

Abstract
This review covers the initial discovery of the marine actinomycete genus Salinispora through its development as a model for natural product research. A focus is placed on the novel chemical structures reported with reference to their biological activities and the synthetic and biosynthetic studies they have inspired. The time line of discoveries progresses from more traditional bioassay-guided approaches through the application of genome mining and genetic engineering techniques that target the products of specific biosynthetic gene clusters. This overview exemplifies the extraordinary biosynthetic diversity that can emanate from a narrowly defined genus and supports future efforts to explore marine taxa in the search for novel natural products.
1 Introduction
Microbial-derived natural products represent a major component of today’s pharmaceutical arsenal. Despite their historical importance, the world’s major pharmaceutical companies moved en masse away from microbial natural products in favor of alternative discovery platforms such as combinatorial chemistry 1. Contributing to this paradigm shift was the continued re-discovery of known compounds and a growing belief that microbial resources have been over-exploited. However, increased demand for new drugs to treat antibiotic resistant bacterial infections and other chronic diseases, coupled with the low returns from alternative discovery platforms, have led to a resurgence of interest in natural products research 2. This renewed interest includes the exploration of bacteria from poorly studied environments, a concept based on the premise that adaptations to these environments include the production of new secondary metabolites 3. Marine bacteria have become a particular focus in these efforts and have yielded many interesting new compounds 4, 5.
Actinomycetes are a major source of microbial-derived natural products 6 making marine-derived strains likely targets for natural product discovery 7, 8. Although it was revealed long ago that actinomycetes could be recovered from marine samples, including deep sea sediments 9, it remains unknown to what extent these bacteria are ecologically or evolutionarily distinct from their terrestrial relatives. This uncertainty arises from the fact that spore-forming actinomycetes are abundant in soils and washed into the sea in large numbers where their metabolic activities remain largely unknown 10. Although there is evidence that common soil genera such as Streptomyces can be metabolically active in the sea 11, we have yet to gain a broader perspective on this subject. None-the-less, there is emerging evidence for marine adaptation even among streptomycetes 12, 13 and a number of exclusively marine Streptomyces spp. have been described 14. Furthermore, at least five marine actinomycete genera have been described 15–19 providing clear evidence that marine-derived actinomycetes can be taxonomically distinct from those occurring on land. Among these genera, Salinispora has proven to be a prolific source of novel natural products 4 and a model organism with which to address correlations between bacterial diversity and secondary metabolite production 20, 21.
Here we review the discovery of the marine actinomycete genus Salinispora and its development as a model for natural product research. The focus is on new carbon skeletons with the discoveries presented largely in chronological manner. Some of these molecules have important biological activities, which have been summarized. Many have inspired synthetic, biosynthetic, and mechanistic studies, which have been highlighted. Early discovery efforts employed more traditional bioassay-guided approaches while some of the more recent discoveries result from the application of genome mining and genetic engineering approaches. We have also summarized the known compounds and new derivatives thereof that have been reported from this taxon. The major aim of this review is to encapsulate the remarkable biosynthetic capacities of a single marine actinomycete taxon and to emphasize how natural products chemistry has been merged with biological and biochemical studies in an interdisciplinary effort to develop more informed approaches to natural product discovery.
2 Discovery of the genus
The cultivation of Salinispora strains was first reported in 1989 as part of a study addressing actinomycete distributions in marine sediments 22. At the time, their morphological and chemotaxonomic characteristics indicated they were close relatives of the genus Micromonospora, and it was proposed they represented a new species within this genus based on the observation that they failed to grow when seawater was replaced with deionized water in the growth medium. Subsequent phylogenetic studies placed these bacteria in a clade that was distinct from the Micromonosporae, and it was suggested they represent a new genus for which the name “Salinospora” was originally proposed 23. This taxon was formally described in 2005 as the first obligate marine actinomycete genus with the name revised to Salinispora to meet nomenclatural standards 19. The original description included the species S. tropica and S. arenicola while a third species, S. pacifica, was subsequently proposed 24 and formally described 25. The three species share approximately 99% 16S rRNA gene sequence identity and are not well resolved using this conserved phylogenetic marker 25. However, less conserved loci have been used to generate well-supported phylogenies that clearly delineate the three species and reveal the sister relationship between S. tropica and S. pacifica relative to the more ancestral S. arenicola lineage 24–27.
Salinispora spp. are most frequently reported from marine sediments, however this may represent sampling bias. They have also been reported from an ascidian 28, seaweeds 13, and marine sponges 27, 29. To date, there is no evidence that plant or invertebrate-associated populations are ecologically or evolutionarily distinct from those that occur in sediments. Salinispora strains have been cultured from depths as great as 1100 m 30 but have been detected using culture independent methods from much greater depths, the current record being 5669 m 31. They have been cultured from tropical and sub-tropical sites around the globe 32, with the most northern report coming from samples collected off Japan 33 (Figure 1). The lack of reports from more northern and southern latitudes may be due to limited sampling from these regions or yet to be determined environmental variables that limit their distributions.
Figure 1.

Global locations from which the genus Salinispora has been reported. These reports originate from multiple research groups and are based on GenBank (http://www.ncbi.nlm.nih.gov/genbank/) 16S rRNA sequence deposits.
To date, reports of S. tropica have been restricted to the Caribbean, S. pacifica has been reported from numerous global sites except for the Caribbean, while S. arenicola has the broadest distribution and has been reported from all sites from which the genus has been recovered 32, 34. Salinispora spp. are heavily invested in secondary metabolism, with ca. 10% of their genomes devoted to this process 35. The majority of their secondary metabolite biosynthetic gene clusters are located in genomic islands, which was used to suggest the products provide ecologically relevant adaptive traits 36. The genus is unique among the Micromonosporaceae in that all strains tested to date fail to grow when seawater is replaced with deionized water in the growth medium, which was subsequently linked to a variety of marine adaptation genes using both bioinformatic 37 and experimental approaches 38. However, the primary interest in this taxon has focused on its ability to produce unique and biologically active secondary metabolites.
3 Salinispora natural products
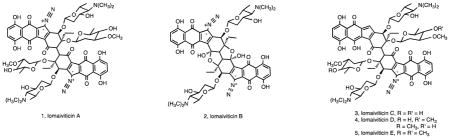
The secondary metabolites reported to date from Salinispora spp., are predominantly new (Table 1). This supports the concept that new taxa from poorly studied environments represent an important resource for secondary metabolite discovery. While not widely recognized, the first compounds described from the genus Salinispora were lomaiviticins A and B (1, 2) 28, the structures of which were published in 2001. At the time, the producing strain was reported as a new Micromonospora species with the proposed name “Micromonospora lomaivitiensis”. However, subsequent 16S rRNA gene sequence analysis identified this strain as S. pacifica 39. The lomaiviticins were isolated by researchers at Wyeth (now Pfizer) as part of efforts to identify enediyne-producing bacteria from the marine ascidian Polysyncraton lithostrotum. Although the lomaiviticins do not belong to this structural class, they none-the-less possess powerful antibiotic activities and, in the case of 1, nanomolar to picomolar cancer cell cytotoxicities that were ultimately linked to the induction of double-strand DNA breaks 40, a mechanism of action similar to that exerted by enediynes 41. Further studies by the Herzon group led to the isolation of additional compounds in this series (lomaiviticins C–E) (3-5) and the complete relative and absolute stereochemistry of 1 42. The gene cluster responsible for lomaiviticin biosynthesis (lom) was initially identified in S. tropica by deleting the beta-ketosynthase gene in the ST_PKS2 pathway and correlating its loss to the loss of biological activity associated with lomaiviticin 43. The lom locus was subsequently shown to occur in most strains of S. pacifica in addition to all S. tropica strains for which genome sequences are available 44. It was independently characterized in S. pacifica by the Balskus group 39 who established that the associated type II polyketide synthase (PKS) supports a new strategy for propionyl starter unit generation previously observed in type I PKS pathways 45. Numerous groups have also established synthetic routes to different portions of the lomaiviticin aglycone with Herzon and coworkers completing the first enantioselective synthesis of the aglycone 46. To date, the total synthesis of lomaiviticin has not been reported.
Table 1.
Secondary metabolites reported from Salinispora spp.
| No. | Speciesa | Compound | Biosynthetic origin | Novelty | Activity (target) | Ref. |
|---|---|---|---|---|---|---|
| 1 | S. tropica | salinosporamide A | PKS-NRPS | new | proteasome | 47 |
| 2 | S. tropica | sporolide A | ePKS | new | reverse transcriptased | 85 |
| 3 | S. tropica | salinilactam | type I PKS | new | ND | 35 |
| 4 | S. tropica | sioxanthin | terpene | new | ND | 123 |
| 5 | S. tropica | antiprotealide | PKS-NRPS | new | proteasome | 57 |
| 6 | S. pacifica | pacificanone A | type I PKS | new | ND | 121 |
| 7 | S. pacifica | salinipyrone A | type I PKS | new | ND | 121 |
| 8 | S. pacifica | cyanosporoside A | PKSe | new | ND | 93 |
| 9 | S. pacifica | lomaiviticin A | type II PKS | new | cytotoxic (DNA) | 28 |
| 10 | S. pacifica | enterocin | type II PKS | known | antibiotic | 129 |
| 11 | S. arenicola | saliniketal Ab | type I PKS | new | ornithine decarboxylase | 99 |
| 12 | S. arenicola | arenicolide A | type I PKS | new | ND | 96 |
| 13 | S. arenicola | saliniquinone | type II PKS | new | cytotoxic | 116 |
| 14 | S. arenicola | cyclomarin A | NRPS | known | anti-inflammatory | 117 |
| 15 | S. arenicola | cyclomarazinec | NRPS | new | ND | 118 |
| 16 | S. arenicola | arenimycin | NRPS | new | antibiotic | 113 |
| 17 | S. arenicola | arenamide A | type II PKS | new | anti-inflammatory (NFκB) | 98 |
| 18 | S. arenicola | staurosporines | alkaloid | known | protein kinase | 21 |
| 19 | S. arenicola | isopimara-8,15-dien-19-ol | terpene | new | ND | 124 |
| 20 | S. arenicola | rifamycin B | type I PKS | known | RNA polymerase | 104 |
| 21 | S. arenicola | mevinolin | PKS | known | HMG-CoA reductase | 133 |
| 22 | St, Sa, and Sp | desferioxamine B | NRPS | known | iron chelator | 132 |
| 23 | St, Sa, and Sp | lymphostin | NRPS-PKS | known | immunosuppressant | 127 |
Original report of compound detection from Salinispora spp.
Rifamycin synthase intermediate.
Cyclomarin synthetase intermediate.
Predicted,
e = enediyne, ND = not determined.
3.1 Salinosporamides
Two years after the discovery of the lomaiviticins, salinosporamide A (6) was reported from S. tropica strain CNB-392 47. Salinosporamide A garnered immediate attention due to the rarity of the fused γ-lactam-β-lactone bicyclic ring system and its potent activity against the 20S proteasome, which became a validated target for cancer chemotherapy following the approval of bortezomib (Velcade®) for the treatment of multiple myeloma and other cancers 48. At the time, the most closely related compound was clasto-lactacystin-β-lactone (7), also known as omuralide 49, a transformation product of lactacystin, which was originally discovered by Ōmura and coworkers from a Streptomyces sp. 50. These compounds share the same ring system however 4 lacks the methyl group at the C-3 ring junction, has a methyl instead of a chloroethyl at C-2, and an isopropyl instead of a cyclohexene at C-5. A crystal structure of 6 bound to the yeast 20S proteasome revealed that the β-lactone carbonyl reacts with the catalytic N-terminal threonine to form an irreversible, covalent adduct 51. A subsequent intra-molecular reaction between the C3-O and the C-2 side chain of 1 yields a cyclic tetrahydrofuran ring that blocks access to nucleophilic water into the binding pocket thus contributing to the irreversible binding of the compound to the proteasome. Subsequent studies revealed a redundant proteasome β-subunit within the salinosporamide gene cluster that confers resistance to this compound in the native organism 52. Salinisporamide A was developed by Nereus Pharmaceuticals (San Diego) under the names NPI-0052 and marizomib, undergoing extensive pre-clinical evaluation 53 and a variety of phase I and phase Ib clinical trails 54. It is currently undergoing additional phase I clinical trials via a license from the University of California San Diego to Triphase Accelerator Corp. (http://triphaseco.com/pipeline/). Total syntheses were reported by both the Corey and Danishefsky groups in 2005 55, 56, and these were followed by numerous other synthetic routes. Despite the synthetic tractability of salinosporamide A, Nereus Pharmaceuticals produced the material used for clinical trails via fermentation 57. The development of optimized fermentation protocols resulted in a number of publications addressing the ionic requirements for Salinispora growth and salinosporamide A production 58–61.
Subsequent studies of S. tropica strain CNB-392 led to the isolation salinosporamides B (8) and C (9) along with five related compounds that were determined to be artifacts of the isolation process 62. The structure of salinosporamide B differs from A simply by the loss of chlorine, however the >500-fold loss in cytotoxicity associated with 8 provided the first evidence that the chloroethyl substituent plays a major role in the biological activity of 6. During the course of purifying multi-gram quantities of 6 for clinical trails, researchers at Nereus Pharmaceuticals isolated seven additional compounds in the salinosporamide series (salinosporamides D–J, 10-16) from S. tropica strain NPS000465 (CNB-476) 63. These compounds largely represent modifications to the C-2 chloroethyl substituent and include bromosalinosporamide (17), which was produced when synthetic sea salts were replaced with sodium bromide in the fermentation medium. A more detailed analysis of salinosporamide structure activity relationships revealed that replacement of the chloroethyl group with non-halogenated substituents was associated with a marked reduction in potency while halogen exchange was well tolerated 64.
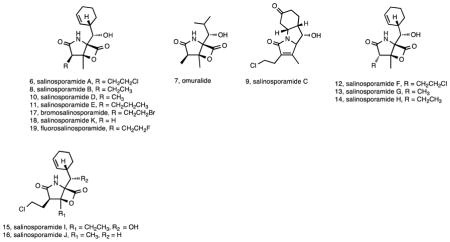
As of this writing, the most recent natural product reported in the salinosporamide series from a Salinispora spp. is salinosporamide K (18). This compound was discovered by genome mining following the surprising observation of a biosynthetic pathway related to that reported for salinosporamide A in S. pacifica strain CNT-133 65. This pathway lacked the genes associated with the biosynthesis of the chloroethyl substituent in salinosporamide A and as predicted yielded a product that lacked substitution at the C-2 position. In a follow-up analysis of 61 S. pacifica strains, 15 tested positive for the sal pathway and salinosporamide K production was confirmed in one additional strain 66. Phylogenetic analyses were used to infer that the sal pathway was acquired prior to the S. tropica - S. pacifica split and subsequently evolved independently in these two species with gene deletion accounting for the loss of the chloroethylmalonyl-CoA pathway in S. pacifica 66. These studies, along with the discovery of the related compounds cinnabaramides A–G and the associated biosynthetic pathway from a Streptomyces sp. 67, 68, provided some of the first evidence of the evolutionary complexity associated with secondary metabolism in Salinispora species. The sal pathway has more recently been detected in a limited number of S. arenicola strains 33, 44 although compound production appears to be very low in this species (unpublished data).
The structure of salinosporamide A belies its biosynthetic complexity. While the C-2 ethyl group in the deschloro-analog originates from butyrate via ethylbutyryl-CoA 69, the chloroethyl group in salinosporamide A is derived from a new chlorination mechanism driven by the S-adenosyl-L-methionine-dependent chlorinase SalL 70. The halogenated product 5-chloro-5-deoxyadenosine is then converted in a seven-step route to chloroethylmalonyl-CoA, which acts as an unprecedented halogenated PKS extender unit in salinosporamide A biosynthesis 71, 72. The biosynthetic pathway to chloroethylmalonyl-CoA is unique to salinosproamide A and has not yet been observed in public DNA sequence databases, thereby supporting the notion that new microbial genera harbor novel biosynthetic processes. Subsequent biosynthetic studies revealed that salinosporamides D and E are alternatively accessed from methylmalonyl-CoA and propylmalonyl-CoA substrates, respectively, with the latter representing a new PKS extender unit derived from an α,β-unsaturated fatty acid 73. Realizing that chloroethylmalonyl-CoA is a dedicated substrate in salinosporamide A biosynthesis, its selective overproduction was achieved by the genetic manipulation of the pathway specific regulatory gene salR2 to increase the production yield of salinosporamide A 74. Salinosporamide’s cyclohexenylalanine residue is also unique among natural products and originates via a newly realized pathway from prephenic acid involving the prephenate decarboxylase SalX 75. How dihydro-4-hydroxyphenylpyruvate is converted into cyclohexenylalanine and how salinosporamide’s β-lactone-γ-lactam bicyclic ring system is enzymatically constructed remain outstanding questions. Resolving the complexities of salinosporamide biosynthesis has provided new insight into the mechanisms of natural product assembly and opportunities to generate new structural diversity via metabolic engineering.
3.2 Engineered salinosporamides
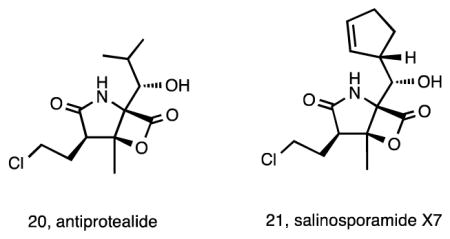
Metabolic engineering has provided unprecedented opportunities to generate new chemical diversity outside of the inherent capabilities of wild-type bacteria 76. The first such efforts with a Salinispora spp. involved a combination of genetic engineering and precursor-directed biosynthesis to yield fluorosalinosporamide (19) 77. By inactivating the SalL chlorinase in S. tropica, which does not accept flouride, and adding synthetic 5′-fluorodeoxyadenosine (5′-FDA), a precursor of fluoroacetate production in Streptomyces cattleya 78, it was possible to isolate 19 from a fermentation of the mutant strain. The proteasome inhibition of this compound was intermediate between that of salinosporamide A and the deschloro-analogue, with the increased energy required to break the C–F bond resulting in a reversible interaction with the active site threonine 77. In subsequent studies, it was possible to generate 19 by replacing the salL chlorinase gene in S. tropica with the S. cattleya fluorinase responsible for generating the C–F bond in 5′-FDA 79. Additional bioengineering efforts led to the production of antiprotealide (20) 80, originally produced as a synthetic hybrid between salinosporamide A and omuralide 55. By deleting the salX prephenate decarboxylase, a series of salinosporamide derivatives with diverse natural and unnatural amino acid residues were engineered 81, including antiprotealide and salinosporamide X7 (21), the later of which displayed equal to slightly improved cytotoxic potency compared to salinosporamide A (6). Interestingly, 20 was subsequently shown to be produced as a natural product by S. tropica during the large-scale production of salinosporamide A for clinical trials 57. Engineering approaches continue to hold great promise for the generation of additional new compounds in the salinosporamide series 82. Detailed reviews covering various methods to produce salinosporamides including traditional fermentation, precursor-directed biosynthesis, mutasynthesis, semi-synthesis, and total synthesis provide detail on much of the work that has been done on these compounds 83, 84.
3.3 Other new compounds
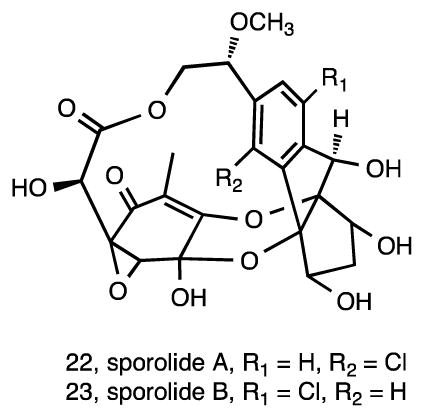
Subsequent studies of S. tropica strain CNB-392 led to the isolation of sporolides A (22) and B (23) 85. It was proposed that these compounds are non-enzymatically derived via a Bergman cyclization reaction from an unstable nine-membered enediyne precursor 86, which was subsequently supported by the analysis of the sporolide (spo) gene cluster 87. Nine-membered enediynes are notoriously difficult to isolate in the absence of an apoprotein due to their lack of stability 88. Although there was no biological activity reported for sporolides A and B, in silico target prediction showed a maximum docking score with HIV-1 reverse transcriptase, with the activity of sporolide B confirmed in vitro using a fluorescent assay 89. A synthesis of the sporolide ring framework has been achieved 90 along with the total synthesis of sporolide B 91, 92.
S. pacifica is the most diverse of the three species in terms of molecular systematics 26. It also maintains considerably greater PKS and nonribosomal peptide synthetase (NRPS) diversity than the other two species 44. In addition to the lomaiviticins, the second structurally novel series of compounds discovered from S. pacifica were the cyclopenta[a]indene glycosides cyanosporaside A (24) and B (25), with the producing strain CNS-103 isolated from a sediment collected in Palau 93. The similarity of the cyanosporaside aglycone to the cycloaromatization product of the Streptomyces-derived nine-membered enediyne compound C-1027 94 led to the hypothesis that, like sporolides A and B, 24 and 25 are also derived from an enediyne precursor. Subsequent studies using a different S. pacifica strain (CNS-143) yielded four additional compounds in the series (cyanosporosides C–F) (26-29) and the first genetic evidence supporting the cyanosporaside’s enediyne biosynthetic origin 95. As might be expected, none of these enediyne cycloaromatization products have the potent cytotoxic activities associated with the predicted parent molecules, none of which have been isolated to date.
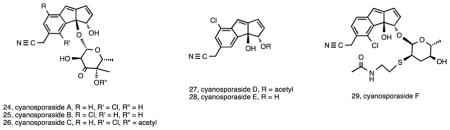
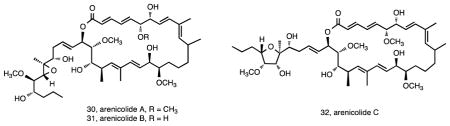
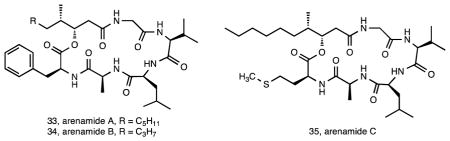
S. arenicola is the most broadly distributed and abundant of the three species 32. It has also been the source of a number of interesting new compounds in addition to some well-known actinomycete secondary metabolites. The first new structures reported from this species were the 26-membered ring macrolides arenicolides A–C (30-32) isolated from strain CNS-005 96. These compounds were discovered using LC-MS based screening with A and B being simple methyl derivatives and C possessing a substituted tetrahydrofuran ring potentially generated from a cyclization of the epoxide. Three additional S. arenicola strains were found to produce arenicolide A and expression studies linked a specific ketosynthase sequence to its biosynthesis 97. All four of the arenicolide-producing strains originated from separate samples collected around the island of Guam, supporting the concept that location plays an important role in secondary metabolism 44. Genome sequence data and molecular networking (Duncan et al., unpublished data) provided strong circumstantial evidence linking these compounds to the gene cluster identified as PKS28, which was only observed in one of 75 Salinispora genome sequences, further supporting the restricted distribution of this pathway among Salinispora strains. Another group of new compounds that also appear to be rare among S. arenicola strains are the arenamides. These cyclohexadepsipeptides (33-35) were discovered by comparative LC-MS analysis of crude extracts, which revealed that strain CNT-088 produced compounds not previously observed from this species 98. Once isolated, arenamides A and B demonstrated NFκB inhibition and anti-inflammatory activity. To date, this is the only strain from which these compounds have been detected.
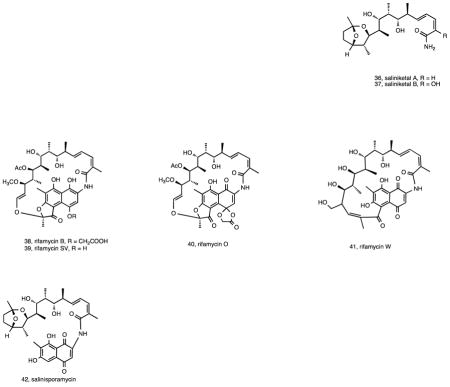
Continued studies of S. arenicola strain CNR-005 along with concurrent studies of strain CNR-059 led to the isolation of the bicyclic polyketides saliniketals A (36) and B (37) 99. Saliniketals A and B were found to inhibit ornithine decarboxylase induction, an important target for the chemoprevention of cancer, with IC50 values of 1.95 and 7.83 μg/mL, respectively. These compounds possess unusual structural features that have inspired at least three total syntheses 100–102. Structural similarities between the saliniketals and the ansa chain of the rifamycin class of antibiotics, which co-occur in the fermentation extract, led to questions about the biosynthetic origin of these compounds, which were ultimately shown to be byproducts of the rifamycin biosynthetic pathway 103. Rifamycins were first reported in Salinispora spp. from the sponge-derived strain M403, which was shown to produce both rifamycin B (38) and SV (39) 104. Subsequent studies included the development of an HPLC-MS-MS method capable of detecting picomolar concentrations of compounds in this class 105 and provided evidence that S. arenicola strains were also capable of producing rifamycins O (40) and W (41) 20. A new antibiotic in the rifamycin series, salinisporamycin (42), was also reported from a sediment-derived S. arenicola strain 106. The ability of rifamycin-producing S. arenicola strains isolated from a marine sponge to inhibit Mycobacterium strains isolated from the same sponge was used to suggest that rifamycins may function in competition against sponge microbial community members 107. Rifamycins were shown to be produced throughout the S. arenicola growth cycle, however the amount produced was time 108 and salinity 109 dependent with rifamycins S and W achieving maximum concentrations after 29 days 110.
As part of our efforts to isolate new compounds from Salinispora species using traditional bioassay-guided approaches, we observed that certain compounds were produced in taxonomic-specific patterns. This concept was addressed in more detail and led to the conclusion that some compounds are “species specific”, i.e., they were consistently produced by members of the same species 21. In the case of S. arenicola, species-specific compounds include rifamycins and staurosporines while S. tropica strains consistently produce salinosporamides 21. Similar patterns have not been detected at the species level for S. pacifica. Subsequent analyses of Salinispora genome sequences support these observations 44 while studies of strains derived from Great Barrier Reef sponges confirmed the association between rifamycin production and S. arenicola 20. The fixation of certain gene clusters at the species level, regardless of geographic origin, provides clear evidence of selection and implies that the small molecule products of these pathways are associated with important ecological functions that may help distinguish the three species. However, this is not to imply that other species cannot also produce either identical or related compounds, as is the case for the rifamycins, staurosporines, and salinosporamides 67, 111, 112.
The consistent production of compounds in the rifamycin class by S. arenicola strains creates challenges when screening for antibiotic activity. In an effort to discover new antibiotics from Salinispora sp., an extract library exceeding 2000 testing units was screened against a rifamycin-resistant MRSA strain. Of the six strains that showed promising activity, S. arenicola strain CNR-647 was investigated further leading to the isolation of the new antibiotic arenimycin A (43) 113. This compound belongs to the benzo[α]naphthacene class of antibiotics with an N-linked 2-O-methyl-L-rhamnose residue and provides another example of a compound that is rarely observed among Salinispora isolates. The arenimycins were subsequently linked to the arn gene cluster in S. arenicola strain CNB-527 using a glycogenomic approach, which also led to the discovery of a second compound in the series, arenimycin B (44) 114. Its structure contains a disaccharide unit that is associated with improved MRSA activity. Experimental support linking the arn cluster to the arenimycins comes from the heterologous expression of the cluster from a desert soil eDNA sample, which led to the discovery of two additional compounds in the series 115. Compounds such as the arenimycins, which are rarely observed among Salinispora strains, were originally termed “accessory” metabolites 21 and there was some evidence that their production is linked to specific geographic locations 97. This ultimately led to the hypothesis that strains acquire pathways from the local gene pool and that sampling from diverse locations would increase the likelihood of discovering new secondary metabolites from otherwise highly similar strains 44.
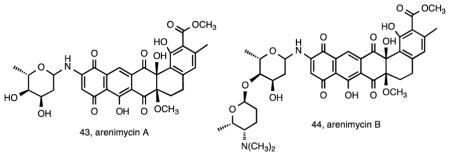
Another example of the metabolic diversity among strains that are clonal at the 16S rRNA level comes from S. arenicola strain CNS-325, which yielded saliniquinones A–F (45-50) 116. These six new anthraquinone-γ-pyrones are highly cytotoxic and represent the first members of the pluramycin class to contain both a terminal olefin and five carbons in the C-2 side chain. A final example of S. arenicola metabolic diversity was the isolation of compounds in the previously described cyclomarin class117 including cyclomarin D (51), a new compound in the series, from strain CNS-205 118. Cyclomarins were produced by only two of 46 Salinispora strains examined as part of a chemotyping study 21 and the pathway was observed in only one of 75 Salinispora genome sequences 44 indicating the rarity of this gene cluster. Interestingly, the cyclomarins were originally reported from a marine-derived Streptomyces sp. 117 suggesting the gene cluster may have been exchanged among these sediment-inhabiting taxa. In the course of studying cyclomarin biosynthesis, two new diketopiperazines cyclomarazines A (52) and B (53) were isolated from S. arenicola CNS-205 and shown to share a common biogenesis with the cyclomarins 118. Functional characterization of the prenyltransferase CymD in the cyclomarin (cym) pathway 119 provided a mechanism to generate unnatural N-alkylated tryptophan derivatives in the cyclomarin series 120.
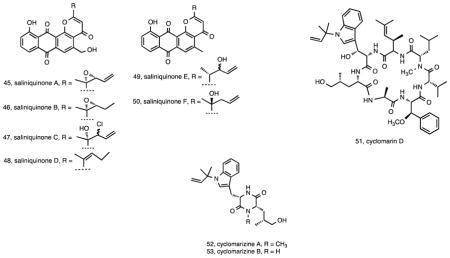
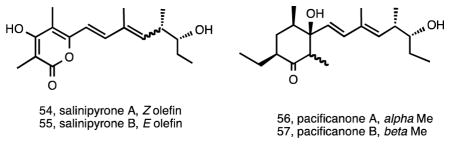
While the 16S rRNA gene has proven too conservative to delineate the three Salinispora spp. using phylogenetic approaches, it is well known that even minor differences in this locus can correspond to major differences in genome content, and thus detailed analyses have been performed to document all Salinispora 16S rRNA sequence variants reported to date 27, 32. These efforts have required a careful monitoring of the position of all variable nucleotides relative to the level of conservation for that region of the gene and have provided a method to distinguish among 16S rRNA sequence variants or “sequence types” based on single nucleotide polymorphisms, each of which has been assigned a letter. These sequence types have proven of value in terms of targeting strains for secondary metabolite production, with one of the first such applications coming from a study of S. pacifica strain CNS-237, which differed from the cyanosporaside-producing S. pacifica strains by three base pairs. CNS-327 was found to produce the new polyketides salinipyrones A (54) and B (55) and pacificanones A (56) and B (57) 121. The pacificanones bear a uniquely substituted cyclohexanone ring, while the similarity in the two structure classes suggests a common type I PKS biosynthetic origin with the differences potentially due to module skipping 121.
4 Genome-aided natural product discovery
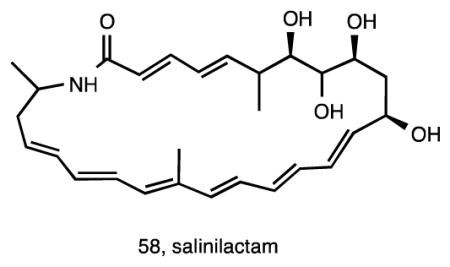
Genome mining has added an important new dimension to the field of natural product discovery 122 and to our understanding of the ecology and evolution of secondary metabolism 44. The first Salinispora genome sequence analyzed was that of S. tropica strain CNB-440, which revealed a total of 17 diverse biosynthetic pathways of which only four had been linked to their respective products 35. Among these was a type I PKS that created problems with the genome assembly due to the highly repetitive nature of the modules comprising the pathway. Using reverse genome mining, the preliminary structure of the macrolactam salinilactam A (58), isolated from this strain, revealed a framework that was consistent with this pathway. Further structure elucidation revealed that salinilactam A was derived from a PKS with at least 10 extension modules, information that ultimately proved critical for the assembly of the pathway and closure of the genome. Once assembled, a bioinformatic analysis of the pathway facilitated the elucidation of the structure, which proved problematic due to its instability and overlapping olefinic NMR signals 35.
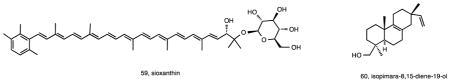
A dominant phenotypic trait associated with Salinispora cultures is their orange pigmentation. While this was assumed to be due to carotenoid production, the biosynthetic origin and structures of these compounds had not been defined. A bioinformatic search of the S. tropica strain CNB-440 genome revealed genes associated with carotenoid biosynthesis in four distinct chromosomal regions 123. Genetic investigations confirmed that, contrary to what is typically observed in bacteria, carotenoid biosynthesis in Salinispora spp. is not due to a single gene cluster. The structure of this pigment was assigned as a new, C-40 carotenoid called sioxanthin (59), which is glycosylated on one end of the molecule and contains an aryl moiety on the other. Glycosylation is unusual among actinomycete carotenoids, and sioxanthin joins a rare group of compounds that possess both polar and non-polar head groups. Additional genome mining efforts targeted a second terpenoid gene cluster that was shared between CNB-440 and CNS-205 36. Called terp1 in S. arenicola strain CNS-205, recombinant expression studies were used demonstrate that this three-gene cluster produces the new diterpenoid isopimara-8,15-diene-19-ol (60) 124. This compound was not observed in cultures of the native strain suggesting the cluster is either inactive or expressed only under certain conditions.
5 Previously described secondary metabolites and new derivatives
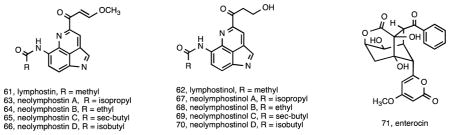
As already mentioned, previously described compounds reported from Salinispora strains include staurosporines and rifamycins, which have been discussed above in the context of species-specific production 21. It is interesting to note that these two secondary metabolites, which are consistently observed from S. arenicola, are among the most potent, biologically active compounds reported from the genus. Additional known compounds reported from Salinispora spp. include lymphostin (61), a potent immunosuppressant originally isolated from a Streptomyces sp. 125. Lymphostin shares structural similarities with the ammosamides, reported from a marine-derived Streptomyces spp. 126, and belongs to the diverse class of pyrroloquinoline natural products. The molecular basis for lymphostin biosynthesis has been determined via interrogation of the lym gene cluster, which includes a uniquely organized modular synthetase 127. Fermentation studies designed to induce lymphostin production also yielded the new derivative lymphostinol (62) along with the eight additional analogues neolymphostin A–D (63-66) and neolymphostinol A–D (67-70). The lym pathway represents a rare example of one that is common to the vast majority of Salinispora strains 44. Another previously described secondary metabolite was predicted based on the detection of a gene cluster with a high level of homology to the enterocin pathway 44 in three S. pacifica strains. Interestingly, and as in the case of the cyclomarins, this pathway has also been observed in a marine-derived Streptomyces sp. 128. Given that enterocin (71) had not been previously reported from Salinispora strains, the pathway was instead heterologously expressed in two different Streptomyces hosts using the recently developed yeast-mediated transformation-associated recombination technique known as TAR cloning 129. This experiment represents the first successful heterologous expression of a Salinispora secondary metabolite gene cluster and opens the door for future studies targeting orphan biosynthetic gene clusters in this genus.
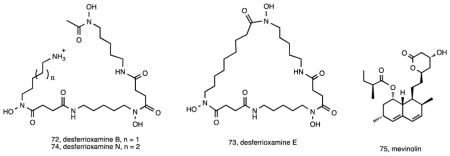
Salinispora genomes are also enriched in biosynthetic gene clusters predicted to encode the biosynthesis of siderophores 36, small, high affinity, iron-chelating compounds secreted by bacteria 130. Gene inactivation experiments suggested that the siderophore associated with the des pathway was the primary iron chelator in both S. tropica and S. arenicola 131. Bioassay-guided fractionation confirmed the production of desferrioxamines B (72) and E (73) in both species, which were formally linked to the des pathway via gene inactivation. Subsequent studies using Ni(II)-based immobilized metal ion chromatography led to the isolation of six additional desferrioxamine siderophores including the new analogue desferrioxamine N (74) 132. A recent report also describes the isolation of the known fungal metabolite mevinolin (75) from two different S. arenicola strains 133, which led the authors to suggest the possibility of horizontal gene transfer between fungi and bacteria. Given that this compound appears to be a common fungal metabolite 134, the most parsimonious explanation is that the gene cluster was acquired by S. arenicola, a hypothesis that can be readily tested by determining its relationship to the iterative type I PKS responsible for mevinolin (lovastatin) biosynthesis in fungi.
6 Conclusions
Since its discovery 23 years ago 22, the genus Salinispora has become a robust model for natural product research. It speaks to the value of assigning formal taxonomic names, which have provided opportunities to address species level differences in secondary metabolism, and the associated deposition of type strains, which have been accessed by researchers worldwide. The acquisition of thousands of strains from global collection sites over a twenty-plus year research endeavor at both the Scripps Institution of Oceanography and other universities around the world has created a resource that may truly be unparalleled in terms of creating opportunities to compare natural product biosynthesis among closely related environmental bacteria. These types of comparisons have begun to reveal the enormous complexities associated with natural product gene evolution 44 and will continue to provide insight into the mechanisms by which bacteria generate new structural diversity. Research on Salinispora spp. has helped clarify the concept that new microbial taxa, especially those inhabiting poorly studied environments such as the world’s oceans, represent a promising resource for natural product discovery. Certainly the ratio of new to known compounds discovered from this genus (Table 1) supports this concept. Interestingly, the first two discoveries, lomaiviticins A and B and salinisporamide A, represent two of the most promising biomedical leads discovered to date from the genus. While this may be largely due to chance, it does suggest that the discovery of new taxa can bring an initial wealth of new chemical structures.
The development of new methodologies in genome sequencing, bioinformatics, molecular genetics, and a better understanding of the biosynthetic principles that govern natural product assembly have driven the ongoing resurgence in natural product research. Coupled with improved mass spectral-based analytical approaches such as peptidogenomics 135, glycogenomics 114, and molecular networking 136, it has become possible to interrogate strains using highly informed approaches that eliminate some of the randomness traditionally associated with natural product discovery. While the initial Salinispora discoveries were largely based on traditional cultivation and screening approaches, the more recent discoveries have been driven by genomics and genetic manipulations. These advances speak to the value of interdisciplinary collaboration and the importance of developing new approaches to natural product discovery. Certainly one of the challenges that remains in the exploitation of this genus, and this is by no means limited to Salinispora spp., is translating the unrealized biosynthetic potential observed in genome sequence data into new chemical discoveries.
Supplementary Material
{kind=link}
Acknowledgments
All authors acknowledge support from the National Institutes of Health (NIH) under grant R01GM085770. PRJ and WF additionally acknowledge NIH grant U01 TW0007401, WF acknowledges NIH grant R37 CA044848, and BM acknowledges NIH grant R01CA127622.
References
- 1.Koehn FE, Carter GT. Nat Rev Drug Discov. 2005;4:206–220. doi: 10.1038/nrd1657. [DOI] [PubMed] [Google Scholar]
- 2.Li JW, Vederas JC. Science. 2009;325:161–165. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- 3.Letzel AC, Pidot SJ, Hertweck C. Nat Prod Rep. 2013;30:392–428. doi: 10.1039/c2np20103h. [DOI] [PubMed] [Google Scholar]
- 4.Fenical W, Jensen PR. Nat Chem Biol. 2006;2:666–673. doi: 10.1038/nchembio841. [DOI] [PubMed] [Google Scholar]
- 5.Williams PG. Trends Biotechnol. 2009;27:45–52. doi: 10.1016/j.tibtech.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 6.Berdy J. J Antibiot. 2005;58:1–26. doi: 10.1038/ja.2005.1. [DOI] [PubMed] [Google Scholar]
- 7.Bull AT, Stach JEM. Trends Microbiol. 2007;15:491–499. doi: 10.1016/j.tim.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 8.Lam KS. Curr Opin Microbiol. 2006;9:245–251. doi: 10.1016/j.mib.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 9.Weyland H. Nature. 1969;223:858–858. doi: 10.1038/223858a0. [DOI] [PubMed] [Google Scholar]
- 10.Takizawa M, Colwell RR, Hill RT. Appl Environ Microbiol. 1993;59:997–1002. doi: 10.1128/aem.59.4.997-1002.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moran MA, Rutherford LT, Hodson RE. Appl Environ Microbiol. 1995;61:3695–3700. doi: 10.1128/aem.61.10.3695-3700.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prieto-Davó A, Fenical W, Jensen PR. Aquat Microbial Ecol. 2008;52:1–11. [Google Scholar]
- 13.Jensen PR, Gontang E, Mafnas C, Mincer TJ, Fenical W. Environ Microbiol. 2005;7:1039–1048. doi: 10.1111/j.1462-2920.2005.00785.x. [DOI] [PubMed] [Google Scholar]
- 14.Khan ST, Tamura T, Takagi M, Shinya K. Int J Syst Evol Microbiol. 2010;60:2775–2779. doi: 10.1099/ijs.0.019869-0. [DOI] [PubMed] [Google Scholar]
- 15.Tian XP, Tang SK, Dong JD, Zhang YQ, Xu LH, Zhang S, Li WJ. Int J Syst Evol Microbiol. 2009;59:948–952. doi: 10.1099/ijs.0.005231-0. [DOI] [PubMed] [Google Scholar]
- 16.Yi H, Schumann P, Sohn K, Chun J. Int J Syst Evol Microbiol. 2004;54:1585–1589. doi: 10.1099/ijs.0.03036-0. [DOI] [PubMed] [Google Scholar]
- 17.Han SK, Nedashkovskaya OI, Mikhailov VV, Kim SB, Bae KS. Int J Syst Evol Microbiol. 2003;53:2061–2066. doi: 10.1099/ijs.0.02627-0. [DOI] [PubMed] [Google Scholar]
- 18.Tian XP, Zhi XY, Qiu YQ, Zhang YQ, Tang SK, Xu LH, Zhang S, Li WJ. Int J Syst Evol Microbiol. 2009;59:222–228. doi: 10.1099/ijs.0.001982-0. [DOI] [PubMed] [Google Scholar]
- 19.Maldonado LA, Fenical W, Jensen PR, Kauffman CA, Mincer TJ, Ward AC, Bull AT, Goodfellow M. Int J Syst Evol Microbiol. 2005;55:1759–1766. doi: 10.1099/ijs.0.63625-0. [DOI] [PubMed] [Google Scholar]
- 20.Bose U, Hewavitharana AK, Vidgen ME, Ng YK, Shaw PN, Fuerst JA, Hodson MP. PloS one. 2014;9:e91488. doi: 10.1371/journal.pone.0091488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jensen PR, Williams PG, Oh DC, Zeigler L, Fenical W. Appl Environ Microbiol. 2007;73:1146–1152. doi: 10.1128/AEM.01891-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jensen P, Dwight R, Fenical W. Appl Environ Microbiol. 1991;57:1102–1108. doi: 10.1128/aem.57.4.1102-1108.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mincer TJ, Jensen PR, Kauffman CA, Fenical W. Appl Environ Microbiol. 2002;68:5005–5011. doi: 10.1128/AEM.68.10.5005-5011.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jensen PR, Mafnas C. Environ Microbiol. 2006;8:1881–1888. doi: 10.1111/j.1462-2920.2006.01093.x. [DOI] [PubMed] [Google Scholar]
- 25.Ahmed L, Jensen P, Freel K, Brown R, Jones A, Kim BY, Goodfellow M. Antonie Van Leeuwenhoek. 2013;103:1069–1078. doi: 10.1007/s10482-013-9886-4. [DOI] [PubMed] [Google Scholar]
- 26.Freel KC, Millan-Aguinaga N, Jensen PR. Appl Environ Microbiol. 2013;79:5997–6005. doi: 10.1128/AEM.00880-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vidgen ME, Hooper JNA, Fuerst JA. Antonie Van Leeuwenhoek. 2012;101:603–618. doi: 10.1007/s10482-011-9676-9. [DOI] [PubMed] [Google Scholar]
- 28.He H, Ding WD, Bernan VS, Richardson AD, Ireland CM, Greenstein M, Ellestad GA, Carter GT. J Am Chem Soc. 2001;123:5362–5363. doi: 10.1021/ja010129o. [DOI] [PubMed] [Google Scholar]
- 29.Kim TK, Garson MJ, Fuerst JA. Environ Microbiol. 2005;7:509–518. doi: 10.1111/j.1462-2920.2005.00716.x. [DOI] [PubMed] [Google Scholar]
- 30.Mincer TJ, Fenical W, Jensen PR. Appl Environ Microbiol. 2005;71:7019–7028. doi: 10.1128/AEM.71.11.7019-7028.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prieto-Davó A, Villarreal-Gómez LJ, Forschner-Dancause S, Bull AT, Stach JE, Smith DC, Rowley DC, Jensen PR. FEMS Microbiol Ecol. 2013;84:510–518. doi: 10.1111/1574-6941.12082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Freel KC, Edlund A, Jensen PR. Environ Microbiol Rep. 2012;14:480–493. doi: 10.1111/j.1462-2920.2011.02641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goo KS, Tsuda M, Ulanova D. Antonie Van Leeuwenhoek. 2014;105:207–219. doi: 10.1007/s10482-013-0067-2. [DOI] [PubMed] [Google Scholar]
- 34.Jensen PR. Microbiol Today. 2013:112–115. [Google Scholar]
- 35.Udwary DW, Zeigler L, Asolkar RN, Singan V, Lapidus A, Fenical W, Jensen PR, Moore BS. Proc Natl Acad Sci. 2007;104:10376–10381. doi: 10.1073/pnas.0700962104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Penn K, Jenkins C, Nett M, Udwary DW, Gontang EA, McGlinchey RP, Foster B, Lapidus A, Podell S, Allen EE, Moore BS, Jensen PR. ISME J. 2009;3:1193–1203. doi: 10.1038/ismej.2009.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Penn K, Jensen P. BMC Genomics. 2012;13:86. doi: 10.1186/1471-2164-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bucarey SA, Penn K, Paul L, Fenical W, Jensen PR. Appl Environ Microbiol. 2012;78:4175–4182. doi: 10.1128/AEM.00577-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Janso JE, Haltli BA, Eustáquio AS, Kulowski K, Waldman AJ, Zha L, Nakamura H, Bernan VS, He H, Carter GT, Koehn FE, Balskus EP. Tetrahedron. 2014;70:4156–4164. doi: 10.1016/j.tet.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Colis LC, Woo CM, Hegan DC, Li Z, Glazer PM, Herzon SB. Nat Chem. 2014;6:504–510. doi: 10.1038/nchem.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nicolaou K, Dai WM. Angewandte Chemie Inter Ed. 1991;30:1387–1416. [Google Scholar]
- 42.Woo CM, Beizer NE, Janso JE, Herzon SB. J Am Chem Soc. 2012;134:15285–15288. doi: 10.1021/ja3074984. [DOI] [PubMed] [Google Scholar]
- 43.Kersten RD, Lane AL, Nett M, Richter TKS, Duggan BM, Dorrestein PC, Moore BS. ChemBioChem. 2013;14:955–962. doi: 10.1002/cbic.201300147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ziemert N, Lechner A, Wietz M, Millan-Aguinaga N, Chavarria KL, Jensen PR. Proc Natl Acad Sci. 2014;111:E1130–E1139. doi: 10.1073/pnas.1324161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Waldman AJ, Balskus EP. Organic Letters. 2014;16:640–643. doi: 10.1021/ol403714g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herzon SB, Lu L, Woo CM, Gholap SL. J Am Chem Soc. 2011;133:7260–7263. doi: 10.1021/ja200034b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W. Angew Chem Int Ed. 2003;42:355–357. doi: 10.1002/anie.200390115. [DOI] [PubMed] [Google Scholar]
- 48.Orlowski RZ, Kuhn DJ. Clin Cancer Res. 2008;14:1649–1657. doi: 10.1158/1078-0432.CCR-07-2218. [DOI] [PubMed] [Google Scholar]
- 49.Corey E, Li WDZ. Chem Pharm Bull Tokyo. 1999;47:1–10. doi: 10.1248/cpb.47.1. [DOI] [PubMed] [Google Scholar]
- 50.Smura, Matsuzaki K, Tomoko F, Kosuge K, Furuya T, Fujita S, Nakagawa A. J Antibiot. 1991;44:117–118. doi: 10.7164/antibiotics.44.117. [DOI] [PubMed] [Google Scholar]
- 51.Groll M, Huber R, Potts BCM. J Am Chem Soc. 2006;128:5136–5141. doi: 10.1021/ja058320b. [DOI] [PubMed] [Google Scholar]
- 52.Kale AJ, McGlinchey RP, Lechner A, Moore BS. ACS Chemical Biol. 2011;6:1257–1264. doi: 10.1021/cb2002544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Potts BC, Albitar MX, Anderson KC, Baritaki S, Berkers C, Bonavida B, Chandra J, Chauhan D, Cusack JC, Fenical W, Ghobrial IM, Groll M, Jensen PR, Lam KS, Lloyd GK, McBride W, McConkey DJ, Miller CP, Neuteboom STC, Oki Y, Ovaa H, Pajonk F, Richardson PG, Roccaro AM, Sloss CM, Spear MA, Valashi E, Younes A, Palladino MA. Curr Cancer Drug Targets. 2011;11:254–284. doi: 10.2174/156800911794519716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fenical W, Jensen PR, Palladino MA, Lam KS, Lloyd GK, Potts BC. Bioorg Med Chem. 2009;17:2175–2180. doi: 10.1016/j.bmc.2008.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reddy LR, Fournier J-F, Subba Reddy BV, Corey EJ. J Am Chem Soc. 2005;127:8974–8976. doi: 10.1021/ja052376o. [DOI] [PubMed] [Google Scholar]
- 56.Endo A, Danishefsky SJ. J Am Chem Soc. 2005;127:8298–8299. doi: 10.1021/ja0522783. [DOI] [PubMed] [Google Scholar]
- 57.Manam RR, Macherla VR, Tsueng G, Dring CW, Weiss J, Neuteboom STC, Lam KS, Potts BC. J Nat Prod. 2009;72:295–297. doi: 10.1021/np800578e. [DOI] [PubMed] [Google Scholar]
- 58.Tsueng G, Lam KS. Appl Microbiol Biotechnol. 2010;86:1525–1534. doi: 10.1007/s00253-009-2424-7. [DOI] [PubMed] [Google Scholar]
- 59.Tsueng G, Lam K. Appl Microbiol Biotechnol. 2008;78:821–826. doi: 10.1007/s00253-008-1357-x. [DOI] [PubMed] [Google Scholar]
- 60.Tsueng G, Lam KS. Appl Microbiol Biotechnol. 2008;80:873–880. doi: 10.1007/s00253-008-1614-z. [DOI] [PubMed] [Google Scholar]
- 61.Tsueng G, Teisan S, Lam KS. Appl Microbiol Biotechnol. 2008;78:827–832. doi: 10.1007/s00253-008-1358-9. [DOI] [PubMed] [Google Scholar]
- 62.Williams PG, Buchanan GO, Feling RH, Kauffman CA, Jensen PR, Fenical W. J Org Chem. 2005;70:6196–6203. doi: 10.1021/jo050511+. [DOI] [PubMed] [Google Scholar]
- 63.Reed KA, Manam RR, Mitchell SS, Xu J, Teisan S, Chao TH, Deyanat-Yazdi G, Neuteboom STC, Lam KS, Potts BCM. J Nat Prod. 2007;70:269–276. doi: 10.1021/np0603471. [DOI] [PubMed] [Google Scholar]
- 64.Macherla VR, Mitchell SS, Manam RR, Reed KA, Chao TH, Nicholson B, Deyanat-Yazdi G, Mai B, Jensen PR, Fenical W, Neuteboom STC, Lam KS, Palladino MA, Potts BCM. J Med Chem. 2005;48:3684–3687. doi: 10.1021/jm048995+. [DOI] [PubMed] [Google Scholar]
- 65.Eustáquio AS, Nam SJ, Penn K, Lechner A, Wilson MC, Fenical W, Jensen PR, Moore BS. ChemBioChem. 2011;12:61–64. doi: 10.1002/cbic.201000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Freel KC, Nam SJ, Fenical W, Jensen PR. Appl Environ Microbiol. 2011;77:7261–7270. doi: 10.1128/AEM.05943-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stadler M, Bitzer J, Mayer-Bartschmid A, Müller H, Benet-Buchholz J, Gantner F, Tichy HV, Reinemer P, Bacon KB. J Nat Prod. 2007;70:246–252. doi: 10.1021/np060162u. [DOI] [PubMed] [Google Scholar]
- 68.Rachid S, Huo L, Herrmann J, Stadler M, Köpcke B, Bitzer J, Müller R. Chem Bio Chem. 2011;12:922–931. doi: 10.1002/cbic.201100024. [DOI] [PubMed] [Google Scholar]
- 69.Beer LL, Moore BS. Org Lett. 2007;9:845–848. doi: 10.1021/ol063102o. [DOI] [PubMed] [Google Scholar]
- 70.Eustaquio AS, Pojer F, Noel JP, Moore BS. Nat Chem Biol. 2008;4:69–74. doi: 10.1038/nchembio.2007.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Eustáquio AS, McGlinchey RP, Liu Y, Hazzard C, Beer LL, Florova G, Alhamadsheh MM, Lechner A, Kale AJ, Kobayashi Y, Reynolds KA, Moore BS. Proc Nat Acad Sci. 2009;106:12295–12300. doi: 10.1073/pnas.0901237106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kale AJ, McGlinchey RP, Moore BS. J Biol Chem. 2010;285:33710–33717. doi: 10.1074/jbc.M110.153833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu Y, Hazzard C, Eustáquio AS, Reynolds KA, Moore BS. J Am Chem Soc. 2009;131:10376–10377. doi: 10.1021/ja9042824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lechner A, Eustáquio A, Gulder TAM, Hafner M, Moore BS. Chem Biol. 2011;18:1527–1536. doi: 10.1016/j.chembiol.2011.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mahlstedt S, Fielding EN, Moore BS, Walsh CT. Biochemistry. 2010;49:9021–9023. doi: 10.1021/bi101457h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Khosla C, Keasling JD. Nat Rev Drug Discov. 2003;2:1019–1025. doi: 10.1038/nrd1256. [DOI] [PubMed] [Google Scholar]
- 77.Eustáquio AS, Moore BS. Angew Chem Int Ed. 2008;47:3936–3938. doi: 10.1002/anie.200800177. [DOI] [PubMed] [Google Scholar]
- 78.Dong C, Huang F, Deng H, Schaffrath C, Spencer JB, O’Hagan D, Naismith JH. Nature. 2004;427:561–565. doi: 10.1038/nature02280. [DOI] [PubMed] [Google Scholar]
- 79.Eustáquio AS, O’Hagan D, Moore BS. J Nat Prod. 2010;73:378–382. doi: 10.1021/np900719u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McGlinchey RP, Nett M, Eustáquio AS, Asolkar RN, Fenical W, Moore BS. J Am Chem Soc. 2008;130:7822–7823. doi: 10.1021/ja8029398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nett M, Gulder TA, Kale AJ, Hughes CC, Moore BS. J Med Chem. 2009;52:6163–6167. doi: 10.1021/jm901098m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nett M, Moore BS. Pure Appl Chem. 2009;81:1075–1081. [Google Scholar]
- 83.Potts BC, Lam KS. Mar Drugs. 2010;8:835–880. doi: 10.3390/md8040835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gulder TAM, Moore BS. Angewandte Chemie Inter Ed. 2010;49:9346–9367. doi: 10.1002/anie.201000728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Buchanan GO, Williams PG, Feling RH, Kauffman CA, Jensen PR, Fenical W. Org Lett. 2005;7:2731–2734. doi: 10.1021/ol050901i. [DOI] [PubMed] [Google Scholar]
- 86.Perrin CL, Rodgers BL, O’Connor JM. J Am Chem Soc. 2007;129:4795–4799. doi: 10.1021/ja070023e. [DOI] [PubMed] [Google Scholar]
- 87.McGlinchey RP, Nett M, Moore BS. J Am Chem Soc. 2008;130:2406–2407. doi: 10.1021/ja710488m. [DOI] [PubMed] [Google Scholar]
- 88.Jean M, Tomasi S, Van De Weghe P. Org Biomol Chem. 2012;10:7453–7456. doi: 10.1039/c2ob26033f. [DOI] [PubMed] [Google Scholar]
- 89.Dineshkumar K, Aparna V, Madhuri KZ, Hopper W. Chemical Biol Drug Design. 2014;83:350–361. doi: 10.1111/cbdd.12252. [DOI] [PubMed] [Google Scholar]
- 90.Nicolaou KC, Wang J, Tang Y. Angew Chem Inter Ed. 2008;47:1432–1435. doi: 10.1002/anie.200705334. [DOI] [PubMed] [Google Scholar]
- 91.Nicolaou KC, Tang Y, Wang J. Angew Chem Inter Ed. 2009;121:3501–3505. [Google Scholar]
- 92.Nicolaou KC, Wang J, Tang Y, Botta L. J Am Chem Soc. 2010;132:11350–11363. doi: 10.1021/ja1048994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Oh DC, Williams PG, Kauffman CA, Jensen PR, Fenical W. Org Lett. 2006;8:1021–1024. doi: 10.1021/ol052686b. [DOI] [PubMed] [Google Scholar]
- 94.Kenichiro Y, Minami Y, Azuma R, Saeki M, Otani T. Tetrahedron Lett. 1993;34:2637–2640. [Google Scholar]
- 95.Lane AL, Nam SJ, Fukuda T, Yamanaka K, Kauffman CA, Jensen PR, Fenical W, Moore BS. J Am Chem Soc. 2013;135:4171–4174. doi: 10.1021/ja311065v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Williams PG, Miller ED, Asolkar RN, Jensen PR, Fenical W. J Org Chem. 2007;72:5025–5034. doi: 10.1021/jo061878x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Edlund A, Loesgen S, Fenical W, Jensen PR. Appl Environ Microbiol. 2011:611–611. doi: 10.1128/AEM.00611-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Asolkar RN, Freel KC, Jensen PR, Fenical W, Kondratyuk TP, Park EJ, Pezzuto JM. J Nat Prod. 2009;72:396–402. doi: 10.1021/np800617a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Williams PG, Asolkar RN, Kondratyuk T, Pezzuto JM, Jensen PR, Fenical W. J Nat Prod. 2007;70:83–88. doi: 10.1021/np0604580. [DOI] [PubMed] [Google Scholar]
- 100.Liu J, De Brabander JK. J Am Chem Soc. 2009;131:12562–12563. doi: 10.1021/ja9061757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Paterson I, Razzak M, Anderson EA. Org Lett. 2008;10:3295–3298. doi: 10.1021/ol801148d. [DOI] [PubMed] [Google Scholar]
- 102.Yadav JS, Samad Hossain S, Madhu M, Mohapatra DK. J Org Chem. 2009;74:8822–8825. doi: 10.1021/jo901913h. [DOI] [PubMed] [Google Scholar]
- 103.Wilson MC, Gulder TAM, Mahmud T, Moore BS. J Am Chem Soc. 2010;132:12757–12765. doi: 10.1021/ja105891a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kim TK, Hewavitharana AK, Shaw PN, Fuerst JA. Appl Environ Microbiol. 2006;72:2118–2125. doi: 10.1128/AEM.72.3.2118-2125.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hewavitharana AK, Shaw PN, Kim TK, Fuerst JA. J Chrom B. 2007;852:362–366. doi: 10.1016/j.jchromb.2007.01.042. [DOI] [PubMed] [Google Scholar]
- 106.Matsuda S, Adachi K, Matsuo Y, Nukina M, Shizuri Y. J Antibiot. 2009;62:519–526. doi: 10.1038/ja.2009.75. [DOI] [PubMed] [Google Scholar]
- 107.Izumi H, Gauthier MEA, Degnan BM, Ng YK, Hewavitharana AK, Shaw PN, Fuerst JA. FEMS Microbiol Lett. 2010;313:33–40. doi: 10.1111/j.1574-6968.2010.02118.x. [DOI] [PubMed] [Google Scholar]
- 108.Ng Y, Hewavitharana A, Webb R, Shaw PN, Fuerst J. Appl Microbiol Biotechnol. 2013;97:3097–3108. doi: 10.1007/s00253-012-4479-0. [DOI] [PubMed] [Google Scholar]
- 109.Ng YK, Hodson MP, Hewavitharana AK, Bose U, Shaw PN, Fuerst J. J Appl Microbiol. 2014 doi: 10.1111/jam.12507. [DOI] [PubMed] [Google Scholar]
- 110.Bose U, Hewavitharana AK, Ng YK, Shaw PN, Fuerst JA, Hodson MP. Mar Drugs. 2015;13:249–266. doi: 10.3390/md13010249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Furusaki A, Hashiba N, Matsumoto T, Hirano A, Iwai Y, Omura S. J Chem Soc Chem Comm. 1978:800–801. [Google Scholar]
- 112.Floss H, Yu T. Chem Rev. 2005;105:621–632. doi: 10.1021/cr030112j. [DOI] [PubMed] [Google Scholar]
- 113.Asolkar RN, Kirkland TN, Jensen PR, Fenical W. J Antibiot. 2010;63:37–39. doi: 10.1038/ja.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kersten RD, Ziemert N, Gonzalez DJ, Duggan BM, Nizet V, Dorrestein PC, Moore BS. Proc Natl Acad Sci. 2013;110:4407–4416. doi: 10.1073/pnas.1315492110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kang H-S, Brady SF. J Am Chem Soc. 2014 doi: 10.1021/ja510606j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Murphy BT, Narender T, Kauffman CA, Woolery M, Jensen PR, Fenical W. Aust J Chem. 2010;63:929–934. doi: 10.1071/CH10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Renner MK, Shen YC, Cheng XC, Jensen PR, Frankmoelle W, Kauffman CA, Fenical W, Lobkovsky E, Clardy J. J Am Chem Soc. 1999;121:11273–11276. [Google Scholar]
- 118.Schultz AW, Oh DC, Carney JR, Williamson RT, Udwary DW, Jensen PR, Gould SJ, Fenical W, Moore BS. J Am Chem Soc. 2008;130:4507–4516. doi: 10.1021/ja711188x. [DOI] [PubMed] [Google Scholar]
- 119.Qian Q, Schultz AW, Moore BS, Tanner ME. Biochemistry. 2012;51:7733–7739. doi: 10.1021/bi3009054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Schultz AW, Lewis CA, Luzung MR, Baran PS, Moore BS. J Nat Prod. 2010;73:373–377. doi: 10.1021/np9006876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Oh DC, Gontang EA, Kauffman CA, Jensen PR, Fenical W. J Nat Prod. 2008;71:570–575. doi: 10.1021/np0705155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Corre C, Challis GL. Nat Prod Rep. 2009;26:977–986. doi: 10.1039/b713024b. [DOI] [PubMed] [Google Scholar]
- 123.Richter TKS, Hughes CC, Moore BS. Environ Microbiol. 2014 doi: 10.1111/1462-2920.12669. [DOI] [Google Scholar]
- 124.Xu M, Hillwig ML, Lane AL, Tiernan MS, Moore BS, Peters RJ. J Nat Prod. 2014;77:2144–2147. doi: 10.1021/np500422d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Aotani Y, Nagata H, Yoshida M. J Antibiot. 1997;50:543–545. doi: 10.7164/antibiotics.50.543. [DOI] [PubMed] [Google Scholar]
- 126.Hughes CC, MacMillan JB, Gaudencio SP, Jensen PR, Fenical W. Angew Chem Int Ed. 2009;48:725–727. doi: 10.1002/anie.200804890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Miyanaga A, Janso JE, McDonald L, He M, Liu H, Barbieri L, Eustaquio AS, Fielding EN, Carter GT, Jensen PR, Feng X, Leighton M, Koehn FE, Moore BS. J Am Chem Soc. 2011;133:13311–13313. doi: 10.1021/ja205655w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Piel J, Hertweck C, Shipley PR, Hunt DM, Newman MS, Moore BS. Chem & Biol. 2000;7:943–955. doi: 10.1016/s1074-5521(00)00044-2. [DOI] [PubMed] [Google Scholar]
- 129.Bonet B, Teufel R, Crüsemann M, Ziemert N, Moore BS. J Nat Prod. 2014 doi: 10.1021/np500664q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hider RC, Kong X. Nat Prod Rep. 2010;27:637–657. doi: 10.1039/b906679a. [DOI] [PubMed] [Google Scholar]
- 131.Roberts AA, Schultz AW, Kersten RD, Dorrestein PC, Moore BS. FEMS Microbiol Lett. 2012;335:95–103. doi: 10.1111/j.1574-6968.2012.02641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ejje N, Soe CZ, Gu J, Codd R. Metallomics. 2013;5:1519–1528. doi: 10.1039/c3mt00230f. [DOI] [PubMed] [Google Scholar]
- 133.Bose U, Hodson MP, Shaw PN, Fuerst JA, Hewavitharana AK. Biomed Chrom. 2014;28:1163–1166. doi: 10.1002/bmc.3138. [DOI] [PubMed] [Google Scholar]
- 134.Shindia A. Folia Microbiol. 1997;42:477–480. doi: 10.1007/BF02826557. [DOI] [PubMed] [Google Scholar]
- 135.Kersten RD, Yang YL, Xu Y, Cimermancic P, Nam SJ, Fenical W, Fischbach MA, Moore BS, Dorrestein PC. Nat Chem Biol. 2011;7:794–802. doi: 10.1038/nchembio.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bouslimani A, Sanchez LM, Garg N, Dorrestein PC. Nat Prod Rep. 2014;31:718–729. doi: 10.1039/c4np00044g. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.