection was only observed in +/+ fetal brain (Chrna7, p < 0.01; Il6, p < 0.05)(Fig. 7A–B). The basal level of Il6 mRNA was higher in the +/− and −/− fetal brain than in the wild-type fetal brain (p < 0.05) (Fig. 7B). Poly(I:C) injection did not produce additional effect on Il6 expression in +/− and −/− fetal brain (Fig. 7B).
Fig. 7.
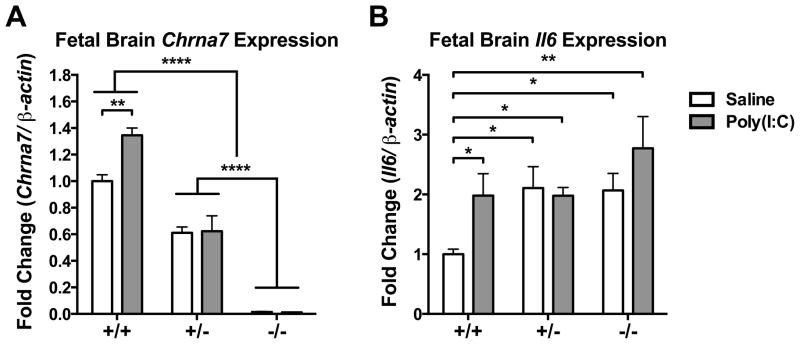
Decreased Chrna7 in fetal brain led to the elevation of Il6 expression. (A) MIA increased Chrna7 mRNA level in wild-type fetal brain, but not in Chrna7+/− and Chrna7−/− fetal brain. (B) MIA increased Il6 mRNA in wild-type fetal brains but not in Chrna7+/− and Chrna7−/− fetal brains. Baseline Il6 levels were elevated in Chrna7+/− and Chrna7−/− fetal brains. Gene expression was normalized by β-actin.Saline +/+ n = 3 litters, +/− n = 3 litters, −/− n = 3 litters; Poly(I:C) +/+ n = 3 litters, +/− n = 3 litters, −/− n = 2 litters. Chrna7: nicotinic acetylcholine receptor alpha 7 subunit, Il6: interleukin-6. Data are presented as Mean ± SEM. Significant difference between groups is labeled as * p < 0.05, ** p < 0.01, **** p < 0.0001.
3.7. Chrna7 heterozygous mouse were more susceptible to MIA induced autistic- and schizophrenia-like behaviors
Combining MIA with genetic mutation of Chrna7 produced synergistic effects on several offspring behaviors. In the open-field test, there was no genotype effect in the offspring who did not receive MIA (Fig. 8A–B). But for the offspring who received MIA, Chrna7+/− mice showed reduction of the time spent in center zone (Poly(I:C) +/+ v.s. Poly(I:C) +/−, p = 0.0637; Saline +/+ v.s. Poly(I:C) +/−, p < 0.05; Saline +/− v.s. Poly(I:C) +/−, p < 0.05)(Fig. 8B). MIA had an effect on marble burying behavior in the wild-type littermates of Chrna7 mutant line (Saline +/+ v.s. Poly(I:C) +/+, p < 0.01)(Fig. 8C). Chrna7+/− offspring showed higher marble burying behavior compared to the wild-type offspring who were maternally injected with saline (Saline +/+ v.s. Saline +/−, p < 0.05; Saline +/+ v.s. Poly(I:C) +/−, p < 0.05). There was no additional effect of MIA on the marble burying behavior in Chrna7+/− offspring (Fig. 8C). Genotype effect was also found in PPI test. Chrna7+/− offspring who had not received MIA exhibited higher inhibition of PPI 5 db than wild-type littermates (Saline +/+ v.s. Saline +/−, p < 0.05). However, MIA decreased the inhibition of PPI 5db in Chrna7+/− offspring (Saline +/− v.s. Poly(I:C) +/−, p < 0.001)(Fig. 8D). The inhibition of PPI15 showed a higher trend in Chrna7+/− than in the wild-type littermates (Fig. 8E). Wild-type and Chrna7+/− offspring who received MIA showed lower inhibition of PPI15 than Chrna7+/− offspring who had not received MIA (Saline+/− v.s. Poly(I:C) +/+, p < 0.05; Saline +/− v.s. Poly(I:C) +/−, p < 0.05)(Fig. 8E).
Fig. 8.

Compared to wild type, Chrna7+/− offspring were more susceptible to MIA, generally exhibiting more autistic- and schizophrenia-like behaviors. (A–B) Chrna7+/− MIA offspring displayed anxiety-like behavior in the open field, while wild-type MIA offspring did not. (C) Wild-type MIA offspring displayed more repetitive behavior in the marble burying test compared to saline offspring. The baseline of repetitive behavior in the Chrna7+/− offspring was higher than in the wild-type saline offspring and there was no effect of MIA in the Chrna7+/− offspring. (D–E) Chrna7+/− MIA offspring displayed a PPI deficit at PPI5 and PPI15, while wild-type MIA offspring exhibited normal PPI5 and PPI15. Saline: n = 4–5 litters, Poly(I:C): n = 4–6 litters. Data are presented as Mean ± SEM. Significant difference between groups is labeled as * p < 0.05, ** p < 0.01, *** p < 0.001.
4. Discussion
Our study used several strategies to investigate the role of both the maternal and fetal α7nAChR in the development of MIA-induced behavioral abnormalities. Stimulating a7nAChR activation in both dam and fetus by providing the dams a choline-supplemented diet during gestation and lactation prevented several behavioral abnormalities in the offspring caused by MIA. We further demonstrated that gestational choline dietary supplementation suppressed MIA-induced Il6 and Chrna7 mRNA increases in the fetal brain. In a complementary series of experiments, we crossed heterozygous Chrna7 null mutant mice and showed that loss of Chrna7 expression in the resultant homozygous and heterozygous Chrna7 null mutant offspring resulted in increased fetal Il6 response to MIA and increased behavioral deficits in the offspring. Taken together, the results indicate that maternal and fetal a7nAChR activation is critical for modulating the effects of MIA.
Maternal choline supplementation had been shown to ameliorate the phenotypes in different mouse models of autism and schizophrenia (Langley et al., 2014; Ricceri et al., 2011; Stevens et al., 2008), as well as in the MIA model as we show in this study. More specifically, we demonstrated that maternal choline supplementation lessened MIA-induced anxiety-like behavior in open-field test and repetitive behavior in marble burying test. This implies choline in modulating the core symptom and comorbidity of autism and schizophrenia. We also observed that maternal choline supplementation changes the basal level of some behaviors, such as open-field center entering behavior, marble burying behavior and PPI. There are several possible mechanisms through which choline supplementation might influence fetal development. During pregnancy, choline can affect the maternal-placental-fetal axis in several ways (Jiang et al., 2014). Choline is a selective agonist of the α7nAChR, compared to other nicotinic receptors (Alkondon et al., 1997). During embryonic development, choline itself appears to serve as the ligand for fetal α7nAChRs before the development of cholinergic nerve innervation (Miwa et al., 2011; Ross et al., 2010). Choline-derived acetylcholine can also be synthesized and accumulated in the placenta. Acetylcholine in the placenta, as the agonist of its acetylcholine receptors, regulates nutrient transport, fluid volume and blood flow during the development of the placenta (Kawashima and Fujii, 2008; Lips et al., 2005; Sastry, 1997; Sastry and Sadavongvivad, 1978). Choline phospholipid phosphatidylcholine can be synthesized from choline in the maternal liver through the cytidine diphosphate (CDP)-choline pathway and the phosphatidylethanolamine N-methyltransferase (SAM) pathway and affect fetal cell division, cell membrane biogenesis, myelination of nerve axons, and lipid transport (Jiang et al., 2014). Choline’s oxidized metabolite, betaine, is a source for the production of the universal methyl donor SAM, which is a substrate of DNA methyltransferases (DNMTs) and histone methyltransferases (HMTs), and important for the fetal epigenome establishment (Davison et al., 2009; Jiang et al., 2014; Kovacheva et al., 2007). In this study, we found that choline regulates an anti-inflammatory reflex, potentially through the agonism of α7nAChR, to suppress the acute inflammatory response in the fetal brain after MIA.
To understand the impact of α7nAChR level in maternal infection, we induced MIA in mice with mutations in the α7nAChR gene. However, knockout of Chrna7 in mice alters the inflammatory response (Wang et al., 2003), which may affect MIA responses in either the dam or offspring. Therefore, our timed-mating strategy was to cross male Chrna7+/− mice with female wild-type mice. Through the mating, we could obtain heterozygous offspring with reduced expression of α7nAChR in a normal wild-type dam, along with wild-type littermates as the ideal control. Compared to wild-type offspring, the Chrna7+/− offspring were more affected by MIA; they were more anxious and displayed more repetitive behavior during the open field and marble burying tests, respectively. Chrna7−/− mice had been reported to display a partial PPI deficit and anxiety (Azzopardi et al., 2013; Paylor et al., 1998), but we did not observe the PPI deficit and anxiety in Chrna7+/− offspring in the maternal saline injection group.
We found differences in the MIA induced behavioral abnormalities between choline supplementation control mice and wild-type mice in the Chrna7 mutant studies. MIA produced more behavioral abnormalities in the choline supplementation control diet group, including marble burying behavior, PPI deficit, and a trend reduction in center zone entering in open-field (Fig. 1). In contrast, MIA only affected marble burying phenotype in the wild-type mice from the Chrna7 mutant studies (Fig. 8). The reasons for the difference are not clear. Notably, several studies have shown that MIA does not produce robust effects on the wild-type littermates of a transgenic line (Abazyan et al., 2010; Ehninger et al., 2012; Ibi et al., 2010). Strain and source differences of mice could be factors affecting the outcome of rodent behaviors (Bryant et al., 2008; Matsuo et al., 2010; Paylor and Crawley, 1997; Simon et al., 2013).
Analysis of α7nAChR expression in the fetal and adult brains revealed that activation of maternal immune system by poly(I:C) increased Chrna7 mRNA expression in the fetal brain. We propose that the elevation of Chrna7 mRNA expression in the MIA fetal brain is related to the Il6 surge after maternal poly(I:C) injection. Chrna7 elevation might reflect the fetal brain’s acute response to inflammation. Supporting this idea, decreasing Chrna7 in the fetal brain increased the basal level of Il6 expression, suggesting a role for α7nAChR in regulating fetal brain cytokines. Choline supplemented fetal brain showed a higher choline level compared to those fetal brains which were not supplemented with maternal choline (Fig. 5B). Our hypothesis is that maternal choline supplementation acted as an α7nAChR agonist and stimulated an anti-inflammatory reflex, effectively inhibiting IL-6 increase in the fetal brain. Due to the ample availability of choline as an α7nAChR agonist, α7nAChRs did not need to increase in response to the inflammation by poly(I:C). In addition, reduction of α7nAChR in fetal brain raised the IL-6 basal levels. Maternal choline supplementation thus may activate an α7nAChR based anti-inflammatory reflex and decrease the acute inflammation in the fetal brain in response to MIA. Other investigators have investigated anti-purinergic therapy in MIA offspring and found increased α7nAChR level in cerebral synaptosomes (Naviaux et al., 2013), along with other evidence suggests that α7nAChRs serve as an anti-inflammatory regulator of innate immunity in the CNS (Tracey, 2009).
Nicotinic cholinergic receptors are only one mechanism that appears to link genetic risk for autism, schizophrenia and MIA. For example, Disc1 mouse mutants including mutant human DISC1 (mhDISC1), a point mutation of DISC1 gene at L100P (Disc1-L100P+/−) and dominant-negative DISC1 (DN-DISC1) have increased behavioral, pathological, and neurochemical deficits after prenatal poly(I:C) treatment (Abazyan et al., 2010; Ibi et al., 2010; Lipina et al., 2013). Another schizophrenia risk gene NRG1 also produced synergetic effects with MIA causing developmental stage-specific changes in social behavior, spatial memory and PPI (O’Leary et al., 2014). Nurr1 (NR4A2) is an orphan member of steroid hormone receptor and highly essential for dopaminergic development. Prenatal poly(I:C) treatment exerts pronounced effects in the development of dopaminergic system and behavioral changes on Nurr1+/− offspring (Vuillermot et al., 2012). TSC2 is a key regulator for mTOR signaling and a risk gene for autism. Gestational poly(I:C) treatment disrupted adult social behavior in the Tsc2 haploinsufficiency offspring (Ehninger et al., 2012). Overexpression of IL-10 in macrophages modulated several behavioral dysfunctions exhibited in the MIA macIL-10tg offspring (Meyer et al., 2008b).
In summary, viral infection in the pregnant mother activates the maternal immune system, increasing the risk for autism and schizophrenia in the offspring. α7nAChR in the fetal brain suppresses inflammatory response by inhibiting IL-6 production and, consequentially, the loss of α7nAChR in the offspring increases their vulnerability to MIA-induced autistic and schizophrenia-like symptoms. Maternal choline supplementation triggers an anti-inflammatory response in the fetal brain and thus decreases the MIA-induced IL-6 elevation during embryonic stage and the autistic- and schizophrenia-like behaviors in the adults. These findings raise the possibility that the abnormal behaviors in MIA offspring produced by elevated IL-6 are modulated by activation of α7nAChRs in the fetal brain.
Supplementary Material
Acknowledgments
We acknowledge Laura Rodriguez for administrative assistance; Jan Ko, Elaine Y. Hsiao and Natalia Malkova for technical assistance; Lorena C. Sandoval, Kwan F. Lee and Jaime Rodriguez for caring for the animals. This work was supported by NIH Conte Center Award (to P.H.P.; NIH 5P50MH086383-04), Autism Speaks (to P.H.P; #7670), and postdoctoral fellowship from National Science Council of Taiwan (to W.-L.W.; NSC 101-2917-I-564-039).
Footnotes
Conflict of Interest Statement
All authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abazyan B, Nomura J, Kannan G, Ishizuka K, Tamashiro KL, Nucifora F, Pogorelov V, Ladenheim B, Yang C, Krasnova IN, Cadet JL, Pardo C, Mori S, Kamiya A, Vogel MW, Sawa A, Ross CA, Pletnikov MV. Prenatal interaction of mutant DISC1 and immune activation produces adult psychopathology. Biol Psychiatry. 2010;68:1172–1181. doi: 10.1016/j.biopsych.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams CE, Broide RS, Chen Y, Winzer-Serhan UH, Henderson TA, Leslie FM, Freedman R. Development of the alpha7 nicotinic cholinergic receptor in rat hippocampal formation. Brain Res Dev Brain Res. 2002;139:175–187. doi: 10.1016/s0165-3806(02)00547-3. [DOI] [PubMed] [Google Scholar]
- Adams CE, Stevens KE. Evidence for a role of nicotinic acetylcholine receptors in schizophrenia. Front Biosci. 2007;12:4755–4772. doi: 10.2741/2424. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Cortes WS, Maelicke A, Albuquerque EX. Choline is a selective agonist of alpha7 nicotinic acetylcholine receptors in the rat brain neurons. Eur J Neurosci. 1997;9:2734–2742. doi: 10.1111/j.1460-9568.1997.tb01702.x. [DOI] [PubMed] [Google Scholar]
- Azzopardi E, Typlt M, Jenkins B, Schmid S. Sensorimotor gating and spatial learning in alpha7-nicotinic receptor knockout mice. Genes Brain Behav. 2013;12:414–423. doi: 10.1111/gbb.12038. [DOI] [PubMed] [Google Scholar]
- Boksa P. Effects of prenatal infection on brain development and behavior: a review of findings from animal models. Brain Behav Immun. 2010;24:881–897. doi: 10.1016/j.bbi.2010.03.005. [DOI] [PubMed] [Google Scholar]
- Brown AS, Patterson PH. Maternal infection and schizophrenia: implications for prevention. Schizophr Bull. 2011;37:284–290. doi: 10.1093/schbul/sbq146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant CD, Zhang NN, Sokoloff G, Fanselow MS, Ennes HS, Palmer AA, McRoberts JA. Behavioral differences among C57BL/6 substrains: implications for transgenic and knockout studies. J Neurogenet. 2008;22:315–331. doi: 10.1080/01677060802357388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor CM, Dincer A, Straubhaar J, Galler JR, Houston IB, Akbarian S. Maternal immune activation alters behavior in adult offspring, with subtle changes in the cortical transcriptome and epigenome. Schizophr Res. 2012;140:175–184. doi: 10.1016/j.schres.2012.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison JM, Mellott TJ, Kovacheva VP, Blusztajn JK. Gestational choline supply regulates methylation of histone H3, expression of histone methyltransferases G9a (Kmt1c) and Suv39h1 (Kmt1a), and DNA methylation of their genes in rat fetal liver and brain. J Biol Chem. 2009;284:1982–1989. doi: 10.1074/jbc.M807651200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, Sano Y, de Vries PJ, Dies K, Franz D, Geschwind DH, Kaur M, Lee YS, Li W, Lowe JK, Nakagawa JA, Sahin M, Smith K, Whittemore V, Silva AJ. Gestational immune activation and Tsc2 haploinsufficiency cooperate to disrupt fetal survival and may perturb social behavior in adult mice. Mol Psychiatry. 2012;17:62–70. doi: 10.1038/mp.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman R, Coon H, Myles-Worsley M, Orr-Urtreger A, Olincy A, Davis A, Polymeropoulos M, Holik J, Hopkins J, Hoff M, Rosenthal J, Waldo MC, Reimherr F, Wender P, Yaw J, Young DA, Breese CR, Adams C, Patterson D, Adler LE, Kruglyak L, Leonard S, Byerley W. Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc Natl Acad Sci U S A. 1997;94:587–592. doi: 10.1073/pnas.94.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garay PA, Hsiao EY, Patterson PH, McAllister AK. Maternal immune activation causes age- and region-specific changes in brain cytokines in offspring throughout development. Brain Behav Immun. 2013;31:54–68. doi: 10.1016/j.bbi.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbett KA, Hsiao EY, Kalman S, Patterson PH, Mirnics K. Effects of maternal immune activation on gene expression patterns in the fetal brain. Transl Psychiatry. 2012;2:e98. doi: 10.1038/tp.2012.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, Miller J, Fedele A, Collins J, Smith K, Lotspeich L, Croen LA, Ozonoff S, Lajonchere C, Grether JK, Risch N. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry. 2011;68:1095–1102. doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey L, Boksa P. A stereological comparison of GAD67 and reelin expression in the hippocampal stratum oriens of offspring from two mouse models of maternal inflammation during pregnancy. Neuropharmacology. 2012;62:1767–1776. doi: 10.1016/j.neuropharm.2011.11.022. [DOI] [PubMed] [Google Scholar]
- Hsiao EY, Patterson PH. Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav Immun. 2011;25:604–615. doi: 10.1016/j.bbi.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibi D, Nagai T, Koike H, Kitahara Y, Mizoguchi H, Niwa M, Jaaro-Peled H, Nitta A, Yoneda Y, Nabeshima T, Sawa A, Yamada K. Combined effect of neonatal immune activation and mutant DISC1 on phenotypic changes in adulthood. Behav Brain Res. 2010;206:32–37. doi: 10.1016/j.bbr.2009.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, West AA, Caudill MA. Maternal choline supplementation: a nutritional approach for improving offspring health? Trends Endocrinol Metab. 2014;25:263–273. doi: 10.1016/j.tem.2014.02.001. [DOI] [PubMed] [Google Scholar]
- Karvat G, Kimchi T. Acetylcholine elevation relieves cognitive rigidity and social deficiency in a mouse model of autism. Neuropsychopharmacology. 2014;39:831–840. doi: 10.1038/npp.2013.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima K, Fujii T. Basic and clinical aspects of non-neuronal acetylcholine: overview of non-neuronal cholinergic systems and their biological significance. J Pharmacol Sci. 2008;106:167–173. doi: 10.1254/jphs.fm0070073. [DOI] [PubMed] [Google Scholar]
- Kovacheva VP, Mellott TJ, Davison JM, Wagner N, Lopez-Coviella I, Schnitzler AC, Blusztajn JK. Gestational choline deficiency causes global and Igf2 gene DNA hypermethylation by up-regulation of Dnmt1 expression. J Biol Chem. 2007;282:31777–31788. doi: 10.1074/jbc.M705539200. [DOI] [PubMed] [Google Scholar]
- Langley EA, Krykbaeva M, Blusztajn JK, Mellott TJ. High maternal choline consumption during pregnancy and nursing alleviates deficits in social interaction and improves anxiety-like behaviors in the BTBR T+Itpr3tf/J mouse model of autism. Behav Brain Res. 2014;278C:210–220. doi: 10.1016/j.bbr.2014.09.043. [DOI] [PubMed] [Google Scholar]
- Leonard S, Gault J, Hopkins J, Logel J, Vianzon R, Short M, Drebing C, Berger R, Venn D, Sirota P, Zerbe G, Olincy A, Ross RG, Adler LE, Freedman R. Association of promoter variants in the alpha7 nicotinic acetylcholine receptor subunit gene with an inhibitory deficit found in schizophrenia. Arch Gen Psychiatry. 2002;59:1085–1096. doi: 10.1001/archpsyc.59.12.1085. [DOI] [PubMed] [Google Scholar]
- Lipina TV, Zai C, Hlousek D, Roder JC, Wong AH. Maternal immune activation during gestation interacts with Disc1 point mutation to exacerbate schizophrenia-related behaviors in mice. J Neurosci. 2013;33:7654–7666. doi: 10.1523/JNEUROSCI.0091-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lips KS, Bruggmann D, Pfeil U, Vollerthun R, Grando SA, Kummer W. Nicotinic acetylcholine receptors in rat and human placenta. Placenta. 2005;26:735–746. doi: 10.1016/j.placenta.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Malkova NV, Yu CZ, Hsiao EY, Moore MJ, Patterson PH. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav Immun. 2012;26:607–616. doi: 10.1016/j.bbi.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo N, Takao K, Nakanishi K, Yamasaki N, Tanda K, Miyakawa T. Behavioral profiles of three C57BL/6 substrains. Front Behav Neurosci. 2010;4:29. doi: 10.3389/fnbeh.2010.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTighe SM, Neal SJ, Lin Q, Hughes ZA, Smith DG. The BTBR mouse model of autism spectrum disorders has learning and attentional impairments and alterations in acetylcholine and kynurenic acid in prefrontal cortex. PLoS One. 2013;8:e62189. doi: 10.1371/journal.pone.0062189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Engler A, Weber L, Schedlowski M, Feldon J. Preliminary evidence for a modulation of fetal dopaminergic development by maternal immune activation during pregnancy. Neuroscience. 2008a;154:701–709. doi: 10.1016/j.neuroscience.2008.04.031. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J. Epidemiology-driven neurodevelopmental animal models of schizophrenia. Prog Neurobiol. 2010;90:285–326. doi: 10.1016/j.pneurobio.2009.10.018. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Schedlowski M, Yee BK. Towards an immuno-precipitated neurodevelopmental animal model of schizophrenia. Neurosci Biobehav Rev. 2005;29:913–947. doi: 10.1016/j.neubiorev.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Schedlowski M, Yee BK. Immunological stress at the maternal-foetal interface: a link between neurodevelopment and adult psychopathology. Brain Behav Immun. 2006;20:378–388. doi: 10.1016/j.bbi.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Meyer U, Murray PJ, Urwyler A, Yee BK, Schedlowski M, Feldon J. Adult behavioral and pharmacological dysfunctions following disruption of the fetal brain balance between pro-inflammatory and IL-10-mediated anti-inflammatory signaling. Mol Psychiatry. 2008b;13:208–221. doi: 10.1038/sj.mp.4002042. [DOI] [PubMed] [Google Scholar]
- Missault S, Van den Eynde K, Vanden Berghe W, Fransen E, Weeren A, Timmermans JP, Kumar-Singh S, Dedeurwaerdere S. The risk for behavioural deficits is determined by the maternal immune response to prenatal immune challenge in a neurodevelopmental model. Brain Behav Immun. 2014 doi: 10.1016/j.bbi.2014.06.013. [DOI] [PubMed] [Google Scholar]
- Miwa JM, Freedman R, Lester HA. Neural systems governed by nicotinic acetylcholine receptors: emerging hypotheses. Neuron. 2011;70:20–33. doi: 10.1016/j.neuron.2011.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naviaux RK, Zolkipli Z, Wang L, Nakayama T, Naviaux JC, Le TP, Schuchbauer MA, Rogac M, Tang Q, Dugan LL, Powell SB. Antipurinergic therapy corrects the autism-like features in the poly(IC) mouse model. PLoS One. 2013;8:e57380. doi: 10.1371/journal.pone.0057380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary C, Desbonnet L, Clarke N, Petit E, Tighe O, Lai D, Harvey R, Waddington JL, O’Tuathaigh C. Phenotypic effects of maternal immune activation and early postnatal milieu in mice mutant for the schizophrenia risk gene neuregulin-1. Neuroscience. 2014 doi: 10.1016/j.neuroscience.2014.06.028. [DOI] [PubMed] [Google Scholar]
- Patterson PH. Maternal infection: window on neuroimmune interactions in fetal brain development and mental illness. Curr Opin Neurobiol. 2002;12:115–118. doi: 10.1016/s0959-4388(02)00299-4. [DOI] [PubMed] [Google Scholar]
- Patterson PH. Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behav Brain Res. 2009;204:313–321. doi: 10.1016/j.bbr.2008.12.016. [DOI] [PubMed] [Google Scholar]
- Patterson PH. Maternal infection and immune involvement in autism. Trends Mol Med. 2011a;17:389–394. doi: 10.1016/j.molmed.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson PH. Modeling autistic features in animals. Pediatr Res. 2011b;69:34R–40R. doi: 10.1203/PDR.0b013e318212b80f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paylor R, Crawley JN. Inbred strain differences in prepulse inhibition of the mouse startle response. Psychopharmacology (Berl) 1997;132:169–180. doi: 10.1007/s002130050333. [DOI] [PubMed] [Google Scholar]
- Paylor R, Nguyen M, Crawley JN, Patrick J, Beaudet A, Orr-Urtreger A. Alpha7 nicotinic receptor subunits are not necessary for hippocampal-dependent learning or sensorimotor gating: a behavioral characterization of Acra7-deficient mice. Learn Mem. 1998;5:302–316. [PMC free article] [PubMed] [Google Scholar]
- Persico AM, Bourgeron T. Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends Neurosci. 2006;29:349–358. doi: 10.1016/j.tins.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Pratt L, Ni L, Ponzio NM, Jonakait GM. Maternal inflammation promotes fetal microglial activation and increased cholinergic expression in the fetal basal forebrain: role of interleukin-6. Pediatr Res. 2013;74:393–401. doi: 10.1038/pr.2013.126. [DOI] [PubMed] [Google Scholar]
- Ricceri L, De Filippis B, Fuso A, Laviola G. Cholinergic hypofunction in MeCP2-308 mice: beneficial neurobehavioural effects of neonatal choline supplementation. Behav Brain Res. 2011;221:623–629. doi: 10.1016/j.bbr.2011.03.051. [DOI] [PubMed] [Google Scholar]
- Rosas-Ballina M, Olofsson PS, Ochani M, Valdes-Ferrer SI, Levine YA, Reardon C, Tusche MW, Pavlov VA, Andersson U, Chavan S, Mak TW, Tracey KJ. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. 2011;334:98–101. doi: 10.1126/science.1209985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RG, Stevens KE, Proctor WR, Leonard S, Kisley MA, Hunter SK, Freedman R, Adams CE. Research review: Cholinergic mechanisms, early brain development, and risk for schizophrenia. J Child Psychol Psychiatry. 2010;51:535–549. doi: 10.1111/j.1469-7610.2009.02187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry BV. Human placental cholinergic system. Biochem Pharmacol. 1997;53:1577–1586. doi: 10.1016/s0006-2952(97)00017-8. [DOI] [PubMed] [Google Scholar]
- Sastry BV, Sadavongvivad C. Cholinergic systems in non-nervous tissues. Pharmacol Rev. 1978;30:65–132. [PubMed] [Google Scholar]
- Simon MM, Greenaway S, White JK, Fuchs H, Gailus-Durner V, Wells S, Sorg T, Wong K, Bedu E, Cartwright EJ, Dacquin R, Djebali S, Estabel J, Graw J, Ingham NJ, Jackson IJ, Lengeling A, Mandillo S, Marvel J, Meziane H, Preitner F, Puk O, Roux M, Adams DJ, Atkins S, Ayadi A, Becker L, Blake A, Brooker D, Cater H, Champy MF, Combe R, Danecek P, di Fenza A, Gates H, Gerdin AK, Golini E, Hancock JM, Hans W, Holter SM, Hough T, Jurdic P, Keane TM, Morgan H, Muller W, Neff F, Nicholson G, Pasche B, Roberson LA, Rozman J, Sanderson M, Santos L, Selloum M, Shannon C, Southwell A, Tocchini-Valentini GP, Vancollie VE, Westerberg H, Wurst W, Zi M, Yalcin B, Ramirez-Solis R, Steel KP, Mallon AM, de Angelis MH, Herault Y, Brown SD. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol. 2013;14:R82. doi: 10.1186/gb-2013-14-7-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27:10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soumiya H, Fukumitsu H, Furukawa S. Prenatal immune challenge compromises the normal course of neurogenesis during development of the mouse cerebral cortex. J Neurosci Res. 2011;89:1575–1585. doi: 10.1002/jnr.22704. [DOI] [PubMed] [Google Scholar]
- Spandidos A, Wang X, Wang H, Seed B. PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res. 2010;38:D792–799. doi: 10.1093/nar/gkp1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens KE, Adams CE, Yonchek J, Hickel C, Danielson J, Kisley MA. Permanent improvement in deficient sensory inhibition in DBA/2 mice with increased perinatal choline. Psychopharmacology (Berl) 2008;198:413–420. doi: 10.1007/s00213-008-1170-3. [DOI] [PubMed] [Google Scholar]
- Stevens KE, Choo KS, Stitzel JA, Marks MJ, Adams CE. Long-term improvements in sensory inhibition with gestational choline supplementation linked to alpha7 nicotinic receptors through studies in Chrna7 null mutation mice. Brain Res. 2014;1552:26–33. doi: 10.1016/j.brainres.2014.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolp HB, Turnquist C, Dziegielewska KM, Saunders NR, Anthony DC, Molnar Z. Reduced ventricular proliferation in the foetal cortex following maternal inflammation in the mouse. Brain. 2011;134:3236–3248. doi: 10.1093/brain/awr237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey KJ. Reflex control of immunity. Nat Rev Immunol. 2009;9:418–428. doi: 10.1038/nri2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuang M. Schizophrenia: genes and environment. Biol Psychiatry. 2000;47:210–220. doi: 10.1016/s0006-3223(99)00289-9. [DOI] [PubMed] [Google Scholar]
- Vuillermot S, Joodmardi E, Perlmann T, Ogren SO, Feldon J, Meyer U. Prenatal immune activation interacts with genetic Nurr1 deficiency in the development of attentional impairments. J Neurosci. 2012;32:436–451. doi: 10.1523/JNEUROSCI.4831-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- Yasui DH, Scoles HA, Horike S, Meguro-Horike M, Dunaway KW, Schroeder DI, Lasalle JM. 15q11.2–13.3 chromatin analysis reveals epigenetic regulation of CHRNA7 with deficiencies in Rett and autism brain. Hum Mol Genet. 2011;20:4311–4323. doi: 10.1093/hmg/ddr357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XL, Zellweger R, Zhu XH, Ayala A, Chaudry IH. Cytokine gene expression in splenic macrophages and Kupffer cells following haemorrhage. Cytokine. 1995;7:8–14. doi: 10.1006/cyto.1995.1002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.